
Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations

 , ,
, ,  , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
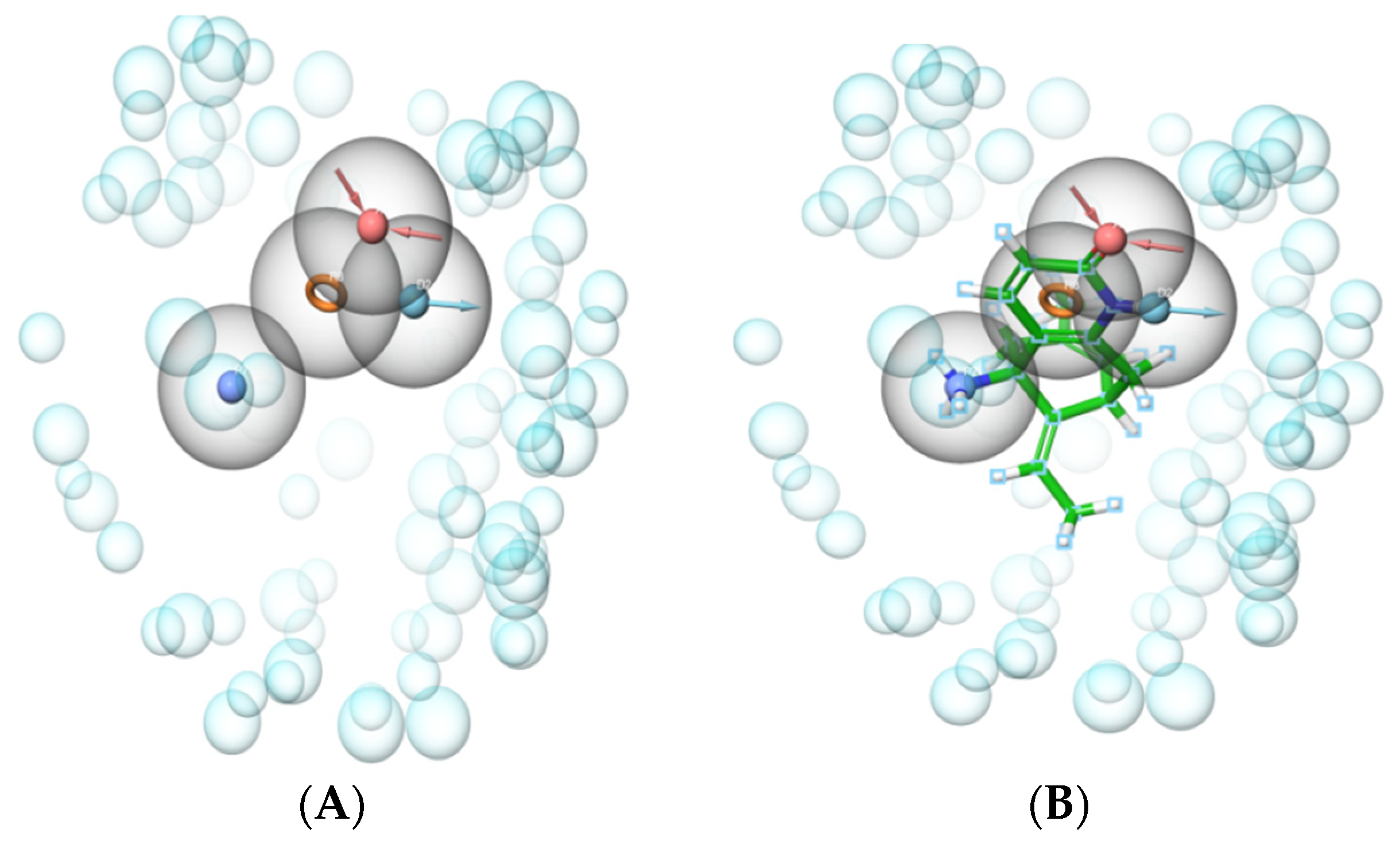
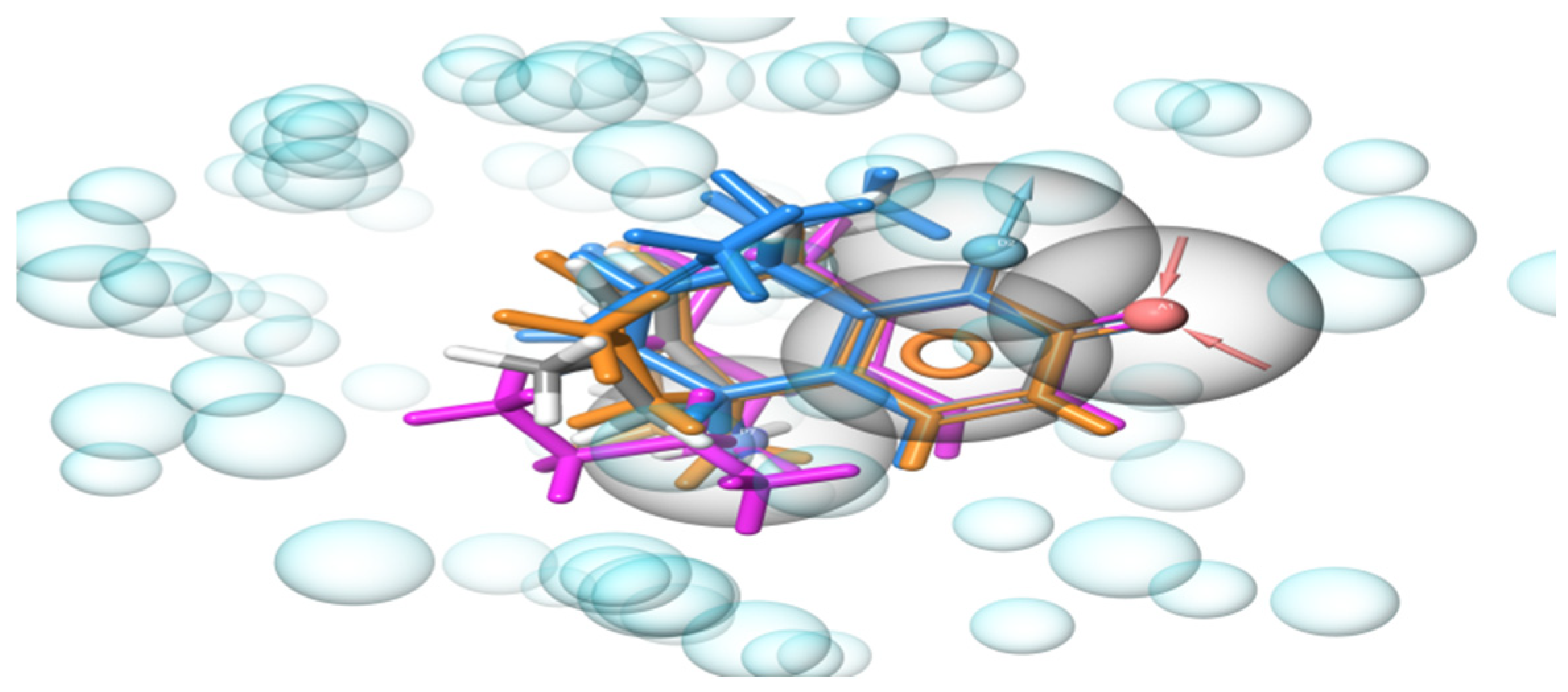
2.1. Development of e-Pharmacophore Hypotheses
2.2. Enrichment Calculations
2.3. Screening e-Pharmacophore-Based Virtual Screening
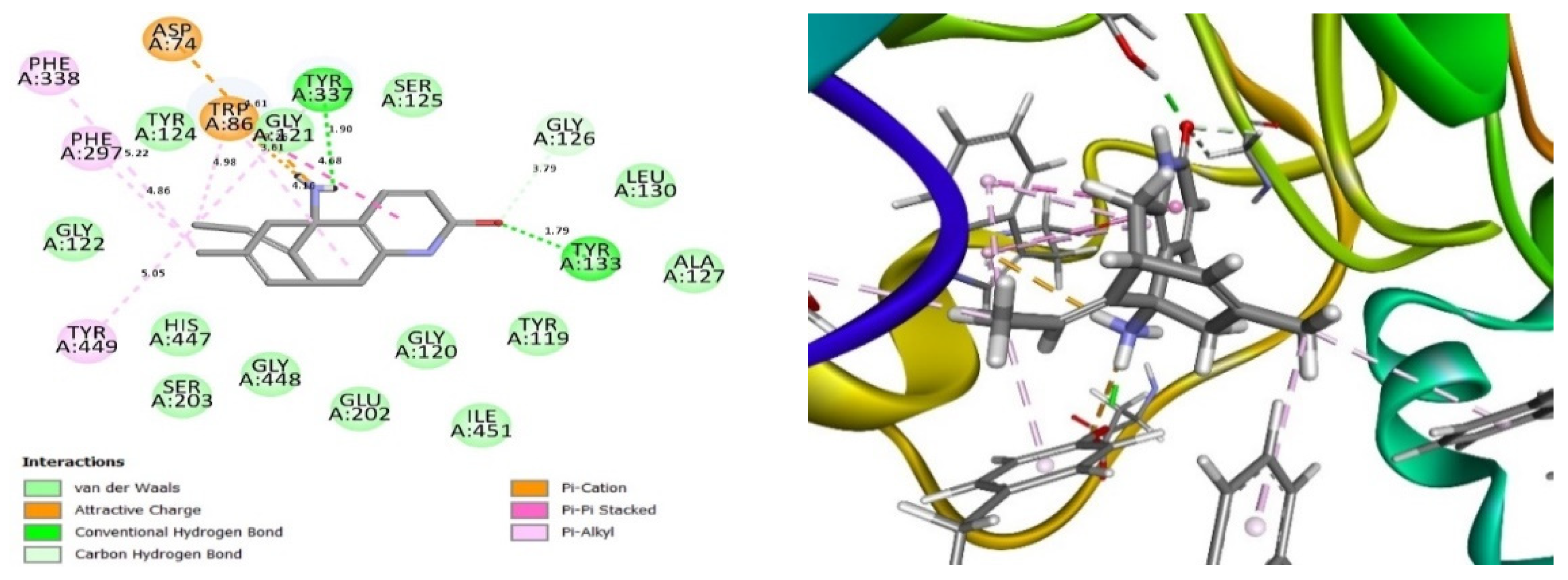
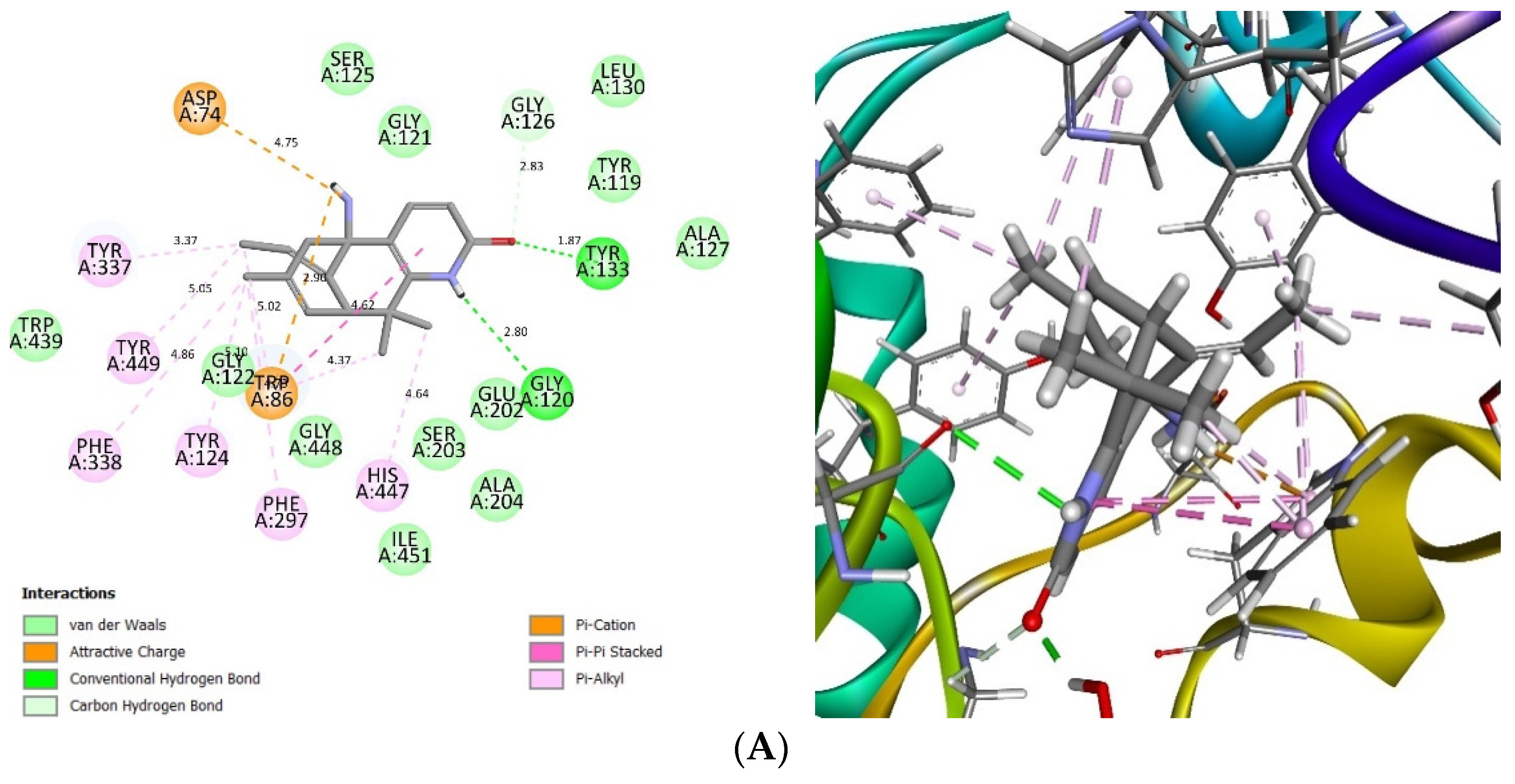
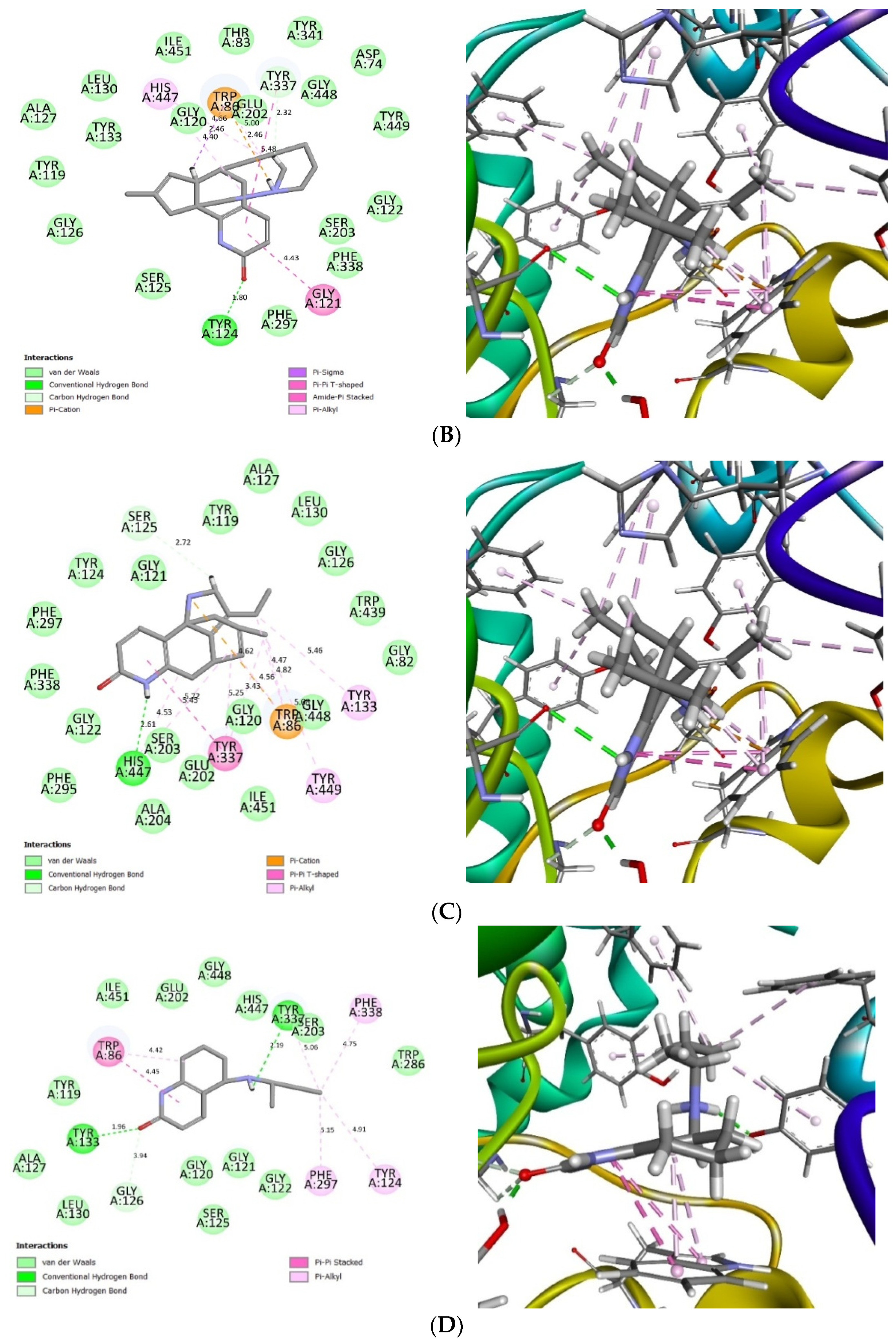
2.4. Glide Ligand Docking
2.5. In Silico ADMET Predictions
2.6. MD Simulations
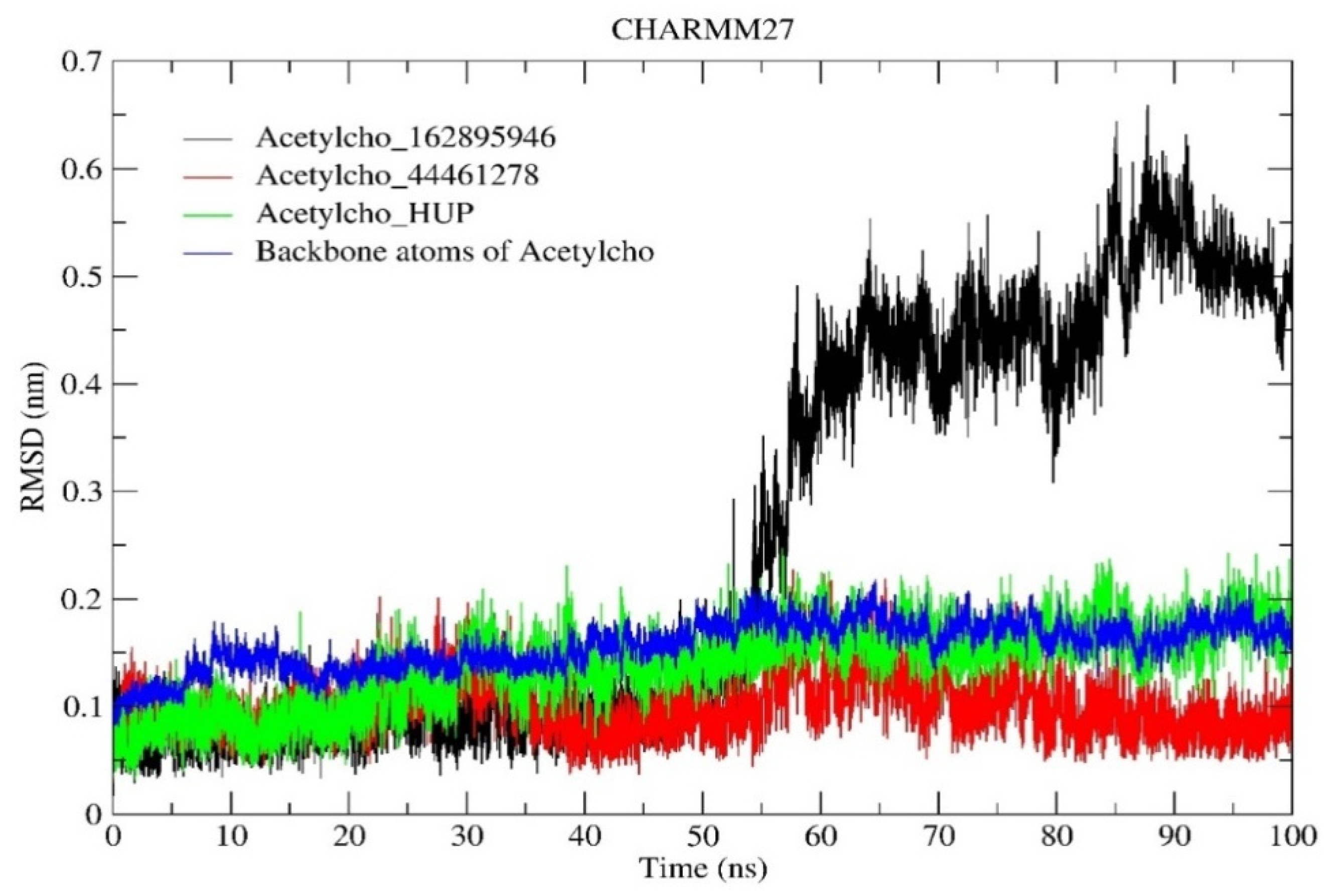
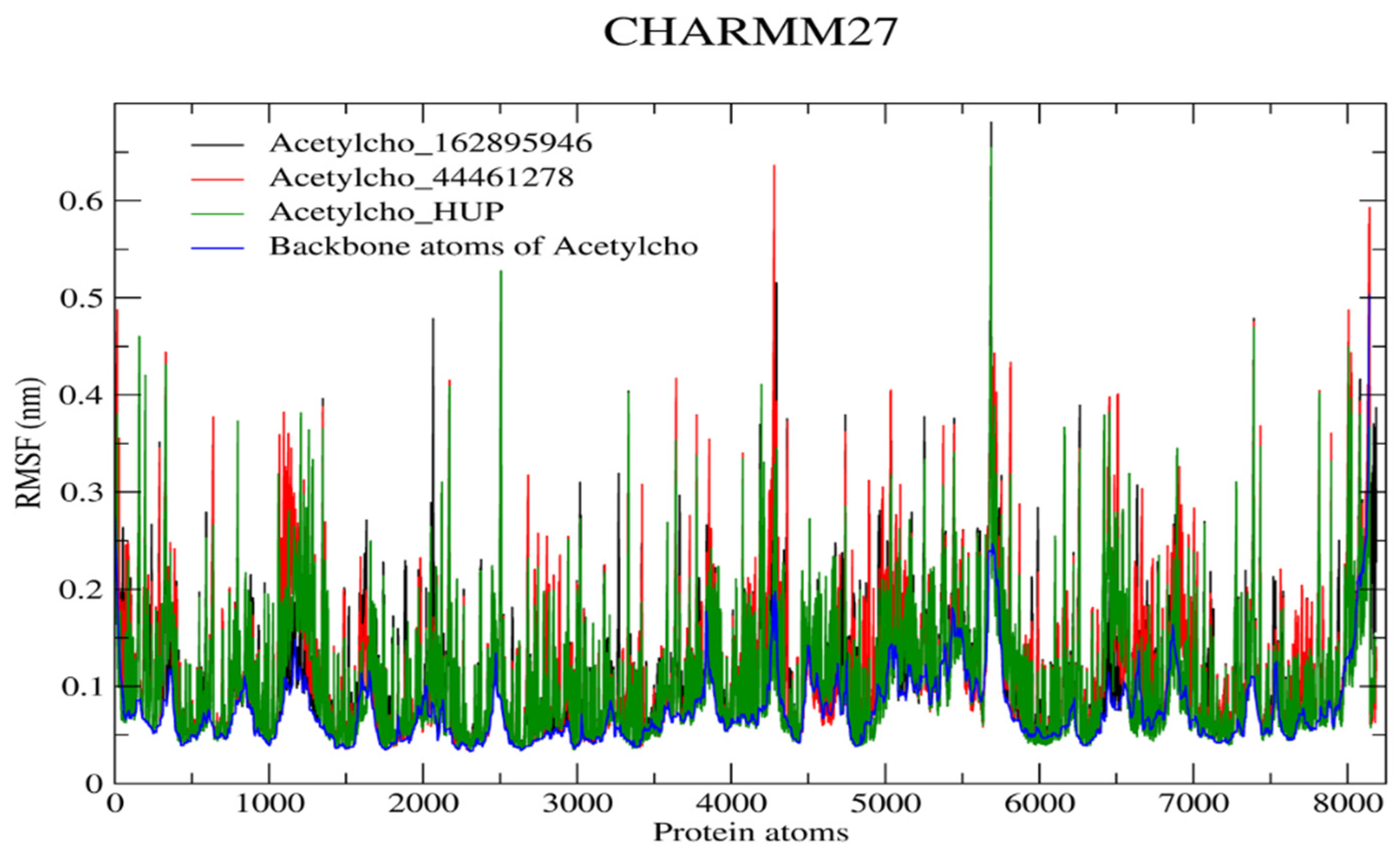
2.6.1. RMSD and RMSF Analysis
2.6.2. Radius of Gyration (Rg) Analysis
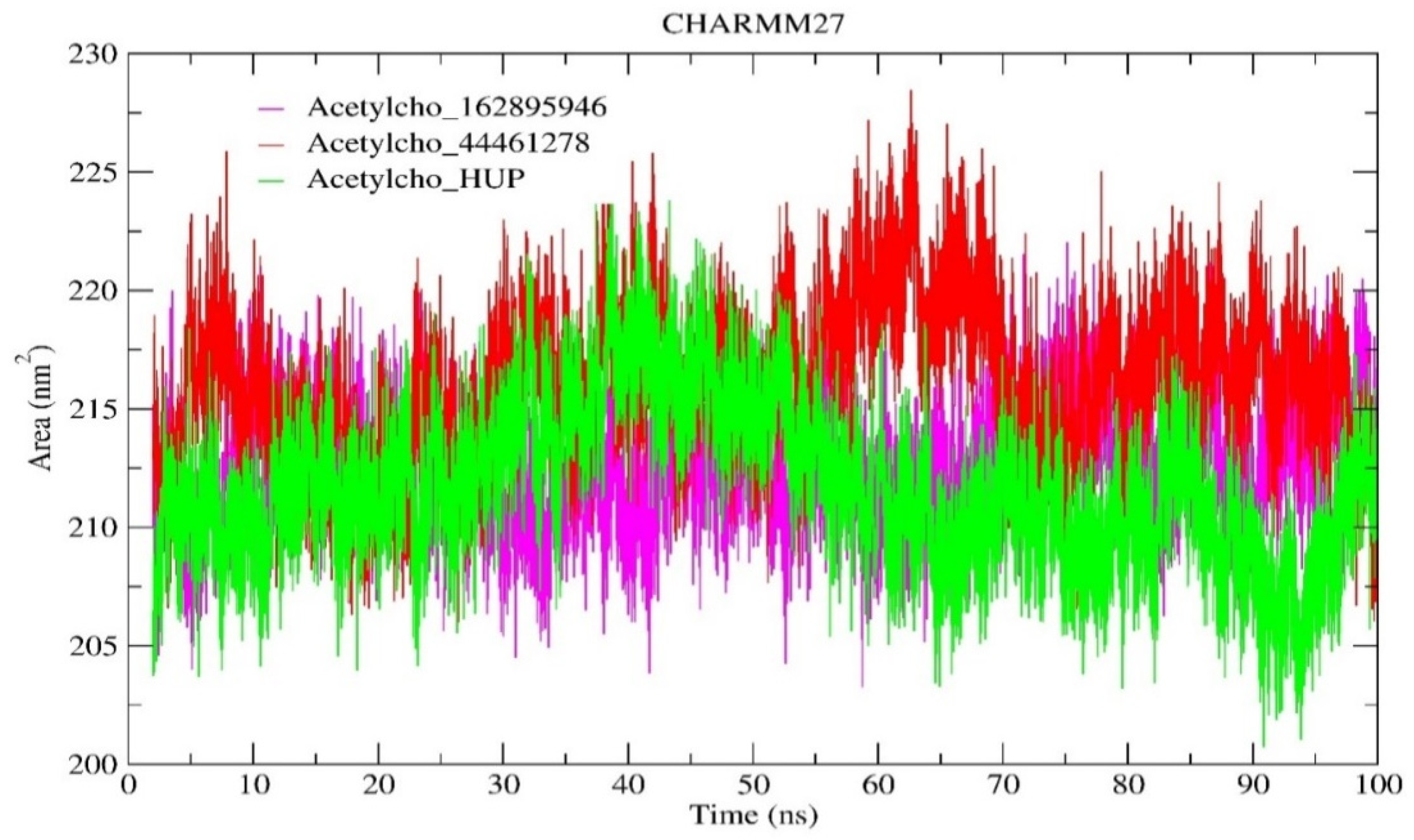
2.6.3. Solvent-Accessible Surface Area (SASA) Analysis
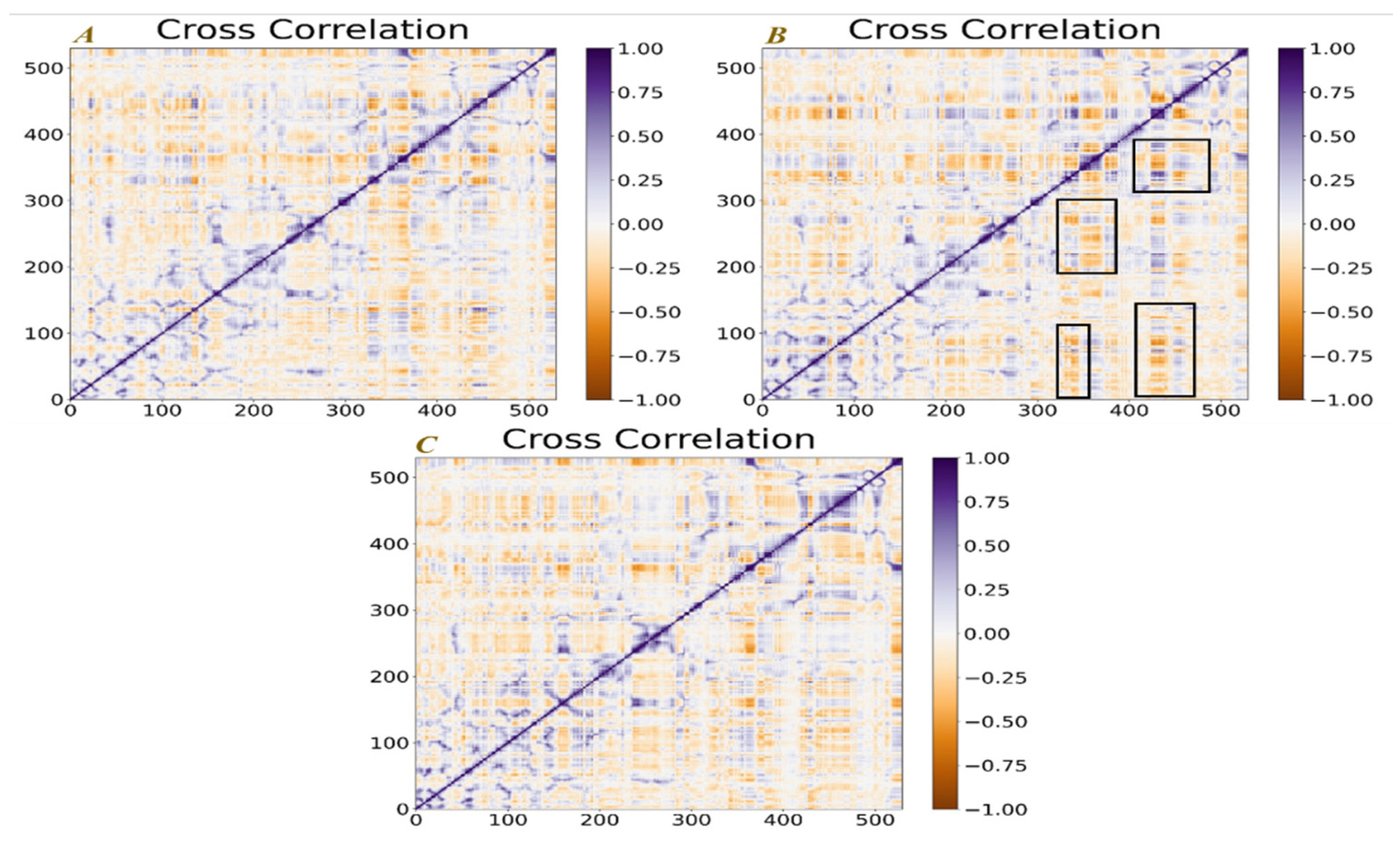
2.7. Dynamic Cross-Correlation Matrix (DCCM) Analysis
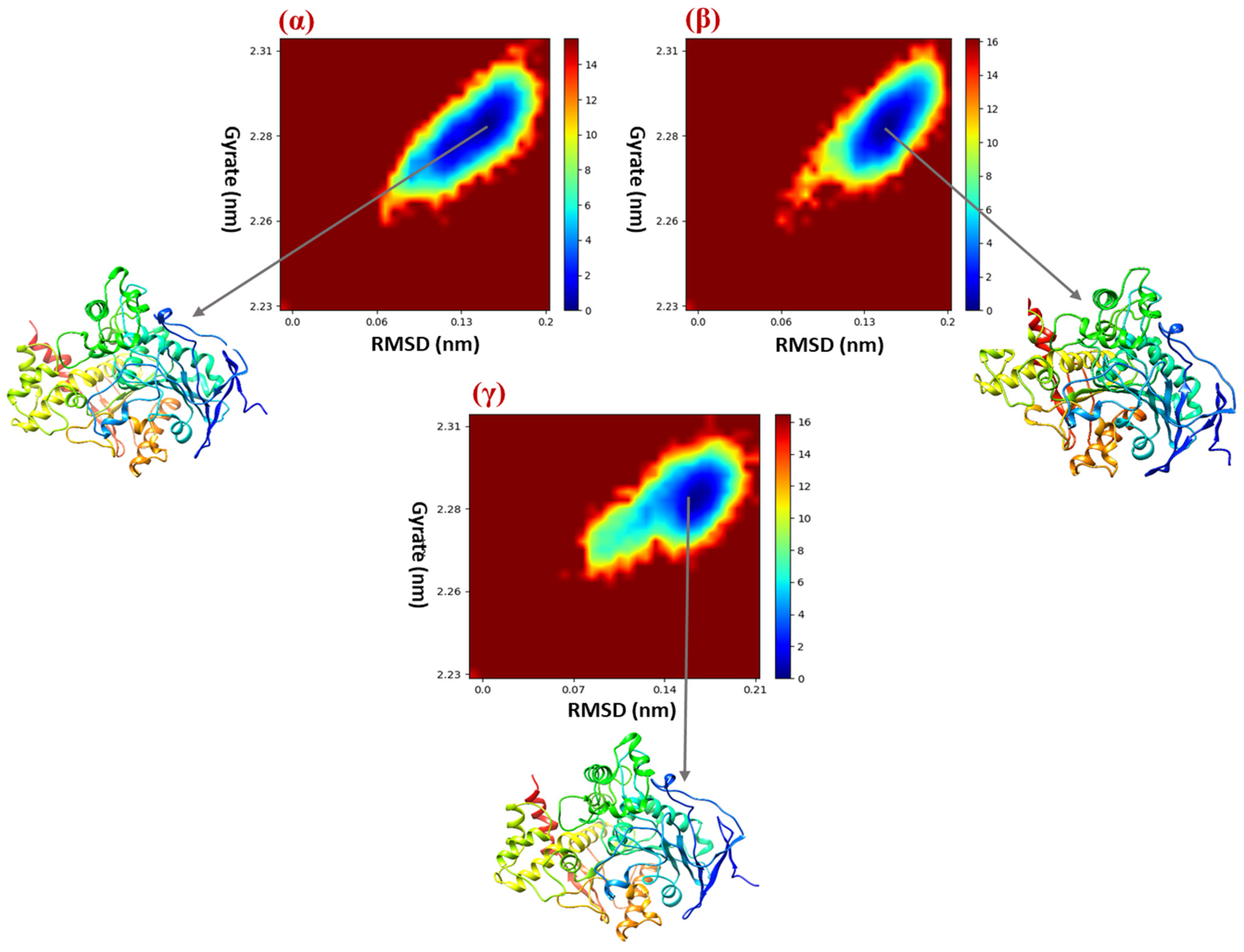
2.8. Free Energy Landscape (FEL)
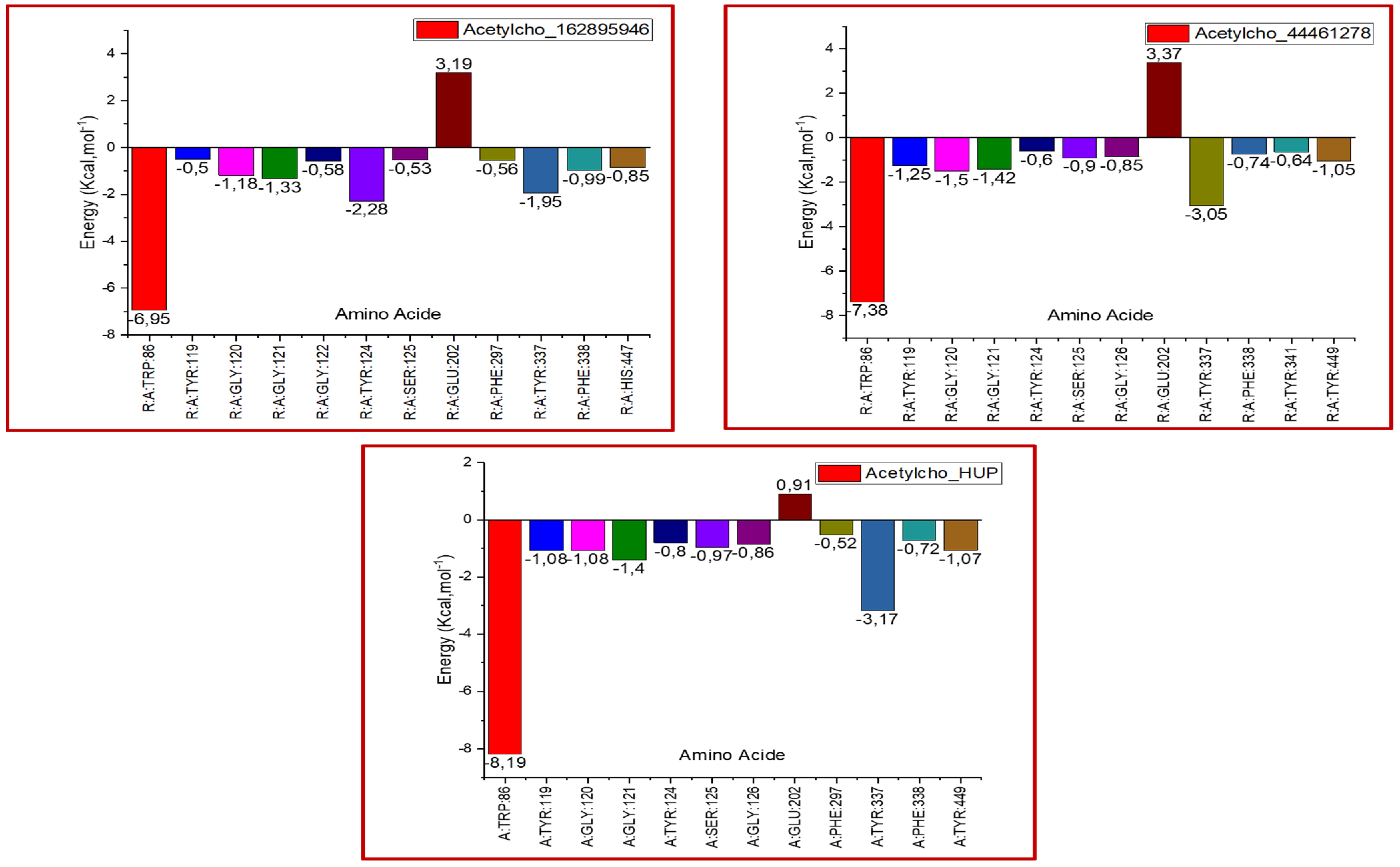
2.9. Binding Free Energy Calculations
3. Materials and Methods
3.1. Dataset
3.2. Preparing the Ligand Structure
3.3. Preparing the Protein Structure
3.4. Receptor Grid Generation
3.5. Development of the e-Pharmacophore Hypothesis
3.6. Enrichment Calculations
3.7. Glide Ligand Docking
3.8. Structure-Based Virtual Screening
