
Current Trends in Sirtuin Activator and Inhibitor Development



Abstract
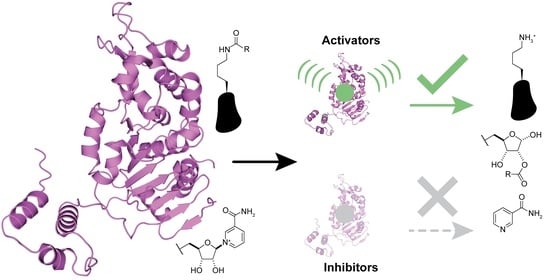
1. Introduction
1.1. Sirtuin Subcellular Localization and Structure Dictate Function
1.2. Differential Modulation of Sirtuin Activity
2. Sirtuin Activators
2.1. Natural Product Sirtuin Activators
2.2. Synthetic Small Molecule Sirt1 Activators
2.3. Synthetic Small Molecule Sirt3 and Sirt5 Activators
2.4. Synthetic Small Molecule Sirt6 Activators
2.5. Alternative Mechanisms of Sirtuin Activation
3. Small Molecule Sirtuin Inhibitors
3.1. Small Molecule Sirtuin Inhibitors Targeting the Acylated Substrate Binding Site
3.2. Small Molecule Sirtuin Inhibitors Targeting the NAD+ Binding Site
3.3. Adenosine Analogs as Small Molecule Sirtuin Inhibitors
3.4. Bivalent Small Molecule Sirtuin Inhibitors Targeting the Acylated Substrate and NAD+ Binding Sites
3.5. Allosteric Small Molecule Inhibitors
4. Mechanism-Based Sirtuin Inhibitors
4.1. Mechanism-Based Sirtuin Inhibition by Nicotinamide
4.2. Mechanism-Based Sirtuin Inhibition by Thioacetyl-Lysine
4.3. Mechanism-Based Sirtuin Inhibition by Thioacyl-Lysine Derivatives
4.4. Mechanism-Based Sirtuin Inhibition by Thiourea-Lysine Derivatives
4.5. Mechanism-Based Sirtuin Inhibition by Alternative Compounds
4.6. Mechanism-Based Sirtuin Inhibition with Cyclic Peptides
5. Peptidic Non-Mechanism-Based Sirtuin Inhibitors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Grozinger, C.M.; Chao, E.D.; Blackwell, H.E.; Moazed, D.; Schreiber, S.L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 2001, 276, 38837–38843. [Google Scholar] [CrossRef] [PubMed]
- Rine, J.; Herskowitz, I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics 1987, 116, 9–22. [Google Scholar] [CrossRef]
- Loo, S.; Rine, J. Silencers and Domains of Generalized Repression. Science 1994, 264, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Michan, S.; Sinclair, D. Sirtuins in mammals: Insights into their biological function. Biochem. J. 2007, 404, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.; Slama, J.T.; Sternglanz, R. Role of NAD(+) in the deacetylase activity of the SIR2-like proteins. Biochem. Biophys. Res. Commun. 2000, 278, 685–690. [Google Scholar] [CrossRef]
- Jackson, M.D.; Denu, J.M. Structural identification of 2′- and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta-NAD+-dependent histone/protein deacetylases. J. Biol. Chem. 2002, 277, 18535–18544. [Google Scholar] [CrossRef]
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic control by sirtuins and other enzymes that sense NAD+, NADH, or their ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The Substrate Specificity of Sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.J.G.; Pereira, J.M.; Impens, F.; Hamon, M.A. Active nuclear import of the deacetylase Sirtuin-2 is controlled by its C-terminus and importins. Sci. Rep. 2020, 10, 2034. [Google Scholar] [CrossRef] [PubMed]
- Cooper, H.M.; Spelbrink, J.N. The human SIRT3 protein deacetylase is exclusively mitochondrial. Biochem. J. 2008, 411, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Albaugh, B.N.; Denu, J.M. Where in the cell is SIRT3? Functional localization of an NAD+-dependent protein deacetylase. Biochem. J. 2008, 411, e11–e13. [Google Scholar] [CrossRef] [PubMed]
- Scher, M.B.; Vaquero, A.; Reinberg, D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev. 2007, 21, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef]
- Sanders, B.D.; Jackson, B.; Marmorstein, R. Structural basis for sirtuin function: What we know and what we don’t. Biochim. Biophys. Acta 2010, 1804, 1604–1616. [Google Scholar] [CrossRef]
- Jin, L.; Wei, W.; Jiang, Y.; Peng, H.; Cai, J.; Mao, C.; Dai, H.; Choy, W.; Bemis, J.E.; Jirousek, M.R.; et al. Crystal Structures of Human SIRT3 Displaying Substrate-induced Conformational Changes. J. Biol. Chem. 2009, 284, 24394–24405. [Google Scholar] [CrossRef]
- Smith, B.C.; Settles, B.; Hallows, W.C.; Craven, M.W.; Denu, J.M. SIRT3 substrate specificity determined by peptide arrays and machine learning. ACS Chem. Biol. 2011, 6, 146–157. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef]
- Wang, M.; Lin, H. Understanding the Function of Mammalian Sirtuins and Protein Lysine Acylation. Annu. Rev. Biochem. 2021, 90, 245–285. [Google Scholar] [CrossRef]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Kudo, N.; Thelen, J.N.; Ito, A.; Yoshida, M.; Denu, J.M. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD+ Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry 2015, 54, 3037–3050. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J. Biol. Chem. 2007, 282, 37256–37265. [Google Scholar] [CrossRef]
