
Utilizing Extracellular Vesicles for Eliminating ‘Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies

 , , , , , and
, , , , , and
Abstract
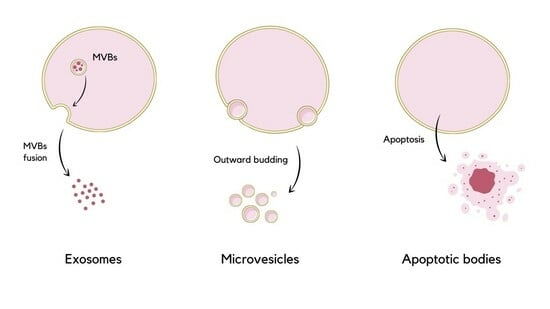
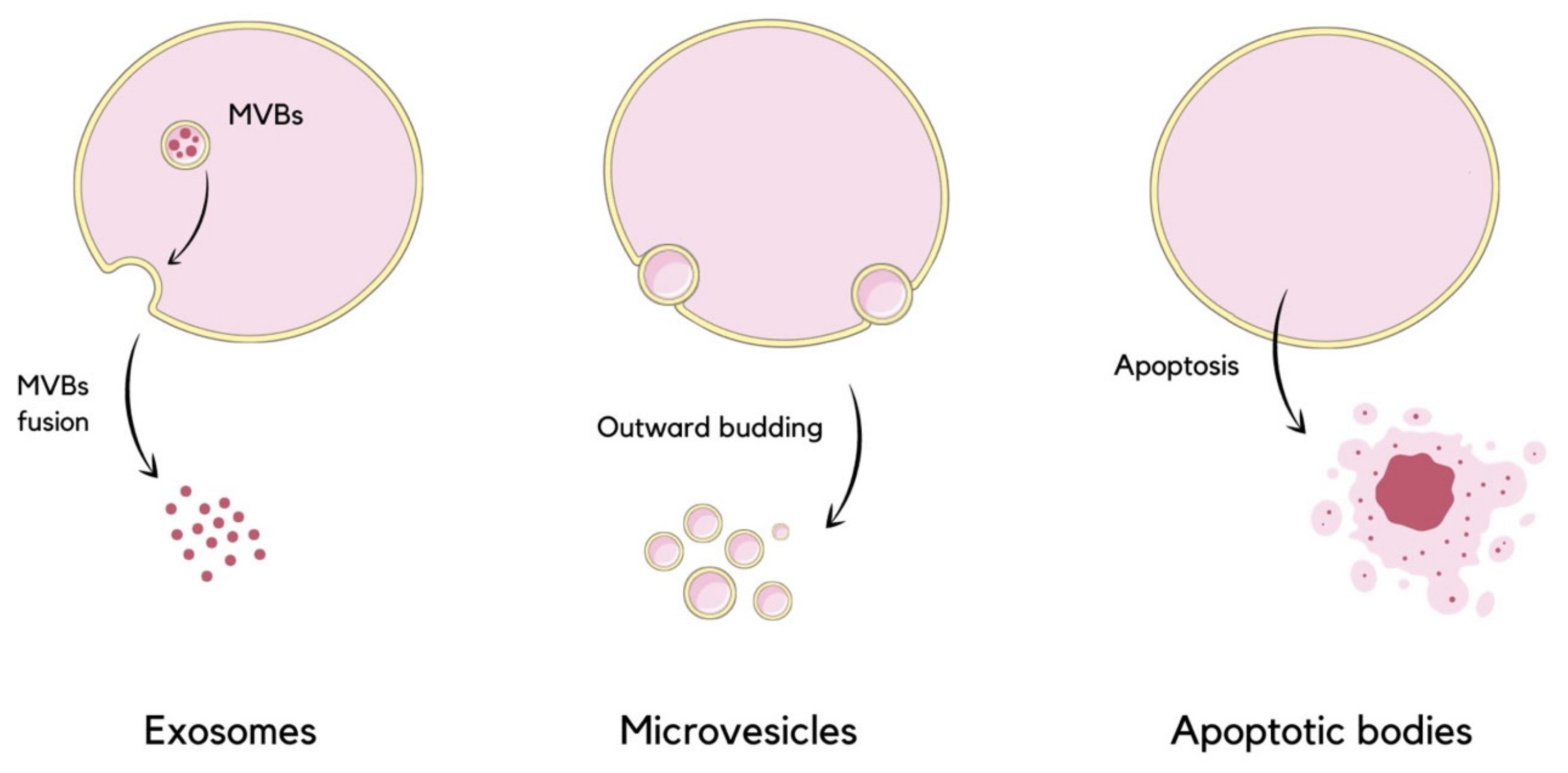
1. Introduction
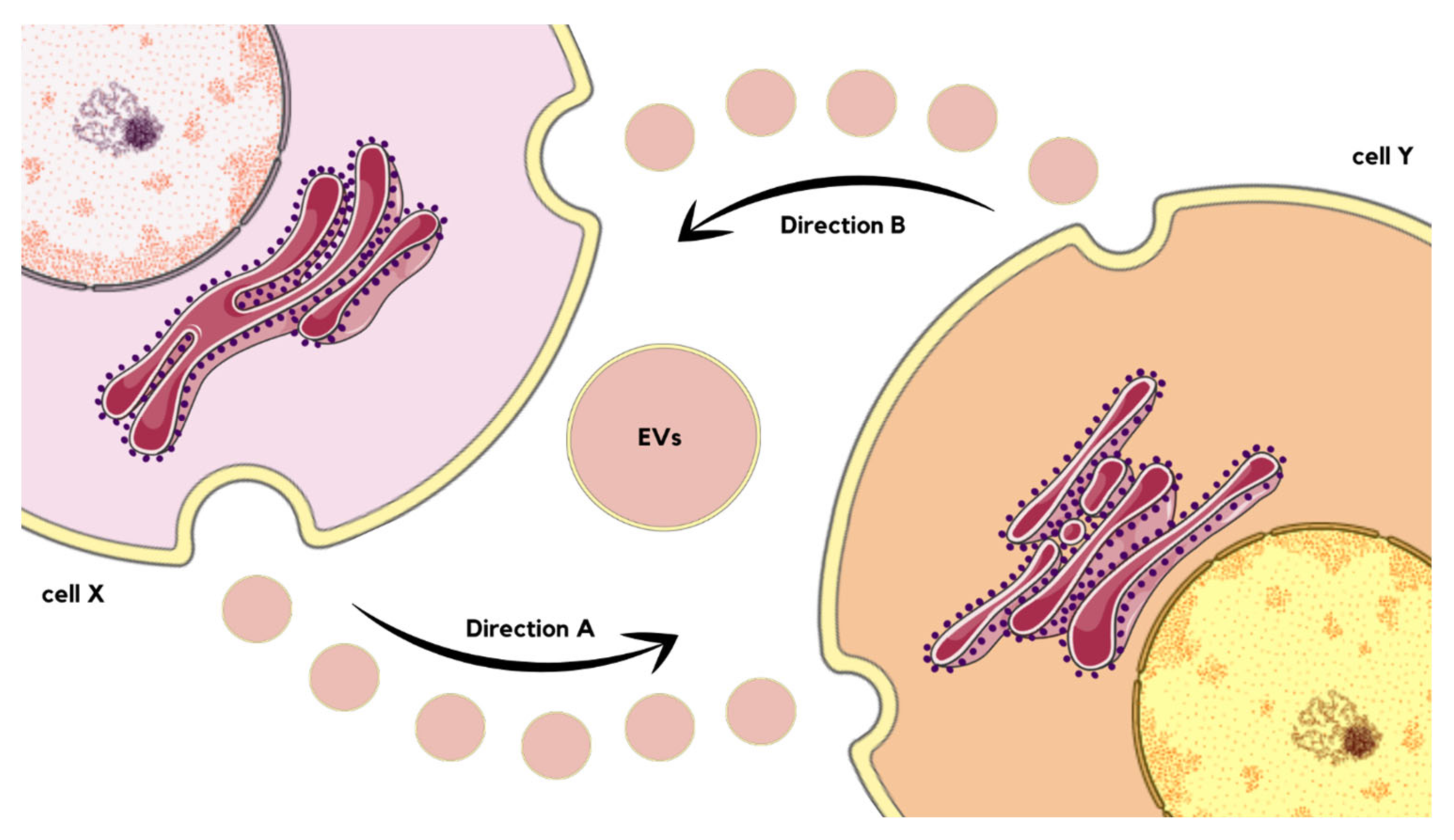
2. EVs as Integrators of Homeostasis
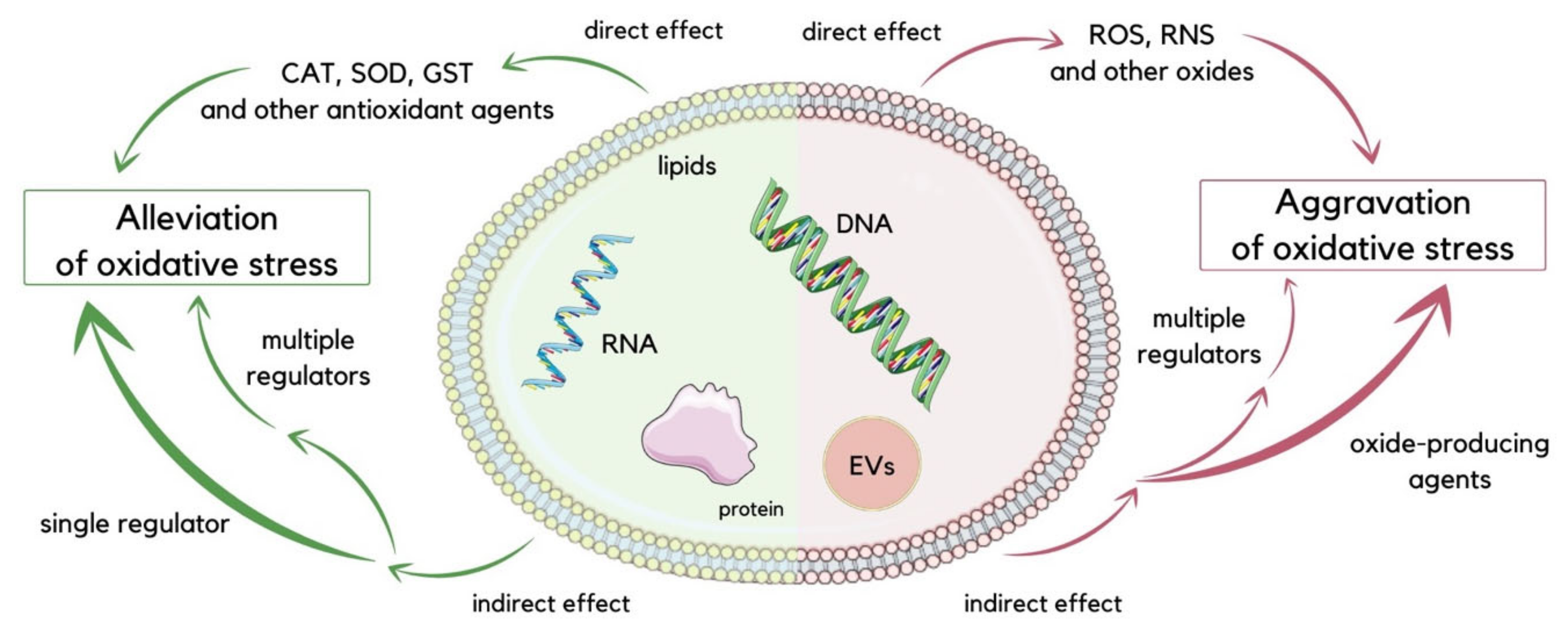
2.1. EVs under Oxidative Stress Conditions
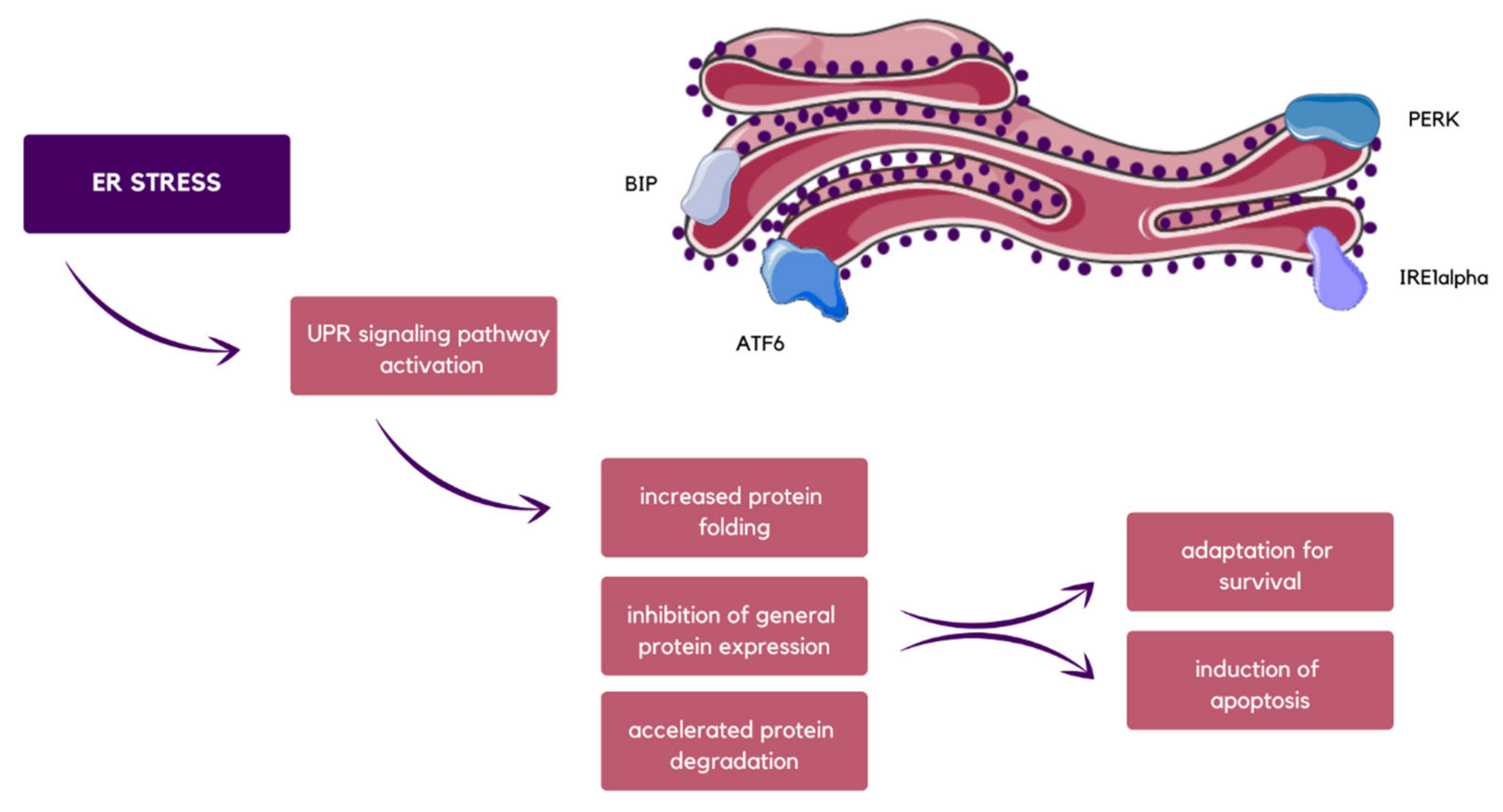
2.2. Interactions between Endoplasmic Reticulum Stress and EVs
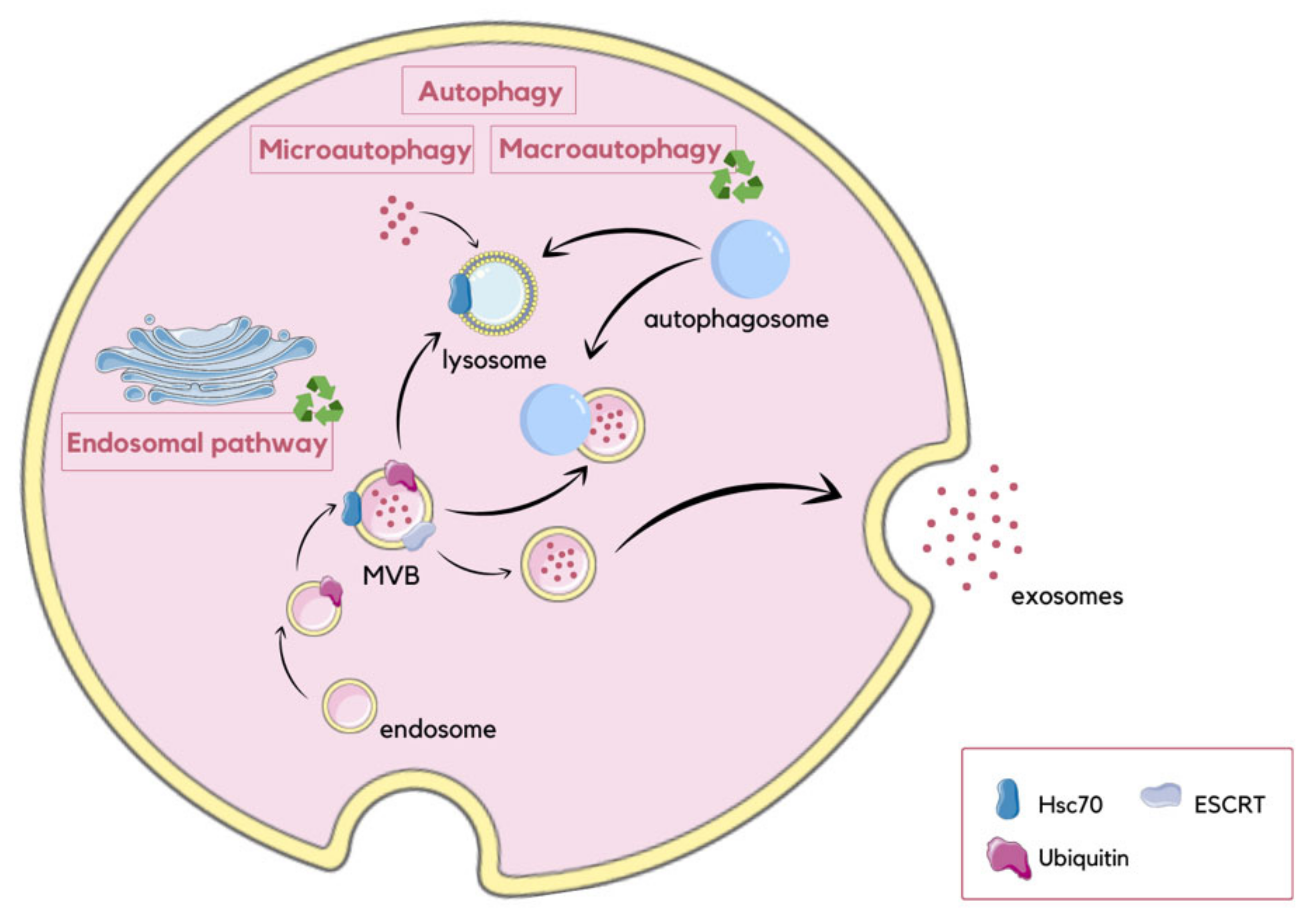
2.3. Interactions between Autophagy and Exosomes
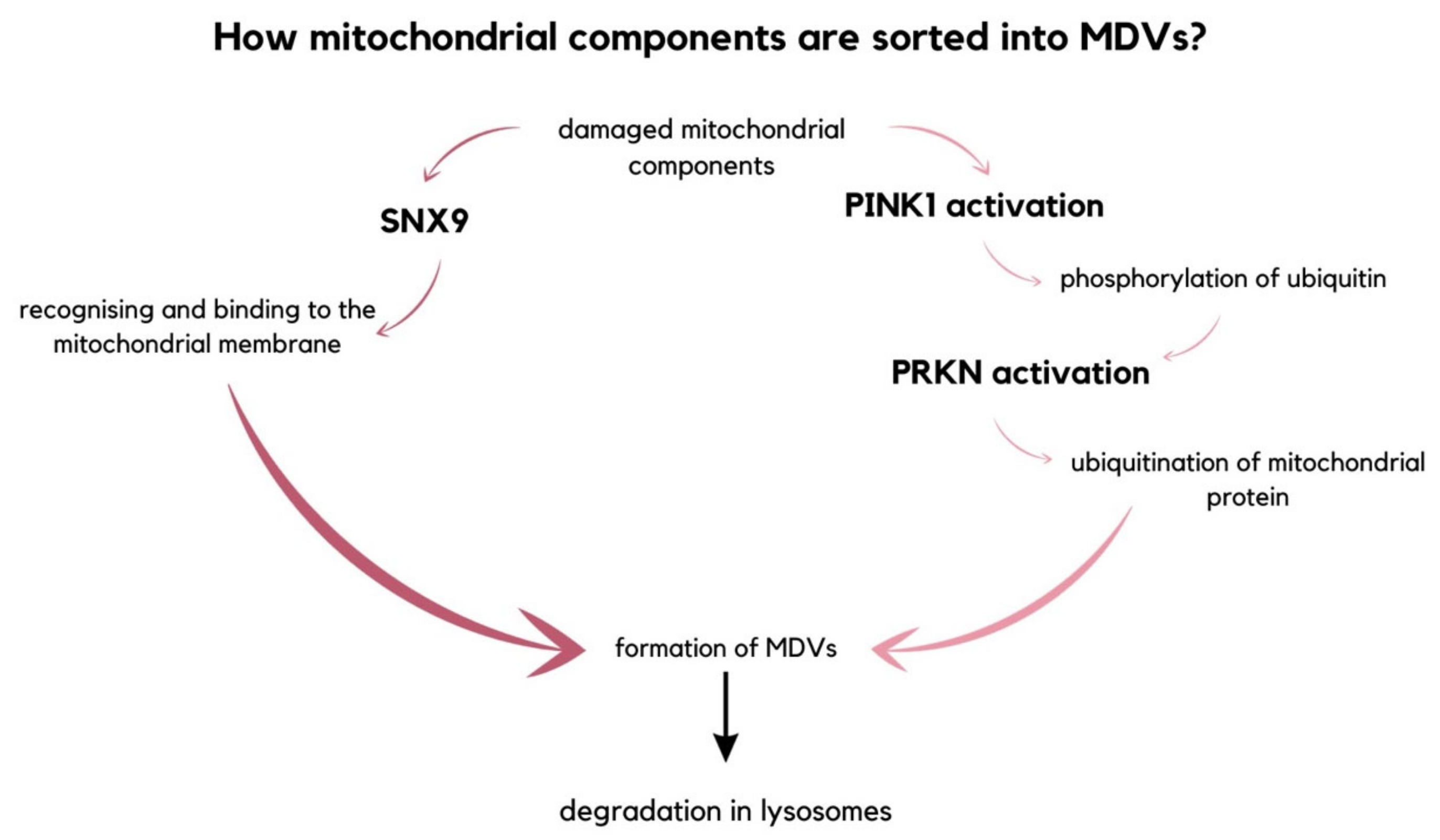
2.4. Mitochondrial EVs and Inflammation
2.5. Extracellular Vesicles as Ferroptosis, Pyroptosis, and Necroptosis Mediators
2.6. Extracellular Vesicles in the Aging Process
3. Future Development, Practical Applications, and Possible Limitations of the EVs-Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Kalra, H.; Drummen, G.P.C.; Mathivanan, S. Focus on extracellular vesicles: Introducing the next small big thing. Int. J. Mol. Sci. 2016, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Margolis, L.; Sadovsky, Y. The biology of extracellular vesicles: The known unknowns. PLoS Biol. 2019, 17, e3000363. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Drapkina, O.; Tonevitsky, A. Transcriptome of extracellular vesicles: State-of-the-art. Front. Immunol. 2019, 10, 202. [Google Scholar] [CrossRef]
- Krylova, S.V.; Feng, D. The Machinery of Exosomes: Biogenesis, Release, and Uptake. Int. J. Mol. Sci. 2023, 24, 1337. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Booth, A.M.; Hildreth, J.E.K. The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 10592–10597. [Google Scholar] [CrossRef]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef]
- Sheta, M.; Taha, E.A.; Lu, Y.; Eguchi, T. Extracellular Vesicles: New Classification and Tumor Immunosuppression. Biology 2023, 12, 110. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Meldolesi, J. Exosomes and Ectosomes in Intercellular Communication. Curr. Biol. 2018, 28, R435–R444. [Google Scholar] [CrossRef]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Szwedowicz, U.; Łapińska, Z.; Gajewska-Naryniecka, A.; Choromańska, A. Exosomes and Other Extracellular Vesicles with High Therapeutic Potential: Their Applications in Oncology, Neurology, and Dermatology. Molecules 2022, 27, 1303. [Google Scholar] [CrossRef] [PubMed]
- Cable, J.; Witwer, K.W.; Coffey, R.J.; Milosavljevic, A.; von Lersner, A.K.; Jimenez, L.; Pucci, F.; Barr, M.M.; Dekker, N.; Barman, B.; et al. Exosomes, microvesicles, and other extracellular vesicles—A Keystone Symposia report. Ann. N. Y. Acad. Sci. 2023, 1523, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Meldolesi, J. Extracellular vesicles, news about their role in immune cells: Physiology, pathology and diseases. Clin. Exp. Immunol. 2019, 196, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.G.; Pink, R.C.; Erdbrügger, U.; Siljander, P.R.; Dellar, E.R.; Pantazi, P.; Akbar, N.; Cooke, W.R.; Vatish, M.; Dias-Neto, E.; et al. In sickness and in health: The functional role of extracellular vesicles in physiology and pathology in vivo: Part I: Health and Normal Physiology: Part I: Health and Normal Physiology. J. Extracell. Vesicles 2022, 11, e12151. [Google Scholar] [CrossRef]
- Latifkar, A.; Hur, Y.H.; Sanchez, J.C.; Cerione, R.A.; Antonyak, M.A. New insights into extracellular vesicle biogenesis and function. J. Cell Sci. 2019, 132, jcs222406. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Khan, Y.S. Histology, Extracellular Vesicles. StatPearls. 2020. Available online: http://www.ncbi.nlm.nih.gov/pubmed/32965927 (accessed on 15 December 2023).
- Popov, L.D. Mitochondrial-derived vesicles: Recent insights. J. Cell. Mol. Med. 2022, 26, 3323–3328. [Google Scholar] [CrossRef]
- Liu, Y.J.; Wang, C. A review of the regulatory mechanisms of extracellular vesicles-mediated intercellular communication. Cell Commun. Signal. 2023, 21, 77. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Hajam, Y.A.; Rani, R.; Ganie, S.Y.; Sheikh, T.A.; Javaid, D.; Qadri, S.S.; Pramodh, S.; Alsulimani, A.; Alkhanani, F.M.; Harakeh, S.; et al. Oxidative Stress in Human Pathology and Aging: Molecular Mechanisms and Perspectives. Cells 2022, 11, 552. [Google Scholar] [CrossRef]