3.9. In Silico ADMET Prediction
3.10. Molecular Dynamic Simulation (MDS)
3.11. Dynamic Cross-Correlation Matrix (DCCM)
3.12. Free Energy Landscape (FEL)
3.13. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Čolović, M.B.; Krstić, D.Z.; Lazarević-Pašti, T.D.; Bondžić, A.M.; Vasić, V.M. Acetylcholinesterase Inhibitors: Pharmacology and Toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.L.; Nielsen, J.; Hedberg, K.; Dunaiskis, A.; Jones, S.; Russo, L.; Johnson, J.; Ives, J.; Liston, D. Syntheses, Resolution, and Structure-Activity Relationships of Potent Acetylcholinesterase Inhibitors: 8-Carbaphysostigmine Analogs. J. Med. Chem. 1992, 35, 1429–1434. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lai, L. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model 2006, 46, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Belaidi, S.; Lotfy, K.; Daoud, I.; Belaidi, H. Molecular docking studies and ADMET properties of new 1.2. 3 triazole derivatives for anti-breast cancer activity. J. Bionanoscience 2018, 12, 26–36. [Google Scholar] [CrossRef]
- Ouassaf, M.; Belaidi, S.; Chtita, S.; Lanez, T.; Abul Qais, F.; Md Amiruddin, H. Combined Molecular Docking and Dynamics Simulations Studies of Natural Compounds as Potent Inhibitors against SARS-CoV-2 Main Protease. J. Biomol. Struct. Dyn. 2022, 40, 11264–11273. [Google Scholar] [CrossRef] [PubMed]