- Zhang, X.; Cao, R.; Niu, J.; Yang, S.; Ma, H.; Zhao, S.; Li, H. Molecular basis for hierarchical histone de-β-hydroxybutyrylation by SIRT3. Cell Discov. 2019, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Wang, Y.; Li, X.; Li, X.M.; Liu, Z.; Yang, T.; Wong, C.F.; Zhang, J.; Hao, Q.; Li, X.D. Identification of ‘erasers’ for lysine crotonylated histone marks using a chemical proteomics approach. Elife 2014, 3, e02999. [Google Scholar] [CrossRef]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Anderson, K.A.; Huynh, F.K.; Fisher-Wellman, K.; Stuart, J.D.; Peterson, B.S.; Douros, J.D.; Wagner, G.R.; Thompson, J.W.; Madsen, A.S.; Green, M.F.; et al. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017, 25, 838–855.e815. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Greco, T.M.; Oberstein, A.; Budayeva, H.G.; Chakrabarti, R.; Rowland, E.A.; Kang, Y.; Shenk, T.; Cristea, I.M. Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 2014, 159, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Khan, S.; Wang, Y.; Charron, G.; He, B.; Sebastian, C.; Du, J.; Kim, R.; Ge, E.; Mostoslavsky, R.; et al. SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Gil, R.; Barth, S.; Kanfi, Y.; Cohen, H.Y. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic Acids Res. 2013, 41, 8537–8545. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Zheng, J.; Feldman, J.L.; Klein, M.A.; Kuznetsov, V.I.; Peterson, C.L.; Griffin, P.R.; Denu, J.M. Multivalent interactions drive nucleosome binding and efficient chromatin deacetylation by SIRT6. Nat. Commun. 2020, 11, 5244. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsov, V.I.; Liu, W.H.; Klein, M.A.; Denu, J.M. Potent Activation of NAD+-Dependent Deacetylase Sirt7 by Nucleosome Binding. ACS Chem. Biol. 2022, 17, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Wang, Y.; Zhang, X.; Kim, D.D.; Sadhukhan, S.; Hao, Q.; Lin, H. SIRT7 Is Activated by DNA and Deacetylates Histone H3 in the Chromatin Context. ACS Chem. Biol. 2016, 11, 742–747. [Google Scholar] [CrossRef]
- Tong, Z.; Wang, M.; Wang, Y.; Kim, D.D.; Grenier, J.K.; Cao, J.; Sadhukhan, S.; Hao, Q.; Lin, H. SIRT7 Is an RNA-Activated Protein Lysine Deacylase. ACS Chem. Biol. 2017, 12, 300–310. [Google Scholar] [CrossRef]
- Dai, H.; Case, A.W.; Riera, T.V.; Considine, T.; Lee, J.E.; Hamuro, Y.; Zhao, H.; Jiang, Y.; Sweitzer, S.M.; Pietrak, B.; et al. Crystallographic structure of a small molecule SIRT1 activator-enzyme complex. Nat. Commun. 2015, 6, 7645. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef]
- Kim, J.E.; Chen, J.; Lou, Z. DBC1 is a negative regulator of SIRT1. Nature 2008, 451, 583–586. [Google Scholar] [CrossRef]
- Verdin, E. AROuSing SIRT1: Identification of a novel endogenous SIRT1 activator. Mol. Cell 2007, 28, 354–356. [Google Scholar] [CrossRef]
- Pan, M.; Yuan, H.; Brent, M.; Ding, E.C.; Marmorstein, R. SIRT1 contains N- and C-terminal regions that potentiate deacetylase activity. J. Biol. Chem. 2012, 287, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Kalous, K.S.; Wynia-Smith, S.L.; Smith, B.C. Sirtuin Oxidative Post-translational Modifications. Front. Physiol. 2021, 12, 763417. [Google Scholar] [CrossRef]
- Flick, F.; Lüscher, B. Regulation of Sirtuin Function by Posttranslational Modifications. Front. Pharmacol. 2012, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The sirtuin family in cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef]
- Bindu, S.; Pillai, V.B.; Gupta, M.P. Role of Sirtuins in Regulating Pathophysiology of the Heart. Trends Endocrinol. Metab. 2016, 27, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Yeong, K.Y.; Berdigaliyev, N.; Chang, Y. Sirtuins and Their Implications in Neurodegenerative Diseases from a Drug Discovery Perspective. ACS Chem. Neuro 2020, 11, 4073–4091. [Google Scholar] [CrossRef]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Karaman Mayack, B.; Sippl, W.; Ntie-Kang, F. Natural Products as Modulators of Sirtuins. Molecules 2020, 25, 3287. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, J.; Chen, D.; Yan, L.; Zheng, W. Sirtuin Inhibition: Strategies, Inhibitors, and Therapeutic Potential. Trends Pharmacol. Sci. 2017, 38, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Loharch, S.; Chhabra, S.; Kumar, A.; Swarup, S.; Parkesh, R. Discovery and characterization of small molecule SIRT3-specific inhibitors as revealed by mass spectrometry. Bioorg. Chem. 2021, 110, 104768. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef]
- Lin, S.-J.; Defossez, P.-A.; Guarente, L. Requirement of NAD and SIR2 for Life-Span Extension by Calorie Restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.P.; Bemis, E.J.; Disch, S.J.; Vu, B.C.; Oalmann, J.C.; Lynch, V.A.; Carney, P.D.; Riera, V.T.; Song, J.; Smith, J.J.; et al. The Identification of the SIRT1 Activator SRT2104 as a Clinical Candidate. Lett. Drug Des. Discov. 2013, 10, 793–797. [Google Scholar] [CrossRef]
- Pillai, V.B.; Samant, S.; Sundaresan, N.R.; Raghuraman, H.; Kim, G.; Bonner, M.Y.; Arbiser, J.L.; Walker, D.I.; Jones, D.P.; Gius, D.; et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat. Commun. 2015, 6, 6656. [Google Scholar] [CrossRef]
- Zhang, J.; Zou, L.; Shi, D.; Liu, J.; Zhang, J.; Zhao, R.; Wang, G.; Zhang, L.; Ouyang, L.; Liu, B. Structure-Guided Design of a Small-Molecule Activator of Sirtuin-3 that Modulates Autophagy in Triple Negative Breast Cancer. J. Med. Chem. 2021, 64, 14192–14216. [Google Scholar] [CrossRef]