- Qi, H.; Wang, Y.; Fa, S.; Yuan, C.; Yang, L. Extracellular Vesicles as Natural Delivery Carriers Regulate Oxidative Stress under Pathological Conditions. Front. Bioeng. Biotechnol. 2021, 9, 810. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
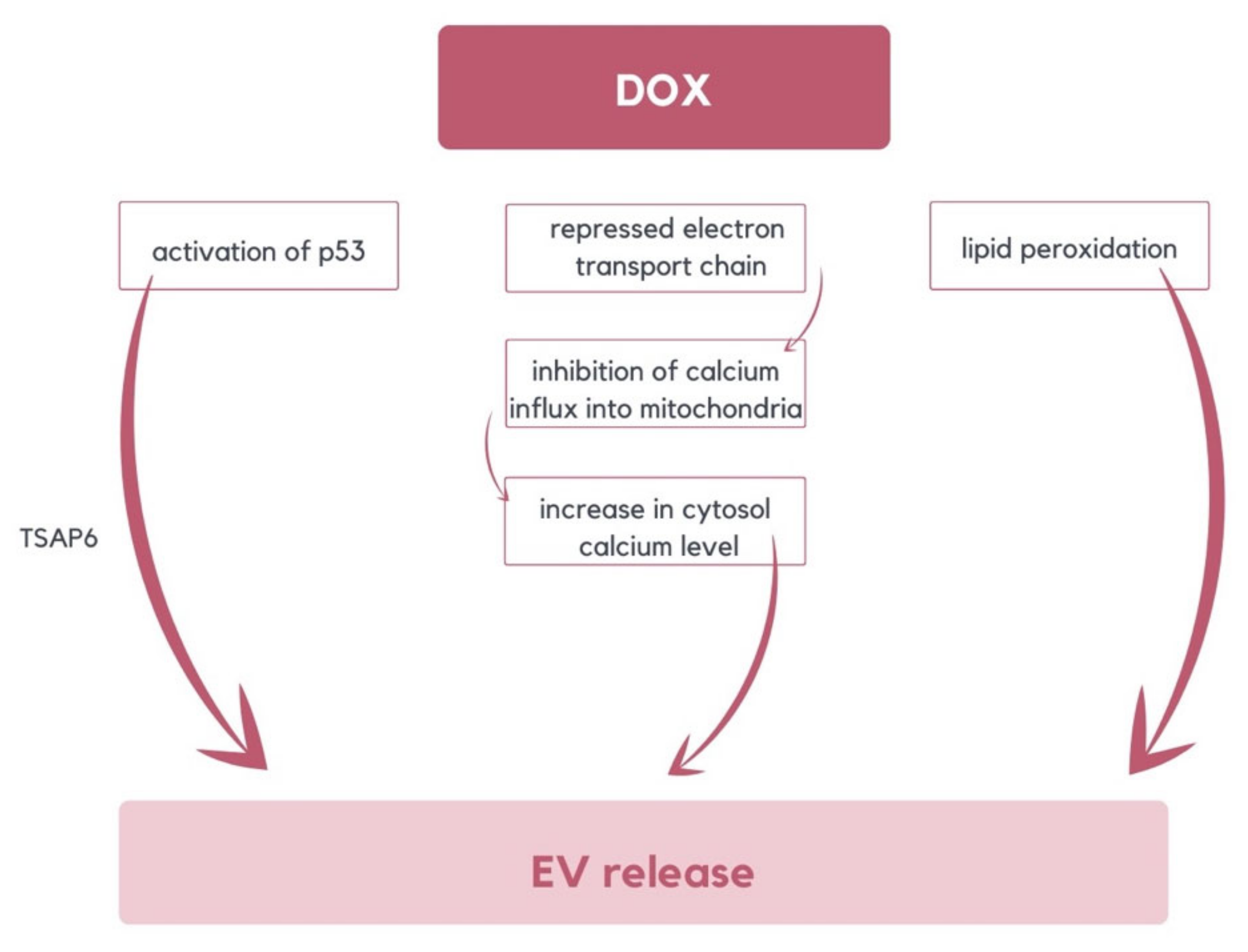
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St. Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef]
- Yarana, C.; Carroll, D.; Chen, J.; Chaiswing, L.; Zhao, Y.; Noel, T.; Alstott, M.; Bae, Y.; Dressler, E.V.; Moscow, J.A.; et al. Extracellular vesicles released by cardiomyocytes in a doxorubicin-induced cardiac injury mouse model contain protein biomarkers of early cardiac injury. Clin. Cancer Res. 2018, 24, 1644–1653. [Google Scholar] [CrossRef]
- Connolly, K.D.; Rees, D.A.; James, P.E. Role of adipocyte-derived extracellular vesicles in vascular inflammation. Free Radic. Biol. Med. 2021, 172, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Chiaradia, E.; Tancini, B.; Emiliani, C.; Delo, F.; Pellegrino, R.M.; Tognoloni, A.; Urbanelli, L.; Buratta, S. Extracellular Vesicles under Oxidative Stress Conditions: Biological Properties and Physiological Roles. Cells 2021, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular vesicles in vascular calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Akbar, N.; Paget, D.; Choudhury, R.P. Extracellular Vesicles in Innate Immune Cell Programming. Biomedicines 2021, 9, 713. [Google Scholar] [CrossRef]
- Yarana, C.; St. Clair, D.K. Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses. Antioxidants 2017, 6, 75. [Google Scholar] [CrossRef]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramírez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef]
- Soleti, R.; Lauret, E.; Andriantsitohaina, R.; Martínez, M.C. Internalization and induction of antioxidant messages by microvesicles contribute to the antiapoptotic effects on human endothelial cells. Free Radic. Biol. Med. 2012, 53, 2159–2170. [Google Scholar] [CrossRef]
- Yao, J.; Zheng, J.; Cai, J.; Zeng, K.; Zhou, C.; Zhang, J.; Li, S.; Li, H.; Chen, L.; He, L.; et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate rat hepatic ischemia-reperfusion injury by suppressing oxidative stress and neutrophil inflammatory response. FASEB J. 2019, 33, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Bodart-Santos, V.; de Carvalho, L.R.P.; de Godoy, M.A.; Batista, A.F.; Saraiva, L.M.; Lima, L.G.; Abreu, C.A.; De Felice, F.G.; Galina, A.; Mendez-Otero, R.; et al. Extracellular vesicles derived from human Wharton’s jelly mesenchymal stem cells protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-β oligomers. Stem Cell Res. Ther. 2019, 10, 332. [Google Scholar] [CrossRef]
- Mahmoud, A.M.; Wilkinson, F.L.; McCarthy, E.M.; Moreno-Martinez, D.; Langford-Smith, A.; Romero, M.; Duarte, J.; Alexander, M.Y. Endothelial microparticles prevent lipid-induced endothelial damage via Akt/eNOS signaling and reduced oxidative stress. FASEB J. 2017, 31, 4636–4648. [Google Scholar] [CrossRef]
- Wang, T.; Jian, Z.; Baskys, A.; Yang, J.; Li, J.; Guo, H.; Hei, Y.; Xian, P.; He, Z.; Liet, Z.; et al. MSC-derived exosomes protect against oxidative stress-induced skin injury via adaptive regulation of the NRF2 defense system. Biomaterials 2020, 257, 120264. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, H.; Xu, W.; Wang, B.; Wu, H.; Tao, Y.; Zhang, B.; Wang, M.; Mao, F.; Yan, Y. Exosomes released by human umbilical cord mesenchymal stem cells protect against cisplatin-induced renal oxidative stress and apoptosis in vivo and in vitro. Stem Cell Res. Ther. 2013, 4, 34. [Google Scholar] [CrossRef] [PubMed]
- Extracellular Vesicles Derived from Human Placental Mesenchymal Stem Cells Alleviate Experimental Colitis in Mice by Inhibiting Inflammation and Oxidative Stress. Available online: https://www.spandidos-publications.com/10.3892/ijmm.2020.4679 (accessed on 15 December 2023).