- Ouassaf, M.; Bourougaa, L.; Al-Mijalli, S.H.; Abdallah, E.M.; Bhat, A.R.; Kawsar, S.M.A. Marine-Derived Compounds as Potential Inhibitors of Hsp90 for Anticancer and Antimicrobial Drug Development: A Comprehensive In Silico Study. Molecules 2023, 28, 8074. [Google Scholar] [CrossRef]
- Yang, L.-Q.; Ji, X.-L.; Liu, S.-Q. The Free Energy Landscape of Protein Folding and Dynamics: A Global View. J. Biomol. Struct. Dyn. 2013, 31, 982–992. [Google Scholar] [CrossRef]
- Son, M.; Park, C.; Rampogu, S.; Zeb, A.; Lee, K. Discovery of Novel Acetylcholinesterase Inhibitors as Potential Candidates for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1000. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.; Rudolph, M.J.; Fiana, B.; Michael, S.C.; Ebony, N.G.; James, L.; Matthew, C.F.; Jude, J.H. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. Available online: https://pubs.acs.org/doi/10.1021/jm300871x (accessed on 10 December 2023). [CrossRef] [PubMed]
- Sahayarayan, J.J.; Rajan, K.S.; Vidhyavathi, R.; Nachiappan, M.; Prabhu, D.; Alfarraj, S.; Arokiyaraj, S.; Daniel, A.N. In-Silico Protein-Ligand Docking Studies against the Estrogen Protein of Breast Cancer Using Pharmacophore Based Virtual Screening Approaches. Saudi J. Biol. Sci. 2021, 28, 400–407. [Google Scholar] [CrossRef]