- Suenkel, B.; Valente, S.; Zwergel, C.; Weiss, S.; Di Bello, E.; Fioravanti, R.; Aventaggiato, M.; Amorim, J.A.; Garg, N.; Kumar, S.; et al. Potent and Specific Activators for Mitochondrial Sirtuins Sirt3 and Sirt5. J. Med. Chem. 2022, 65, 14015–14031. [Google Scholar] [CrossRef]
- You, W.; Rotili, D.; Li, T.M.; Kambach, C.; Meleshin, M.; Schutkowski, M.; Chua, K.F.; Mai, A.; Steegborn, C. Structural Basis of Sirtuin 6 Activation by Synthetic Small Molecules. Angew. Chem. Int. Ed. Engl. 2017, 56, 1007–1011. [Google Scholar] [CrossRef]
- Klein, M.A.; Liu, C.; Kuznetsov, V.I.; Feltenberger, J.B.; Tang, W.; Denu, J.M. Mechanism of activation for the sirtuin 6 protein deacylase. J. Biol. Chem. 2020, 295, 1385–1399. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Shang, J.; Gao, C.; Guan, X.; Chen, Y.; Zhu, L.; Zhang, L.; Zhang, C.; Zhang, J.; Pang, T. A novel SIRT6 activator ameliorates neuroinflammation and ischemic brain injury via EZH2/FOXC1 axis. Acta Pharm. Sin. B 2021, 11, 708–726. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, W.; Huang, S.; Zhang, H.; Lin, G.; Li, H.; Qiao, J.; Li, L.; Yang, S. Discovery of Potent Small-Molecule SIRT6 Activators: Structure–Activity Relationship and Anti-Pancreatic Ductal Adenocarcinoma Activity. J. Med. Chem. 2020, 63, 10474–10495. [Google Scholar] [CrossRef]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer Chemopreventive Activity of Resveratrol, a Natural Product Derived from Grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; McDonagh, T.; Heltweg, B.; Hixon, J.; Westman, E.A.; Caldwell, S.D.; Napper, A.; Curtis, R.; DiStefano, P.S.; Fields, S.; et al. Substrate-specific activation of sirtuins by resveratrol. J. Biol. Chem. 2005, 280, 17038–17045. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Ray, U.; Roy, S.S.; Chowdhury, S.R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell Physiol. Biochem. 2017, 41, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Pirola, L.; Fröjdö, S. Resveratrol: One molecule, many targets. IUBMB Life 2008, 60, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wilk, S.A.; Wang, A.; Zhou, L.; Wang, R.H.; Ogawa, W.; Deng, C.; Dong, L.Q.; Liu, F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J. Biol. Chem. 2010, 285, 36387–36394. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Nguyen, G.T.; Fischer, F.; Suenkel, B.; Schlicker, C.; Fränzel, B.; Tomaschewski, J.; Aladini, F.; Becker, C.; Wolters, D.; et al. A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 2012, 7, e49761. [Google Scholar] [CrossRef]
- Yang, H.; Baur, J.A.; Chen, A.; Miller, C.; Adams, J.K.; Kisielewski, A.; Howitz, K.T.; Zipkin, R.E.; Sinclair, D.A. Design and synthesis of compounds that extend yeast replicative lifespan. Aging Cell 2007, 6, 35–43. [Google Scholar] [CrossRef]
- Biasutto, L.; Mattarei, A.; Marotta, E.; Bradaschia, A.; Sassi, N.; Garbisa, S.; Zoratti, M.; Paradisi, C. Development of mitochondria-targeted derivatives of resveratrol. Bioorg. Med. Chem. Lett. 2008, 18, 5594–5597. [Google Scholar] [CrossRef]
- Nayagam, V.M.; Wang, X.; Tan, Y.C.; Poulsen, A.; Goh, K.C.; Ng, T.; Wang, H.; Song, H.Y.; Ni, B.; Entzeroth, M.; et al. SIRT1 modulating compounds from high-throughput screening as anti-inflammatory and insulin-sensitizing agents. J. Biomol. Screen 2006, 11, 959–967. [Google Scholar] [CrossRef]
- Bemis, J.E.; Vu, C.B.; Xie, R.; Nunes, J.J.; Ng, P.Y.; Disch, J.S.; Milne, J.C.; Carney, D.P.; Lynch, A.V.; Jin, L.; et al. Discovery of oxazolo[4,5-b]pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg. Med. Chem. Lett. 2009, 19, 2350–2353. [Google Scholar] [CrossRef] [PubMed]
- Vu, C.B.; Bemis, J.E.; Disch, J.S.; Ng, P.Y.; Nunes, J.J.; Milne, J.C.; Carney, D.P.; Lynch, A.V.; Smith, J.J.; Lavu, S.; et al. Discovery of Imidazo[1,2-b]thiazole Derivatives as Novel SIRT1 Activators. J. Med. Chem. 2009, 52, 1275–1283. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Schaefer, S.; Gertz, M.; Weyand, M.; Steegborn, C. Structures of human sirtuin 3 complexes with ADP-ribose and with carba-NAD+ and SRT1720: Binding details and inhibition mechanism. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1423–1432. [Google Scholar] [CrossRef]
- Pacholec, M.; Bleasdale, J.E.; Chrunyk, B.; Cunningham, D.; Flynn, D.; Garofalo, R.S.; Griffith, D.; Griffor, M.; Loulakis, P.; Pabst, B.; et al. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J. Biol. Chem. 2010, 285, 8340–8351. [Google Scholar] [CrossRef]
- Dai, H.; Kustigian, L.; Carney, D.; Case, A.; Considine, T.; Hubbard, B.P.; Perni, R.B.; Riera, T.V.; Szczepankiewicz, B.; Vlasuk, G.P.; et al. SIRT1 activation by small molecules: Kinetic and biophysical evidence for direct interaction of enzyme and activator. J. Biol. Chem. 2010, 285, 32695–32703. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Gomes, A.P.; Dai, H.; Li, J.; Case, A.W.; Considine, T.; Riera, T.V.; Lee, J.E.; E, S.Y.; Lamming, D.W.; et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013, 339, 1216–1219. [Google Scholar] [CrossRef]