- Sun, T.; Ding, Z.X.; Luo, X.; Liu, Q.S.; Cheng, Y. Blood Exosomes Have Neuroprotective Effects in a Mouse Model of Parkinson’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 3807476. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Zhang, Y.; Yan, D.Y.; Wang, Y.; Xu, X.; Zhao, Y.C.; Xiao, T.T. Protective Effects of Human Milk-Derived Exosomes on Intestinal Stem Cells Damaged by Oxidative Stress. Cell Transplant. 2020, 29, 963689720912690. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Turner, M.; Munkonda, M.N.; Touyz, R.M. Endothelial Microparticle-Derived Reactive Oxygen Species: Role in Endothelial Signaling and Vascular Function. Oxid. Med. Cell. Longev. 2016, 2016, 5047954. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shang, M.; Zhang, M.; Wang, Y.; Chen, Y.; Wu, Y.; Liu, M.; Song, J.; Liu, Y. Microvesicles derived from hypoxia/reoxygenation-treated human umbilical vein endothelial cells promote apoptosis and oxidative stress in H9c2 cardiomyocytes. BMC Cell Biol. 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Hwang, A.; Chan, J.; Wang, J.Y.J. Extracellular vesicles transfer nuclear Abl-dependent and radiation-induced miR-34c into unirradiated cells to cause bystander effects. Mol. Biol. Cell 2018, 29, 2228–2242. [Google Scholar] [CrossRef] [PubMed]
- Xi, X.J.; Zeng, J.J.; Lu, Y.; Chen, S.H.; Jiang, Z.W.; He, P.J.; Mi, H. Extracellular vesicles enhance oxidative stress through P38/NF-kB pathway in ketamine-induced ulcerative cystitis. J. Cell Mol. Med. 2020, 24, 7609–7624. [Google Scholar] [CrossRef]
- Janiszewski, M.; Do Carmo, A.O.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R.M. Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: A novel vascular redox pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Mostefai, H.A.; Meziani, F.; Mastronardi, M.L.; Agouni, A.; Heymes, C.; Sargentini, C.; Asfar, P.; Martinez, M.C.; Andriantsitohaina, R. Circulating Microparticles from Patients with Septic Shock Exert Protective Role in Vascular Function. Am. J. Respir. Crit. Care Med. 2012, 178, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Manèek-Keber, M.; Frank-Bertoncelj, M.; Hafner-Bratkovič, I.; Smole, A.; Zorko, M.; Pirher, N.; Hayer, S.; Kralj-Iglič, V.; Rozman, B.; Ilc, N.; et al. Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles. Sci. Signal 2015, 8, ra60. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef]
- Lespagnol, A.; Duflaut, D.; Beekman, C.; Blanc, L.; Fiucci, G.; Marine, J.C.; Vidal, M.; Amson, R.; Telerman, A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell Death Differ. 2008, 15, 1723–1733. [Google Scholar] [CrossRef]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef]
- Heuvingh, J.; Bonneau, S. Asymmetric oxidation of giant vesicles triggers curvature-associated shape transition and permeabilization. Biophys. J. 2009, 97, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef]
- Almeida, L.M.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. The PERKs of mitochondria protection during stress: Insights for PERK modulation in neurodegenerative and metabolic diseases. Biol. Rev. Camb. Philos. Soc. 2022, 97, 1737–1748. [Google Scholar] [CrossRef] [PubMed]
- Di Conza, G.; Ho, P.C. ER Stress Responses: An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef]
- Furmanik, M.; van Gorp, R.; Whitehead, M.; Ahmad, S.; Bordoloi, J.; Kapustin, A.; Schurgers, L.J.; Shanahan, C.M. Endoplasmic Reticulum Stress Mediates Vascular Smooth Muscle Cell Calcification via Increased Release of Grp78 (Glucose-Regulated Protein, 78 kDa)-Loaded Extracellular Vesicles. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 898. [Google Scholar] [CrossRef] [PubMed]
- Cianciaruso, C.; Phelps, E.A.; Pasquier, M.; Hamelin, R.; Demurtas, D.; Alibashe Ahmed, M.; Piemonti, L.; Hirosue, S.; Swartz, M.A.; De Palma, M.; et al. Primary Human and Rat β-Cells Release the Intracellular Autoantigens GAD65, IA-2, and Proinsulin in Exosomes Together With Cytokine-Induced Enhancers of Immunity. Diabetes 2017, 66, 460–473. [Google Scholar] [CrossRef]