- Anu, K.R.; Sumit, R.B.; Subham, D.; Niraja, R.; Shenoy, G.G.; Varadaraj, B.; Fayaz, S.M.; Jayesh, M.; Alex, J.E. Pharmacophore modeling, molecular docking and dynamics approaches for in silico identification of acetylcholinesterase inhibitors from natural products against Alzheimer’s disease. Res. Sq. 2023; preprint. [Google Scholar] [CrossRef]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel Method for Generating Structure-Based Pharmacophores Using Energetic Analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef]
- Maryam, A.; Siddiqi, A.; Vedithi, S.; Ece, A.; Khalid, R. Identification of Selective Inhibitors for Phosphodiesterase 5A Using E-Pharmacophore Modeling and Large-Scale Virtual Screening-Based Structure Guided Drug Discovery Approaches. J. Biomol. Struct. Dyn. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Loving, K.; Salam, N.; Sherman, W. Energetic Analysis of Fragment Docking and Application to Structure-Based Pharmacophore Hypothesis Generation. J. Comput.-Aided Mol. Des. 2009, 23, 541–554. [Google Scholar] [CrossRef]
- Truchon, J.-F.; Bayly, C. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein–Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Rocha, S.F.D.S.; Olanda, C.G.; Fokoue, H.H.; Sant’Anna, C.M. Virtual screening techniques in drug discovery: Review and recent applications. Curr. Top. Med. Chem. 2019, 19, 1751–1767. [Google Scholar] [CrossRef] [PubMed]