- Minor, R.K.; Baur, J.A.; Gomes, A.P.; Ward, T.M.; Csiszar, A.; Mercken, E.M.; Abdelmohsen, K.; Shin, Y.K.; Canto, C.; Scheibye-Knudsen, M.; et al. SRT1720 improves survival and healthspan of obese mice. Sci. Rep. 2011, 1, 70. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef]
- Park, S.J.; Ahmad, F.; Um, J.H.; Brown, A.L.; Xu, X.; Kang, H.; Ke, H.; Feng, X.; Ryall, J.; Philp, A.; et al. Specific Sirt1 Activator-mediated Improvement in Glucose Homeostasis Requires Sirt1-Independent Activation of AMPK. EBioMedicine 2017, 18, 128–138. [Google Scholar] [CrossRef]
- Hoffmann, E.; Wald, J.; Lavu, S.; Roberts, J.; Beaumont, C.; Haddad, J.; Elliott, P.; Westphal, C.; Jacobson, E. Pharmacokinetics and tolerability of SRT2104, a first-in-class small molecule activator of SIRT1, after single and repeated oral administration in man. Br. J. Clin. Pharmacol. 2013, 75, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Libri, V.; Brown, A.P.; Gambarota, G.; Haddad, J.; Shields, G.S.; Dawes, H.; Pinato, D.J.; Hoffman, E.; Elliot, P.J.; Vlasuk, G.P.; et al. A pilot randomized, placebo controlled, double blind phase I trial of the novel SIRT1 activator SRT2104 in elderly volunteers. PLoS ONE 2012, 7, e51395. [Google Scholar] [CrossRef]
- Venkatasubramanian, S.; Noh, R.M.; Daga, S.; Langrish, J.P.; Joshi, N.V.; Mills, N.L.; Hoffmann, E.; Jacobson, E.W.; Vlasuk, G.P.; Waterhouse, B.R.; et al. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J. Am. Heart Assoc. 2013, 2, e000042. [Google Scholar] [CrossRef]
- Baksi, A.; Kraydashenko, O.; Zalevkaya, A.; Stets, R.; Elliott, P.; Haddad, J.; Hoffmann, E.; Vlasuk, G.P.; Jacobson, E.W. A phase II, randomized, placebo-controlled, double-blind, multi-dose study of SRT2104, a SIRT1 activator, in subjects with type 2 diabetes. Br. J. Clin. Pharmacol. 2014, 78, 69–77. [Google Scholar] [CrossRef]
- van der Meer, A.J.; Scicluna, B.P.; Moerland, P.D.; Lin, J.; Jacobson, E.W.; Vlasuk, G.P.; van der Poll, T. The Selective Sirtuin 1 Activator SRT2104 Reduces Endotoxin-Induced Cytokine Release and Coagulation Activation in Humans. Crit. Care Med. 2015, 43, e199–e202. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Suárez-Fariñas, M.; Cueto, I.; Khacherian, A.; Matheson, R.; Parish, L.C.; Leonardi, C.; Shortino, D.; Gupta, A.; Haddad, J.; et al. A Randomized, Placebo-Controlled Study of SRT2104, a SIRT1 Activator, in Patients with Moderate to Severe Psoriasis. PLoS ONE 2015, 10, e0142081. [Google Scholar] [CrossRef]
- Sands, B.E.; Joshi, S.; Haddad, J.; Freudenberg, J.M.; Oommen, D.E.; Hoffmann, E.; McCallum, S.W.; Jacobson, E. Assessing Colonic Exposure, Safety, and Clinical Activity of SRT2104, a Novel Oral SIRT1 Activator, in Patients with Mild to Moderate Ulcerative Colitis. Inflamm. Bowel Dis. 2016, 22, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Mai, A.; Valente, S.; Meade, S.; Carafa, V.; Tardugno, M.; Nebbioso, A.; Galmozzi, A.; Mitro, N.; De Fabiani, E.; Altucci, L.; et al. Study of 1,4-dihydropyridine structural scaffold: Discovery of novel sirtuin activators and inhibitors. J. Med. Chem. 2009, 52, 5496–5504. [Google Scholar] [CrossRef]
- Valente, S.; Mellini, P.; Spallotta, F.; Carafa, V.; Nebbioso, A.; Polletta, L.; Carnevale, I.; Saladini, S.; Trisciuoglio, D.; Gabellini, C.; et al. 1,4-Dihydropyridines Active on the SIRT1/AMPK Pathway Ameliorate Skin Repair and Mitochondrial Function and Exhibit Inhibition of Proliferation in Cancer Cells. J. Med. Chem. 2016, 59, 1471–1491. [Google Scholar] [CrossRef]
- Zwergel, C.; Aventaggiato, M.; Garbo, S.; Di Bello, E.; Fassari, B.; Noce, B.; Castiello, C.; Lambona, C.; Barreca, F.; Rotili, D.; et al. Novel 1,4-Dihydropyridines as Specific Binders and Activators of SIRT3 Impair Cell Viability and Clonogenicity and Downregulate Hypoxia-Induced Targets in Cancer Cells. J. Med. Chem. 2023, 66, 9622–9641. [Google Scholar] [CrossRef]
- Hu, T.; Shukla, S.K.; Vernucci, E.; He, C.; Wang, D.; King, R.J.; Jha, K.; Siddhanta, K.; Mullen, N.J.; Attri, K.S.; et al. Metabolic Rewiring by Loss of Sirt5 Promotes Kras-Induced Pancreatic Cancer Progression. Gastroenterology 2021, 161, 1584–1600. [Google Scholar] [CrossRef]
- Schlicker, C.; Boanca, G.; Lakshminarasimhan, M.; Steegborn, C. Structure-based development of novel sirtuin inhibitors. Aging 2011, 3, 852–872. [Google Scholar] [CrossRef]
- Iachettini, S.; Trisciuoglio, D.; Rotili, D.; Lucidi, A.; Salvati, E.; Zizza, P.; Di Leo, L.; Del Bufalo, D.; Ciriolo, M.R.; Leonetti, C.; et al. Pharmacological activation of SIRT6 triggers lethal autophagy in human cancer cells. Cell Death Dis. 2018, 9, 996. [Google Scholar] [CrossRef]
- Xu, J.; Shi, S.; Liu, G.; Xie, X.; Li, J.; Bolinger, A.A.; Chen, H.; Zhang, W.; Shi, P.Y.; Liu, H.; et al. Design, synthesis, and pharmacological evaluations of pyrrolo[1,2-a]quinoxaline-based derivatives as potent and selective sirt6 activators. Eur. J. Med. Chem. 2023, 246, 114998. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef]