- Weiss, C.; Kornicka-Grabowska, K.; Mularczyk, M.; Siwinska, N.; Marycz, K. Extracellular Microvesicles (MV’s) Isolated from 5-Azacytidine-and-Resveratrol-Treated Cells Improve Viability and Ameliorate Endoplasmic Reticulum Stress in Metabolic Syndrome Derived Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2020, 16, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiao, P.; Zhong, Y.; Ji, H.; Zhang, Y.; Song, H.; Du, H.; Ding, X.; Wu, H. The Endoplasmic Reticulum-Stressed Head and Neck Squamous Cell Carcinoma Cells Induced Exosomal miR-424-5p Inhibits Angiogenesis and Migration of Humanumbilical Vein Endothelial Cells Through LAMC1-Mediated Wnt/β-Catenin Signaling Pathway. Cell Transplant. 2022, 31, 09636897221083549. [Google Scholar] [CrossRef]
- Osman, A.; Benameur, T.; Korashy, H.M.; Zeidan, A.; Agouni, A. Interplay between Endoplasmic Reticulum Stress and Large Extracellular Vesicles (Microparticles) in Endothelial Cell Dysfunction. Biomedicines 2020, 8, 409. [Google Scholar] [CrossRef]
- Agouni, A.; Osman, A.; El Gamal, H.; Pasha, M. Endoplasmic Reticulum (ER) stress-generated microparticles self-perpetuate ER stress and mediate endothelial cell dysfunction independently of cell survival. FASEB J. 2020, 34 (Suppl. S1), 1. [Google Scholar] [CrossRef]
- Lu, C.; Shi, W.; Hu, W.; Zhao, Y.; Zhao, X.; Dong, F.; Xin, Y.; Peng, T.; Liu, C. Endoplasmic reticulum stress promotes breast cancer cells to release exosomes circ_0001142 and induces M2 polarization of macrophages to regulate tumor progression. Pharmacol. Res. 2022, 177, 106098. [Google Scholar] [CrossRef]
- Kim, T.W.; Ko, S.G. The Herbal Formula JI017 Induces ER Stress via Nox4 in Breast Cancer Cells. Antioxidants 2021, 10, 1881. [Google Scholar] [CrossRef] [PubMed]
- Aydin, Y.; Koksal, A.R.; Reddy, V.; Lin, D.; Osman, H.; Heidari, Z.; Rhadhi, S.M.; Wimley, W.C.; Parsi, M.A.; Dash, S. Extracellular Vesicle Release Promotes Viral Replication during Persistent HCV Infection. Cells 2021, 10, 984. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, X. Interactions between endoplasmic reticulum stress and extracellular vesicles in multiple diseases. Front. Immunol. 2022, 13, 955419. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Sugimoto, H.; O’Connell, J.T.; Kato, N.; Villanueva, A.; Vidal, A.; Qiu, L.; Vitkin, E.; Perelman, L.T.; Melo, C.A.; et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 2014, 26, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Luo, F.; Liu, X.; Lu, L.; Xu, H.; Yang, Q.; Xue, J.; Shi, L.; Li, J.; Zhang, A.; et al. NF-kB-regulated exosomal miR-155 promotes the inflammation associated with arsenite carcinogenesis. Cancer Lett. 2017, 388, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, F.; De Feo, A.; Coscia, C.; Puglisi, R.; Pedini, F.; Pasquini, L.; Bellenghi, M.; Errico, M.C.; Pagani, E.; Carè, A. Exosome-mediated transfer of miR-222 is sufficient to increase tumor malignancy in melanoma. J. Transl. Med. 2016, 14, 56. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, L.; Li, D.; Yin, Y.; Zhang, C.; Li, J.; Zhang, Y. Microvesicle-delivery miR-150 promotes tumorigenesis by up-regulating VEGF, and the neutralization of miR-150 attenuate tumor development. Protein Cell 2013, 4, 932–941. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Choi, D.; Lee, T.H.; Spinelli, C.; Chennakrishnaiah, S.; D’Asti, E.; Rak, J. Extracellular vesicle communication pathways as regulatory targets of oncogenic transformation. Semin. Cell Dev. Biol. 2017, 67, 11–22. [Google Scholar] [CrossRef]
- Wu, C.H.; Silvers, C.R.; Messing, E.M.; Lee, Y.F. Bladder cancer extracellular vesicles drive tumorigenesis by inducing the unfolded protein response in endoplasmic reticulum of nonmalignant cells. J. Biol. Chem. 2019, 294, 3207. [Google Scholar] [CrossRef]
- Jia, L.X.; Zhang, W.M.; Li, T.T.; Liu, Y.; Piao, C.M.; Ma, Y.C.; Lu, Y.; Wang, Y.; Liu, T.T.; Qi, Y.F.; et al. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin. Sci. 2017, 131, 1287–1299. [Google Scholar] [CrossRef]
- Safiedeen, Z.; Rodríguez-Gómez, I.; Vergori, L.; Soleti, R.; Vaithilingam, D.; Douma, I.; Agouni, A.; Leiber, D.; Dubois, S.; Simard, G.; et al. Temporal Cross Talk Between Endoplasmic Reticulum and Mitochondria Regulates Oxidative Stress and Mediates Microparticle-Induced Endothelial Dysfunction. Antioxid. Redox Signal 2017, 26, 15–27. [Google Scholar] [CrossRef]