- Ioakimidis, L.; Thoukydidis, L.; Mirza, A.; Naeem, S.; Reynisson, J. Benchmarking the Reliability of QikProp. Correlation between Experimental and Predicted Values. QSAR Comb. Sci. 2008, 27, 445–456. [Google Scholar] [CrossRef]
- Ouassaf, M.; Belaidi, S.; Khamouli, S.; Belaid, H.; Chtita, S. Combined 3D-QSAR and molecular docking analysis of thienopyrimidine derivatives as Staphylococcus aureus inhibitors. Acta Chim. Slov. 2021, 68, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, H.Y.; Chen, Z.D.; Gong, J.N.; Chen, C.Y.C. A novel artificial intelligence protocol for finding potential inhibitors of acute myeloid leukemia. J. Mater. Chem. B 2020, 8, 2063–2081. [Google Scholar] [CrossRef]
- Zoete, V.; Michel, A.C.; Aurélien, G.; Olivier, M. Swiss Param: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, E.A.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Bourougaa, L.; Ouassaf, M.; Shtaiwi, A. Discovery of Novel Potent Drugs for Influenza by Inhibiting the Vital Function of Neuraminidase via Fragment-Based Drug Design (FBDD) and Molecular Dynamics Simulation Strategies. J. Biomol. Struct. Dyn. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Anbarasu, K.; Jayanthi, S. Identification of Curcumin Derivatives as Human LMTK3Inhibitors for Breast Cancer: A Docking, Dynamics, and MM/PBSA Approach. 3 Biotech 2018, 8, 228. [Google Scholar] [CrossRef]
- Bourougaa, L.; Mebarka, O.; Khan, S.; Htar, T. Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Studies for the Discovery of Novel Neuraminidase Inhibitors. J. Biomol. Struct. Dyn. 2023; online ahead of print. [Google Scholar] [CrossRef]
- Srinivasan, E.; Rajasekaran, R. Effect of β-Cyclodextrin-EGCG Complexion against Aggregated a-Synuclein through Density Functional Theory and Discrete Molecular Dynamics. Chem. Phys. Lett. 2019, 717, 38–46. [Google Scholar] [CrossRef]
- McCammon, J.A. Protein Dynamics. Rep. Prog. Phys. 1984, 47, 1. [Google Scholar] [CrossRef]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-Energy Landscape, Principal Component Analysis, and Structural Clustering to Identify Representative Conformations from Molecular Dynamics Simulations: The Myoglobin Case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Mitra, A.; Biswas, R.; Bagchi, A.; Ghosh, R. Insight into the Binding of a Synthetic Nitro-Flavone Derivative with Human Poly (ADP-Ribose) Polymerase 1. Int. J. Biol. Macromol. 2019, 141, 444–459. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | EF (1%) | BEDROC | ROC | AUAC |