- Shang, J.-l.; Ning, S.-b.; Chen, Y.-y.; Chen, T.-x.; Zhang, J. MDL-800, an allosteric activator of SIRT6, suppresses proliferation and enhances EGFR-TKIs therapy in non-small cell lung cancer. Acta Pharmacol. Sin. 2021, 42, 120–131. [Google Scholar] [CrossRef]
- Shang, J.; Zhu, Z.; Chen, Y.; Song, J.; Huang, Y.; Song, K.; Zhong, J.; Xu, X.; Wei, J.; Wang, C.; et al. Small-molecule activating SIRT6 elicits therapeutic effects and synergistically promotes anti-tumor activity of vitamin D(3) in colorectal cancer. Theranostics 2020, 10, 5845–5864. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, A.; Dölle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011, 286, 21767–21778. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.D.; Schmidt, M.T.; Oppenheimer, N.J.; Denu, J.M. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003, 278, 50985–50998. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A.; Moir, R.D.; Schramm, V.L.; Willis, I.M. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol. Cell 2005, 17, 595–601. [Google Scholar] [CrossRef]
- Kalous, K.S.; Wynia-Smith, S.L.; Summers, S.B.; Smith, B.C. Human sirtuins are differentially sensitive to inhibition by nitrosating agents and other cysteine oxidants. J. Biol. Chem. 2020, 295, 8524–8536. [Google Scholar] [CrossRef]
- Kokkola, T.; Suuronen, T.; Pesonen, M.; Filippakopoulos, P.; Salminen, A.; Jarho, E.M.; Lahtela-Kakkonen, M. BET Inhibition Upregulates SIRT1 and Alleviates Inflammatory Responses. Chembiochem 2015, 16, 1997–2001. [Google Scholar] [CrossRef]
- Tenhunen, J.; Kokkola, T.; Huovinen, M.; Rahnasto-Rilla, M.; Lahtela-Kakkonen, M. Impact of structurally diverse BET inhibitors on SIRT1. Gene 2020, 741, 144558. [Google Scholar] [CrossRef]
- Järvenpää, J.; Rahnasto-Rilla, M.; Lahtela-Kakkonen, M.; Küblbeck, J. Profiling the regulatory interplay of BET bromodomains and Sirtuins in cancer cell lines. Biomed. Pharmacother. 2022, 147, 112652. [Google Scholar] [CrossRef]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef]
- Di Fruscia, P.; Zacharioudakis, E.; Liu, C.; Moniot, S.; Laohasinnarong, S.; Khongkow, M.; Harrison, I.F.; Koltsida, K.; Reynolds, C.R.; Schmidtkunz, K.; et al. The discovery of a highly selective 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidin-4(3H)-one SIRT2 inhibitor that is neuroprotective in an in vitro Parkinson’s disease model. ChemMedChem 2015, 10, 69–82. [Google Scholar] [CrossRef]
- Gertz, M.; Fischer, F.; Nguyen, G.T.T.; Lakshminarasimhan, M.; Schutkowski, M.; Weyand, M.; Steegborn, C. Ex-527 inhibits Sirtuins by exploiting their unique NAD+-dependent deacetylation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, E2772–E2781. [Google Scholar] [CrossRef] [PubMed]
- Trapp, J.; Jochum, A.; Meier, R.; Saunders, L.; Marshall, B.; Kunick, C.; Verdin, E.; Goekjian, P.; Sippl, W.; Jung, M. Adenosine mimetics as inhibitors of NAD+-dependent histone deacetylases, from kinase to sirtuin inhibition. J. Med. Chem. 2006, 49, 7307–7316. [Google Scholar] [CrossRef] [PubMed]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Kudo, N.; Ito, A.; Arata, M.; Nakata, A.; Yoshida, M. Identification of a novel small molecule that inhibits deacetylase but not defatty-acylase reaction catalysed by SIRT2. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170070. [Google Scholar] [CrossRef] [PubMed]
- Disch, J.S.; Evindar, G.; Chiu, C.H.; Blum, C.A.; Dai, H.; Jin, L.; Schuman, E.; Lind, K.E.; Belyanskaya, S.L.; Deng, J.; et al. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. [Google Scholar] [CrossRef]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef]
- Ai, T.; Wilson, D.J.; More, S.S.; Xie, J.; Chen, L. 5-((3-Amidobenzyl)oxy)nicotinamides as Sirtuin 2 Inhibitors. J. Med. Chem. 2016, 59, 2928–2941. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Taylor, D.M.; Balabadra, U.; Xiang, Z.; Woodman, B.; Meade, S.; Amore, A.; Maxwell, M.M.; Reeves, S.; Bates, G.P.; Luthi-Carter, R.; et al. A brain-permeable small molecule reduces neuronal cholesterol by inhibiting activity of sirtuin 2 deacetylase. ACS Chem. Biol. 2011, 6, 540–546. [Google Scholar] [CrossRef]
- Quinti, L.; Casale, M.; Moniot, S.; Pais, T.F.; Van Kanegan, M.J.; Kaltenbach, L.S.; Pallos, J.; Lim, R.G.; Naidu, S.D.; Runne, H.; et al. SIRT2- and NRF2-Targeting Thiazole-Containing Compound with Therapeutic Activity in Huntington’s Disease Models. Cell Chem. Biol. 2016, 23, 849–861. [Google Scholar] [CrossRef]
- Pannek, M.; Alhalabi, Z.; Tomaselli, D.; Menna, M.; Fiorentino, F.; Robaa, D.; Weyand, M.; Puhlmann, M.; Tomassi, S.; Barreca, F.; et al. Specific Inhibitors of Mitochondrial Deacylase Sirtuin 4 Endowed with Cellular Activity. J. Med. Chem. 2024, 67, 1843–1860. [Google Scholar] [CrossRef]
- Yao, J.; Yin, Y.; Han, H.; Chen, S.; Zheng, Y.; Liang, B.; Wu, M.; Shu, K.; Debnath, B.; Lombard, D.B.; et al. Pyrazolone derivatives as potent and selective small-molecule SIRT5 inhibitors. Eur. J. Med. Chem. 2023, 247, 115024. [Google Scholar] [CrossRef]