- Shanahan, C.M. Mechanisms of vascular calcification in CKD-evidence for premature ageing? Nat. Rev. Nephrol. 2013, 9, 661–670. [Google Scholar] [CrossRef]
- Karwowski, W.; Naumnik, B.; Szczepański, M.; Myśliwiec, M. The mechanism of vascular calcification—A systematic review. Med. Sci. Monit. 2012, 18, RA1–RA11. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Tintut, Y.; Demer, L.L. Regulatory mechanisms in vascular calcification. Nat. Rev. Cardiol. 2010, 7, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Wang, Y.; Liu, O.; Jia, L.; Fang, W.; Du, J.; Wei, Y. Tauroursodeoxycholic Acid Attenuates Angiotensin II Induced Abdominal Aortic Aneurysm Formation in Apolipoprotein E-deficient Mice by Inhibiting Endoplasmic Reticulum Stress. Eur. J. Vasc. Endovasc. Surg. 2017, 53, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Baixauli, F.; López-Otín, C.; Mittelbrunn, M. Exosomes and Autophagy: Coordinated Mechanisms for the Maintenance of Cellular Fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Arias, E.; Cuervo, A.M. Chaperone-mediated autophagy in protein quality control. Curr. Opin. Cell Biol. 2011, 23, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Tan, J.; Miao, Y.; Lv, Y.; Zhang, Q. Crosstalk between exosomes and autophagy: A review of molecular mechanisms and therapies. J. Cell. Mol. Med. 2021, 25, 2297. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Humeau, J.; Leduc, M.; Cerrato, G.; Loos, F.; Kepp, O.; Kroemer, G. Phosphorylation of eukaryotic initiation factor-2α (eIF2α) in autophagy. Cell Death Dis. 2020, 11, 433. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Coelho-Júnior, H.J.; Landi, F.; Bucci, C.; Marzetti, E. Mitochondrial-Derived Vesicles: The Good, the Bad, and the Ugly. Int. J. Mol. Sci. 2023, 24, 3835. [Google Scholar] [CrossRef]
- Zecchini, V.; Paupe, V.; Herranz-Montoya, I.; Janssen, J.; Wortel, I.M.N.; Morris, J.L.; Ferguson, A.; Chowdury, S.R.; Segarra-Mondejar, M.; Costa, A.S.H.; et al. Fumarate induces vesicular release of mtDNA to drive innate immunity. Nature 2023, 615, 499–506. [Google Scholar] [CrossRef]
- Terešak, P.; Lapao, A.; Subic, N.; Boya, P.; Elazar, Z.; Simonsen, A. Regulation of PRKN-independent mitophagy. Autophagy 2022, 18, 24–39. [Google Scholar] [CrossRef]
- Faas, M.M.; de Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef]
- Deus, C.M.; Tavares, H.; Beatriz, M.; Mota, S.; Lopes, C. Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 2022, 11, 2364. [Google Scholar] [CrossRef]
- Marchi, S.; Guilbaud, E.; Tait, S.W.G.; Yamazaki, T.; Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 2023, 23, 159. [Google Scholar] [CrossRef]
- Green, D.R. The Mitochondrial Pathway of Apoptosis: Part I: MOMP and Beyond. Cold Spring Harb. Perspect. Biol. 2022, 14, a041038. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of mtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Picca, A.; Guerra, F.; Calvani, R.; Bucci, C.; Lo Monaco, M.R.; Bentivoglio, A.R.; Coelho-Júnior, H.J.; Landi, F.; Bernabei, R.; Marzetti, E. Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. Int. J. Mol. Sci. 2019, 20, 805. [Google Scholar] [CrossRef]
- Picca, A.; Calvani, R.; Coelho-junior, H.J.; Marzetti, E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021, 10, 537. [Google Scholar] [CrossRef]
- Di Mambro, T.; Pellielo, G.; Agyapong, E.D.; Carinci, M.; Chianese, D.; Giorgi, C.; Morciano, G.; Patergnani, S.; Pinton, P.; Rimessi, A.; et al. The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. Int. J. Mol. Sci. 2023, 24, 8181. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxid. Med. Cell. Longev. 2020, 2020, 9423593. [Google Scholar] [CrossRef]