|---|---|---|---|---|
| Values | 2.40 | 0.08 | 0.70 | 0.69 |
| Hits |
MW g/mol | Lipinsky | QPlogPo/w | QPlogS | QPlogHERGd | QPPCaco | QPPMDCK | % of Human Oral Absorption | QPlogBB | Bioavailability Score | Synthetic Accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CID_162895946 | 270.374 | 0 | 2.596 | −2.958 | −4.438 | 633.027 | 333.880 | 92.287 | 0.444 | 0.55 | 4.46 |
| CID_44461278 | 270.374 | 0 | 2.079 | −2.934 | −4.734 | 226.689 | 110.034 | 81.277 | −0.054 | 0.55 | 4.47 |
| CID_44285285 | 268.35 | 0 | 2.265 | −3.199 | −4.952 | 246.804 | 120.624 | 83.028 | −0.046 | 0.55 | 4.46 |
| CID_81108419 | 220.31 | 0 | 1.521 | −2.033 | −4.709 | 313.894 | 156.427 | 80.541 | −0.055 | 0.55 | 3.19 |
| Huperzine, A | 242.320 | 0 | 1.494 | −2.332 | −4.706 | 175.952 | 83.674 | 75.880 | −0.142 | 0.55 | 4.30 |
| Parameters | Acetylcholinesterase_162895946 | Acetylcholinesterase_44461278 | Acetylcholinesterase HUP |
|---|---|---|---|
| RMSD (nm) | 0.254 | 0.100 | 0.138 |
| RMSF (nm) | 0.115 | 0.120 | 0.110 |
| Rg (nm) | 2.311 | 2.315 | 2.309 |
| SASA (nm2) | 212.665 | 216.149 | 211.755 |
| Acetylcholinesterase_162895946 | Acetylcholinesterase_44461278 | Acetylcholinesterase HUP | |
|---|---|---|---|
| ΔEVDW | −38.67 | −36.79 | −35.42 |
| ΔEEEL | −281.00 | −288.37 | −288.42 |
| ΔEGB | 286.30 | 288.64 | 290.01 |