- Lara, E.; Mai, A.; Calvanese, V.; Altucci, L.; Lopez-Nieva, P.; Martinez-Chantar, M.L.; Varela-Rey, M.; Rotili, D.; Nebbioso, A.; Ropero, S.; et al. Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 2009, 28, 781–791. [Google Scholar] [CrossRef]
- Bedalov, A.; Gatbonton, T.; Irvine, W.P.; Gottschling, D.E.; Simon, J.A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl. Acad. Sci. USA 2001, 98, 15113–15118. [Google Scholar] [CrossRef]
- Heltweg, B.; Gatbonton, T.; Schuler, A.D.; Posakony, J.; Li, H.; Goehle, S.; Kollipara, R.; Depinho, R.A.; Gu, Y.; Simon, J.A.; et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006, 66, 4368–4377. [Google Scholar] [CrossRef]
- Mahajan, S.S.; Scian, M.; Sripathy, S.; Posakony, J.; Lao, U.; Loe, T.K.; Leko, V.; Thalhofer, A.; Schuler, A.D.; Bedalov, A.; et al. Development of pyrazolone and isoxazol-5-one cambinol analogues as sirtuin inhibitors. J. Med. Chem. 2014, 57, 3283–3294. [Google Scholar] [CrossRef]
- Chowdhury, S.; Sripathy, S.; Webster, A.A.; Park, A.; Lao, U.; Hsu, J.H.; Loe, T.; Bedalov, A.; Simon, J.A. Discovery of Selective SIRT2 Inhibitors as Therapeutic Agents in B-Cell Lymphoma and Other Malignancies. Molecules 2020, 25, 455. [Google Scholar] [CrossRef] [PubMed]
- Garske, A.L.; Smith, B.C.; Denu, J.M. Linking SIRT2 to Parkinson’s Disease. ACS Chem. Biol. 2007, 2, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Yang, L.; Hou, S.; Wang, B.; Wang, X.; Hu, L.; Deng, J.; Liu, J.; Chen, X.; Jiang, Y.; et al. Structure–Activity Relationship Studies of 2,4,5-Trisubstituted Pyrimidine Derivatives Leading to the Identification of a Novel and Potent Sirtuin 5 Inhibitor against Sepsis-Associated Acute Kidney Injury. J. Med. Chem. 2023, 66, 11517–11535. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Su, H.; Deng, J.; Mou, L.; Wang, H.; Li, R.; Dai, Q.-Q.; Yan, Y.-H.; Qian, S.; Wang, Z.; et al. Discovery of new human Sirtuin 5 inhibitors by mimicking glutaryl-lysine substrates. Eur. J. Med. Chem. 2021, 225, 113803. [Google Scholar] [CrossRef]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006, 26, 28–38. [Google Scholar] [CrossRef]
- Min, J.; Landry, J.; Sternglanz, R.; Xu, R.-M. Crystal Structure of a SIR2 Homolog–NAD Complex. Cell 2001, 105, 269–279. [Google Scholar] [CrossRef]
- Smith, M.R.; Syed, A.; Lukacsovich, T.; Purcell, J.; Barbaro, B.A.; Worthge, S.A.; Wei, S.R.; Pollio, G.; Magnoni, L.; Scali, C.; et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 2995–3007. [Google Scholar] [CrossRef]
- Süssmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef]
- Kundu, A.; Richa, S.; Dey, P.; Kim, K.S.; Son, J.Y.; Kim, H.R.; Lee, S.Y.; Lee, B.H.; Lee, K.Y.; Kacew, S.; et al. Protective effect of EX-527 against high-fat diet-induced diabetic nephropathy in Zucker rats. Toxicol. Appl. Pharmacol. 2020, 390, 114899. [Google Scholar] [CrossRef]
- Spinck, M.; Bischoff, M.; Lampe, P.; Meyer-Almes, F.-J.; Sievers, S.; Neumann, H. Discovery of Dihydro-1,4-Benzoxazine Carboxamides as Potent and Highly Selective Inhibitors of Sirtuin-1. J. Med. Chem. 2021, 64, 5838–5849. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, T.; Schüler, H. Sirtuins are Unaffected by PARP Inhibitors Containing Planar Nicotinamide Bioisosteres. Chem. Biol. Drug Des. 2016, 87, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Chen, C.-Y.; Ho, K.-K.; Di Fruscia, P.; Myatt, S.S.; Coombes, R.C.; Fuchter, M.J.; Hsiao, C.-D.; Lam, E.W.F. SIRT Inhibitors Induce Cell Death and p53 Acetylation through Targeting Both SIRT1 and SIRT2. Mol. Cancer Ther. 2010, 9, 844–855. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.J.; Meares, G.P.; Hansen, P.A.; Corbett, J.A. FoxO1 and SIRT1 regulate beta-cell responses to nitric oxide. J. Biol. Chem. 2011, 286, 8338–8348. [Google Scholar] [CrossRef] [PubMed]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Herisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The sirtuin-2 inhibitor AK7 is neuroprotective in models of Parkinson’s disease but not amyotrophic lateral sclerosis and cerebral ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Kim, J.; Vollor, L.; Narayanan, K.L.; Edgerly, C.; Cipicchio, P.M.; Lauver, M.A.; Choi, S.H.; Silverman, R.B.; et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012, 2, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.D.; Hill, C.H.; Lawton, G.; Nixon, J.S.; Wilkinson, S.E.; Hurst, S.A.; Keech, E.; Turner, S.E. Inhibitors of protein kinase C. 1. 2,3-bisarylmaleimides. J. Med. Chem. 1992, 35, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kamal, Z.; Ai, T.; Xu, Y.; More, S.S.; Wilson, D.J.; Chen, L. Discovery of potent and selective sirtuin 2 (SIRT2) inhibitors using a fragment-based approach. J. Med. Chem. 2014, 57, 8340–8357. [Google Scholar] [CrossRef]