- Hadian, K.; Stockwell, B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 2023, 22, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Xu, D.; Yang, X.; Tang, H.; Tao, X.; Fan, Y.; Ding, Y. The Emerging Roles of Pyroptosis, Necroptosis, and Ferroptosis in Non-Malignant Dermatoses: A Review. J. Inflamm. Res. 2023, 16, 1967–1977. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Jiang, Q.; Yang, K.P.; Wang, L.; Sethi, G.; Ma, Z. Extracellular vesicle-mediated ferroptosis, pyroptosis, and necroptosis: Potential clinical applications in cancer therapy. Cell Death Discov. 2024, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Qiu, J.; Pan, J.; Pan, J.P. Pyroptosis and Its Role in Cervical Cancer. Cancers 2022, 14, 5764. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, M.C.; Frauenstein, A.; Stafford, C.A.; Phulphagar, K.; Mann, M.; Meissner, F. Quantitative and Dynamic Catalogs of Proteins Released during Apoptotic and Necroptotic Cell Death. Cell Rep. 2020, 30, 1260–1270.e5. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Erlich, Z.; Arad, G.; Edry-Botzer, L.; Zargarian, S.; Cohen, H.; Manko, T.; Ofir-Birin, Y.; Cooks, T.; Regev-Rudzki, N.; et al. Proteomic analysis of necroptotic extracellular vesicles. Cell Death Dis. 2021, 12, 1059. [Google Scholar] [CrossRef]
- Saheera, S.; Potnuri, A.G.; Krishnamurthy, P. Nano-Vesicle (Mis)Communication in Senescence-Related Pathologies. Cells 2020, 9, 1974. [Google Scholar] [CrossRef]
- Das, M.; Kale, V. Involvement of extracellular vesicles in aging process and their beneficial effects in alleviating aging-associated symptoms. Cell Biol. Int. 2021, 45, 2403–2419. [Google Scholar] [CrossRef]
- Andjus, P.; Kosanović, M.; Milićević, K.; Gautam, M.; Vainio, S.J.; Jagečić, D.; Kozlova, E.N.; Pivoriūnas, A.; Chachques, J.C.; Sakaj, M.; et al. Extracellular Vesicles as Innovative Tool for Diagnosis, Regeneration and Protection against Neurological Damage. Int. J. Mol. Sci. 2020, 21, 6859. [Google Scholar] [CrossRef]
- Manni, G.; Buratta, S.; Pallotta, M.T.; Chiasserini, D.; Di Michele, A.; Emiliani, C.; Giovagnoli, S.; Pascucci, L.; Romani, R.; Bellezza, I.; et al. Extracellular Vesicles in Aging: An Emerging Hallmark? Cells 2023, 12, 527. [Google Scholar] [CrossRef]
- Pinson, M.R.; Chung, D.D.; Adams, A.M.; Scopice, C.; Payne, E.A.; Sivakumar, M.; Miranda, R.C. Extracellular Vesicles in Premature Aging and Diseases in Adulthood Due to Developmental Exposures. Aging Dis. 2021, 12, 1516–1535. [Google Scholar] [CrossRef]
- Zhang, B.; Tian, X.; Hao, J.; Xu, G.; Zhang, W. Mesenchymal Stem Cell-Derived Extracellular Vesicles in Tissue Regeneration. Cell Transplant. 2020, 29, 0963689720908500. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Alique, M. Extracellular Vesicles as ‘Very Important Particles’ (VIPs) in Aging. Int. J. Mol. Sci. 2023, 24, 4250. [Google Scholar] [CrossRef] [PubMed]
- Elzanowska, J.; Semira, C.; Costa-Silva, B. DNA in extracellular vesicles: Biological and clinical aspects. Mol. Oncol. 2021, 15, 1701. [Google Scholar] [CrossRef]
- Khanh, V.C.; Yamashita, T.; Ohneda, K.; Tokunaga, C.; Kato, H.; Osaka, M.; Hiramatsu, Y.; Ohneda, O. Rejuvenation of mesenchymal stem cells by extracellular vesicles inhibits the elevation of reactive oxygen species. Sci. Rep. 2020, 10, 17315. [Google Scholar] [CrossRef]
- Ju, Y.; Hu, Y.; Yang, P.; Xie, X.; Fang, B. Extracellular vesicle-loaded hydrogels for tissue repair and regeneration. Mater. Today Bio 2022, 18, 100522. [Google Scholar] [CrossRef] [PubMed]
- Chennakrishnaiah, S.; Meehan, B.; D’Asti, E.; Montermini, L.; Lee, T.H.; Karatzas, N.; Buchanan, M.; Tawil, N.; Choi, D.; Divangahi, M.; et al. Leukocytes as a reservoir of circulating oncogenic DNA and regulatory targets of tumor-derived extracellular vesicles. J. Thromb. Haemost. 2018, 16, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Chen, H.; Wang, Y.; Zhang, L.; Wang, X. Roles of extracellular vesicles in the aging microenvironment and age-related diseases. J. Extracell. Vesicles 2021, 10, e12154. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, C.; Esteller, M. MicroRNAs and Epigenetics. Adv. Cancer Res. 2017, 135, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar] [CrossRef] [PubMed]
- Fulzele, S.; Mendhe, B.; Khayrullin, A.; Johnson, M.; Kaiser, H.; Liu, Y.; Isales, C.M.; Hamrick, M.W. Muscle-derived miR-34a increases with age in circulating extracellular vesicles and induces senescence of bone marrow stem cells. Aging (Albany NY) 2019, 11, 1791. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Mcilvenna, L.C.; Whitham, M. Exercise, healthy ageing, and the potential role of small extracellular vesicles. J. Physiol. 2022, 601, 4937–4951. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Ren, K. Exosomes in perspective: A potential surrogate for stem cell therapy. Odontology 2019, 107, 271. [Google Scholar] [CrossRef]