| ΔESURF | −4.48 | −4.51 | −4.31 |
| ΔGGAS | −319.66 | −325.15 | −323.85 |
| ΔGSOLV | 281.82 | 284.13 | 285.70 |
| ΔTOTAL | −37.85 | −41.02 | −38.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chennai, H.Y.; Belaidi, S.; Bourougaa, L.; Ouassaf, M.; Sinha, L.; Samadi, A.; Chtita, S. Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations. Molecules 2024, 29, 1232. https://doi.org/10.3390/molecules29061232
Chennai HY, Belaidi S, Bourougaa L, Ouassaf M, Sinha L, Samadi A, Chtita S. Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations. Molecules. 2024; 29(6):1232. https://doi.org/10.3390/molecules29061232
Chicago/Turabian StyleChennai, Hind Yassmine, Salah Belaidi, Lotfi Bourougaa, Mebarka Ouassaf, Leena Sinha, Abdelouahid Samadi, and Samir Chtita. 2024. "Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations" Molecules 29, no. 6: 1232. https://doi.org/10.3390/molecules29061232
APA StyleChennai, H. Y., Belaidi, S., Bourougaa, L., Ouassaf, M., Sinha, L., Samadi, A., & Chtita, S. (2024). Identification of Potent Acetylcholinesterase Inhibitors as New Candidates for Alzheimer Disease via Virtual Screening, Molecular Docking, Dynamic Simulation, and Molecular Mechanics–Poisson–Boltzmann Surface Area Calculations. Molecules, 29(6), 1232. https://doi.org/10.3390/molecules29061232