- Ai, T.; Wilson, D.J.; Chen, L. 5-((3-Amidobenzyl)oxy)nicotinamides as SIRT2 Inhibitors: A Study of Constrained Analogs. Molecules 2023, 28, 7655. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Rosenberg, M.A.; Jeganathan, K.B.; Hafner, A.V.; Michan, S.; Dai, J.; Baker, D.J.; Cen, Y.; Wu, L.E.; Sauve, A.A.; et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J. 2014, 33, 1438–1453. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 1599–1612. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-Based Development of an Affinity Probe for Sirtuin 2. Angew. Chem. Int. Ed. Engl. 2016, 55, 2252–2256. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Xu, Y.; Ai, T.; Chen, L.; Verdin, E.; Kapila, Y.L. A Novel Sirtuin-3 Inhibitor, LC-0296, Inhibits Cell Survival and Proliferation, and Promotes Apoptosis of Head and Neck Cancer Cells. Anticancer. Res. 2016, 36, 49–60. [Google Scholar]
- Wood, M.; Rymarchyk, S.; Zheng, S.; Cen, Y. Trichostatin A inhibits deacetylation of histone H3 and p53 by SIRT6. Arch. Biochem. Biophys. 2018, 638, 8–17. [Google Scholar] [CrossRef]
- You, W.; Steegborn, C. Structural Basis of Sirtuin 6 Inhibition by the Hydroxamate Trichostatin A: Implications for Protein Deacylase Drug Development. J. Med. Chem. 2018, 61, 10922–10928. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C. Chapter 32—Pharmacoepigenomics. In Medical Epigenetics; Tollefsbol, T.O., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 585–617. [Google Scholar]
- Ni, D.; Chai, Z.; Wang, Y.; Li, M.; Yu, Z.; Liu, Y.; Lu, S.; Zhang, J. Along the allostery stream: Recent advances in computational methods for allosteric drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1585. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.; Ni, D.; Huang, Z.; Wei, J.; Feng, L.; Su, J.-C.; Wei, Y.; Ning, S.; Yang, X.; et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm. Sin. B 2021, 12, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Guan, X.; Zhang, S.; Wang, Y.; Wang, X.; Lu, Z.; Chong, D.; Wang, J.Y.; Yu, R.; Yu, W.; et al. Discovery of a pyrrole-pyridinimidazole derivative as novel SIRT6 inhibitor for sensitizing pancreatic cancer to gemcitabine. Cell Death Dis. 2023, 14, 499. [Google Scholar] [CrossRef]
- Sundriyal, S.; Moniot, S.; Mahmud, Z.; Yao, S.; Di Fruscia, P.; Reynolds, C.R.; Dexter, D.T.; Sternberg, M.J.E.; Lam, E.W.F.; Steegborn, C.; et al. Thienopyrimidinone Based Sirtuin-2 (SIRT2)-Selective Inhibitors Bind in the Ligand Induced Selectivity Pocket. J. Med. Chem. 2017, 60, 1928–1945. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ma, X.; Yuan, C.; He, Y.; Li, L.; Fang, S.; Xia, W.; He, T.; Qian, S.; Xu, Z.; et al. Discovery of 2-((4,6-dimethylpyrimidin-2-yl)thio)-N-phenylacetamide derivatives as new potent and selective human sirtuin 2 inhibitors. Eur. J. Med. Chem. 2017, 134, 230–241. [Google Scholar] [CrossRef]
- Moniot, S.; Forgione, M.; Lucidi, A.; Hailu, G.S.; Nebbioso, A.; Carafa, V.; Baratta, F.; Altucci, L.; Giacché, N.; Passeri, D.; et al. Development of 1,2,4-Oxadiazoles as Potent and Selective Inhibitors of the Human Deacetylase Sirtuin 2: Structure–Activity Relationship, X-ray Crystal Structure, and Anticancer Activity. J. Med. Chem. 2017, 60, 2344–2360. [Google Scholar] [CrossRef]
- Yang, L.-L.; Wang, H.-L.; Zhong, L.; Yuan, C.; Liu, S.-Y.; Yu, Z.-J.; Liu, S.; Yan, Y.-H.; Wu, C.; Wang, Y.; et al. X-ray crystal structure guided discovery of new selective, substrate-mimicking sirtuin 2 inhibitors that exhibit activities against non-small cell lung cancer cells. Eur. J. Med. Chem. 2018, 155, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.L.; Xu, W.; Yan, J.; Su, H.L.; Yuan, C.; Li, C.; Zhang, X.; Yu, Z.J.; Yan, Y.H.; Yu, Y.; et al. Crystallographic and SAR analyses reveal the high requirements needed to selectively and potently inhibit SIRT2 deacetylase and decanoylase. Medchemcomm 2019, 10, 164–168. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Sir2 protein deacetylases: Evidence for chemical intermediates and functions of a conserved histidine. Biochemistry 2006, 45, 272–282. [Google Scholar] [CrossRef]
- Borra, M.T.; Langer, M.R.; Slama, J.T.; Denu, J.M. Substrate Specificity and Kinetic Mechanism of the Sir2 Family of NAD+-Dependent Histone/Protein Deacetylases. Biochemistry 2004, 43, 9877–9887. [Google Scholar] [CrossRef]
- Avalos, J.L.; Bever, K.M.; Wolberger, C. Mechanism of Sirtuin Inhibition by Nicotinamide: Altering the NAD+ Cosubstrate Specificity of a Sir2 Enzyme. Mol. Cell 2005, 17, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Fatkins, D.G.; Monnot, A.D.; Zheng, W. Nepsilon-thioacetyl-lysine: A multi-facet functional probe for enzymatic protein lysine Nepsilon-deacetylation. Bioorg. Med. Chem. Lett. 2006, 16, 3651–3656. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Mechanism-Based Inhibition of Sir2 Deacetylases by Thioacetyl-Lysine Peptide. Biochemistry 2007, 46, 14478–14486. [Google Scholar] [CrossRef] [PubMed]
- Fatkins, D.G.; Zheng, W. Substituting N(epsilon)-thioacetyl-lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacetylases. Int. J. Mol. Sci. 2008, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kiviranta, P.H.; Suuronen, T.; Wallén, E.A.A.; Leppänen, J.; Tervonen, J.; Kyrylenko, S.; Salminen, A.; Poso, A.; Jarho, E.M. Nϵ-Thioacetyl-Lysine-Containing Tri-, Tetra-, and Pentapeptides as SIRT1 and SIRT2 Inhibitors. J. Med. Chem. 2009, 52, 2153–2156. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, B.M.; Gallo, C.A.; Du, Z.; Wang, Z.; Zheng, W. Discovery of potent, proteolytically stable, and cell permeable human sirtuin peptidomimetic inhibitors containing Nε-thioacetyl-lysine. MedChemComm 2010, 1, 233–238. [Google Scholar] [CrossRef]