- Ryan, S.T.; Ryan, S.T.; Hosseini-Beheshti, E.; Afrose, D.; Ding, X.; Xia, B.; Grau, G.; Little, C.; McClements, L.; Li, J. Extracellular Vesicles from Mesenchymal Stromal Cells for the Treatment of Inflammation-Related Conditions. Int. J. Mol. Sci. 2021, 22, 23. [Google Scholar] [CrossRef]
- Vidal, M. Exosomes: Revisiting their role as ‘garbage bags’. Traffic 2019, 20, 815–828. [Google Scholar] [CrossRef]
- Fowler, C.D.; Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The continuum of aging and age-related diseases: Common mechanisms but different rates. Front. Med. 2018, 5, 349810. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Ruan, L.; Oh, J.; Dong, X.; Zhuge, Q.; Su, D.M. Extracellular vesicles extracted from young donor serum attenuate inflammaging via partially rejuvenating aged T-cell immunotolerance. FASEB J. 2018, 32, 5899. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell–derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542. [Google Scholar] [CrossRef]
- Cavallari, C.; Ranghino, A.; Tapparo, M.; Cedrino, M.; Figliolini, F.; Grange, C.; Giannachi, V.; Garneri, P.; Deregibus, M.C.; Collino, F.; et al. Serum-derived extracellular vesicles (EVs) impact on vascular remodeling and prevent muscle damage in acute hind limb ischemia. Sci. Rep. 2017, 7, 8180. [Google Scholar] [CrossRef]
- Daltro, S.R.T.; Meira, C.S.; Santos, I.P.; Ribeiro dos Santos, R.; Soares, M.B.P. Mesenchymal Stem Cells and Atopic Dermatitis: A Review. Front. Cell Dev. Biol. 2020, 8, 326. [Google Scholar] [CrossRef]
- Leung, D.Y.M. Atopic dermatitis: New insights and opportunities for therapeutic intervention. J. Allergy Clin. Immunol. 2000, 105, 860–876. [Google Scholar] [CrossRef]
- Shin, K.O.; Ha, D.H.; Kim, J.O.; Crumrine, D.A.; Meyer, J.M.; Wakefield, J.S.; Lee, Y.; Kim, B.; Kim, S.; Kim, H.K.; et al. Exosomes from Human Adipose Tissue-Derived Mesenchymal Stem Cells Promote Epidermal Barrier Repair by Inducing de Novo Synthesis of Ceramides in Atopic Dermatitis. Cells 2020, 9, 680. [Google Scholar] [CrossRef]
- Cho, B.S.; Kim, J.O.; Ha, D.H.; Yi, Y.W. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res. Ther. 2018, 9, 187. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, W.; Lian, L.; Xu, Y.; Bai, X.; Xu, S.; Zhang, J. Stroke treatment: Is exosome therapy superior to stem cell therapy? Biochimie 2020, 179, 190–204. [Google Scholar] [CrossRef]
- Mendt, M.; Rezvani, K.; Shpall, E. Mesenchymal stem cell-derived exosomes for clinical use. Bone Marrow Transplant. 2019, 54 (Suppl. S2), 789–792. [Google Scholar] [CrossRef]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef]
- Van Hezel, M.E.; Nieuwland, R.; van Bruggen, R.; Juffermans, N.P. The Ability of Extracellular Vesicles to Induce a Pro-Inflammatory Host Response. Int. J. Mol. Sci. 2017, 18, 1285. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Possible Application of EVs in the Aging Process | Characteristic | Reference |
|---|---|---|
| Genomic instability | EVs have been shown to transport DNA and RNA between cells, which can promote DNA damage repair and maintain genome stability. | [133] |
| Rejuvenation of mesenchymal stem cells | EVs derived from young mesenchymal stem cells (MSCs) were found to possess rejuvenating properties, reversing age-related changes in recipient aged MSCs by transferring bioactive molecules. | [134] |
| Regeneration | According to research, the utilization of EV-loaded hydrogels has demonstrated a promising approach to tissue repair and regeneration, exhibiting efficacy in tissue and organs like: skin, bones, cartilage, heart, nerves, the reproductive system, periodontal, hair, liver, and kidneys. | [135] |
| Anti-tumor regulation | EVs serve as gateways through which cancer cells release their genomic DNA (gDNA) into the extracellular space, which can then be taken up by circulating leukocytes, particularly neutrophils. This changes their functioning, making them more likely to form blood clots and develop inflammation. | [136] |
| Telomere attrition | EVs can induce telomere shortening and DNA damage in recipient cells, leading to aging and genomic instability. However, treatments that remove the RNA or protein content from EVs can reduce this effect. Additionally, EVs can also spread senescence-associated characteristics to nearby cells by inducing hypomethylation, thereby causing genomic instability. | [137] |
| Presentation of abnormal telomeres | The cell-free form of telomeric repeat-containing RNA (cfTERRA) is released in EVs. Research has shown that cancer patients have higher levels of cfTERRA in their blood plasma, and this rise is linked to telomere dysfunction and DNA damage in the parent cells. | [137] |
| Epigenetic changes | The transport of epigenetic regulators, including microRNAs and histones EVs, enables them to modulate gene expression and chromatin structure when transferred between cells. | [138] |
| Dysregulated nutrient sensing | Dysregulated nutrient sensing, a hallmark of aging, involves disrupted pathways related to insulin signaling and mTOR signaling. EVs play a role in this process by transporting hormones and growth factors involved in nutrient sensing, including insulin, and factors related to mTOR signaling. By influencing these pathways, EVs can contribute to the dysregulation of nutrient sensing observed in aging. | [139] |
| Mitochondrial dysfunction | The mitochondrial-lysosomal axis is a cellular system responsible for removing damaged components, particularly dysfunctional mitochondria. Disruption of this system, along with abnormal EV secretion, has been associated with the aging process and various diseases. | [113] |
| Stem cell exhaustion | EVs derived from aged cells can affect mesenchymal stem cells (MSCs) from bone marrow. When young MSCs are exposed to EVs from old MSCs, it activates the mTOR pathway, leads to increased expression of aging markers, and reduces pluripotency marker levels. Similarly, when bone marrow stem cells (BMSCs) are treated with EVs from aged bone marrow fluids, it induces stem cell senescence and impairs their ability to differentiate into bone cells. Additionally, EVs carrying miR-34a, generated by oxidative stress in muscle cells, can suppress a protein called SIRT1 and promote senescence and death in BMSCs. | [137,140] |
| Altered intercellular communication | EVs can transport various signaling molecules, including cytokines and growth factors, between cells. By carrying these signaling molecules, EVs facilitate intercellular communication and contribute to the regulation of tissue homeostasis. | [141] |
| Cellular senescence | Cellular senescence is the loss of replicative potential in a normally dividing cell. EVs can promote or suppress cellular senescence by transferring senescence-associated molecules, such as microRNAs and proteins. Senescent cells are also characterized by an increased amount of pro-inflammatory cytokines, chemokines, tissue-damaging proteases, and other factors that can alter stem and progenitor cell function, hemostatic factors, and growth factors. | [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisielewska, M.; Rakoczy, K.; Skowron, I.; Górczyńska, J.; Kacer, J.; Bocheńska, A.; Choromańska, A. Utilizing Extracellular Vesicles for Eliminating ‘Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies. Molecules 2024, 29, 948. https://doi.org/10.3390/molecules29050948
Kisielewska M, Rakoczy K, Skowron I, Górczyńska J, Kacer J, Bocheńska A, Choromańska A. Utilizing Extracellular Vesicles for Eliminating ‘Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies. Molecules. 2024; 29(5):948. https://doi.org/10.3390/molecules29050948
Chicago/Turabian StyleKisielewska, Monika, Katarzyna Rakoczy, Izabela Skowron, Julia Górczyńska, Julia Kacer, Agata Bocheńska, and Anna Choromańska. 2024. "Utilizing Extracellular Vesicles for Eliminating ‘Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies" Molecules 29, no. 5: 948. https://doi.org/10.3390/molecules29050948
APA StyleKisielewska, M., Rakoczy, K., Skowron, I., Górczyńska, J., Kacer, J., Bocheńska, A., & Choromańska, A. (2024). Utilizing Extracellular Vesicles for Eliminating ‘Unwanted Molecules’: Harnessing Nature’s Structures in Modern Therapeutic Strategies. Molecules, 29(5), 948. https://doi.org/10.3390/molecules29050948