- Chen, B.; Wang, J.; Huang, Y.; Zheng, W. Human SIRT3 tripeptidic inhibitors containing N(ε)-thioacetyl-lysine. Bioorg. Med. Chem. Lett. 2015, 25, 3481–3487. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Asaba, T.; Imai, E.; Tsumoto, H.; Nakagawa, H.; Miyata, N. Identification of a cell-active non-peptide sirtuin inhibitor containing N-thioacetyl lysine. Bioorg. Med. Chem. Lett. 2009, 19, 5670–5672. [Google Scholar] [CrossRef]
- Huhtiniemi, T.; Salo, H.S.; Suuronen, T.; Poso, A.; Salminen, A.; Leppänen, J.; Jarho, E.; Lahtela-Kakkonen, M. Structure-based design of pseudopeptidic inhibitors for SIRT1 and SIRT2. J. Med. Chem. 2011, 54, 6456–6468. [Google Scholar] [CrossRef]
- Mellini, P.; Kokkola, T.; Suuronen, T.; Salo, H.S.; Tolvanen, L.; Mai, A.; Lahtela-Kakkonen, M.; Jarho, E.M. Screen of Pseudopeptidic Inhibitors of Human Sirtuins 1–3: Two Lead Compounds with Antiproliferative Effects in Cancer Cells. J. Med. Chem. 2013, 56, 6681–6695. [Google Scholar] [CrossRef]
- Zang, W.; Hao, Y.; Wang, Z.; Zheng, W. Novel thiourea-based sirtuin inhibitory warheads. Bioorg. Med. Chem. Lett. 2015, 25, 3319–3324. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, B.M.; Hao, Y.; Li, X.; Wesdemiotis, C.; Wang, Z.; Zheng, W. A mechanism-based potent sirtuin inhibitor containing Nε-thiocarbamoyl-lysine (TuAcK). Bioorg. Med. Chem. Lett. 2011, 21, 4753–4757. [Google Scholar] [CrossRef]
- Jing, H.; Hu, J.; He, B.; Negrón Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.-L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Hong, J.Y.; Hu, J.; Jing, H.; Wang, M.; Price, I.R.; Cao, J.; Yang, M.; Zhang, X.; Lin, H. A Small-Molecule SIRT2 Inhibitor That Promotes K-Ras4a Lysine Fatty-Acylation. ChemMedChem 2019, 14, 744–748. [Google Scholar] [CrossRef]
- Nielsen, A.L.; Rajabi, N.; Kudo, N.; Lundø, K.; Moreno-Yruela, C.; Bæk, M.; Fontenas, M.; Lucidi, A.; Madsen, A.S.; Yoshida, M.; et al. Mechanism-based inhibitors of SIRT2: Structure–activity relationship, X-ray structures, target engagement, regulation of α-tubulin acetylation and inhibition of breast cancer cell migration. RSC Chem. Biol. 2021, 2, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Price, I.R.; Bai, J.J.; Lin, H. A Glycoconjugated SIRT2 Inhibitor with Aqueous Solubility Allows Structure-Based Design of SIRT2 Inhibitors. ACS Chem. Biol. 2019, 14, 1802–1810. [Google Scholar] [CrossRef]
- Farooqi, A.S.; Hong, J.Y.; Cao, J.; Lu, X.; Price, I.R.; Zhao, Q.; Kosciuk, T.; Yang, M.; Bai, J.J.; Lin, H. Novel Lysine-Based Thioureas as Mechanism-Based Inhibitors of Sirtuin 2 (SIRT2) with Anticancer Activity in a Colorectal Cancer Murine Model. J. Med. Chem. 2019, 62, 4131–4141. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, J.; Yan, L.; Zheng, W. Simple N(ε)-thioacetyl-lysine-containing cyclic peptides exhibiting highly potent sirtuin inhibition. Bioorg. Med. Chem. Lett. 2016, 26, 1612–1617. [Google Scholar] [CrossRef]
- Li, R.; Yan, L.; Sun, X.; Zheng, W. A bicyclic pentapeptide-based highly potent and selective pan-SIRT1/2/3 inhibitor harboring N(ε)-thioacetyl-lysine. Bioorg. Med. Chem. 2020, 28, 115356. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, W. Cyclic peptide-based potent and selective SIRT1/2 dual inhibitors harboring N(ε)-thioacetyl-lysine. Bioorg. Med. Chem. Lett. 2016, 26, 5234–5239. [Google Scholar] [CrossRef]
- Li, M.; Chiang, Y.-L.; Lyssiotis, C.A.; Teater, M.R.; Hong, J.Y.; Shen, H.; Wang, L.; Hu, J.; Jing, H.; Chen, Z.; et al. Non-oncogene Addiction to SIRT3 Plays a Critical Role in Lymphomagenesis. Cancer Cell 2019, 35, 916–931.e919. [Google Scholar] [CrossRef]
- Troelsen, K.S.; Bæk, M.; Nielsen, A.L.; Madsen, A.S.; Rajabi, N.; Olsen, C.A. Mitochondria-targeted inhibitors of the human SIRT3 lysine deacetylase. RSC Chem. Biol. 2021, 2, 627–635. [Google Scholar] [CrossRef]
- Sauve, A.A.; Schramm, V.L. Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry 2003, 42, 9249–9256. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Du, J.; Lin, H. Thiosuccinyl Peptides as Sirt5-Specific Inhibitors. J. Am. Chem. Soc. 2012, 134, 1922–1925. [Google Scholar] [CrossRef]
- Hu, J.; He, B.; Bhargava, S.; Lin, H. A fluorogenic assay for screening Sirt6 modulators. Org. Biomol. Chem. 2013, 11, 5213–5216. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, N.; Auth, M.; Troelsen, K.R.; Pannek, M.; Bhatt, D.P.; Fontenas, M.; Hirschey, M.D.; Steegborn, C.; Madsen, A.S.; Olsen, C.A. Mechanism-Based Inhibitors of the Human Sirtuin 5 Deacylase: Structure-Activity Relationship, Biostructural, and Kinetic Insight. Angew. Chem. Int. Ed. Engl. 2017, 56, 14836–14841. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Franzini, A.; Pomicter, A.D.; Halverson, B.J.; Antelope, O.; Mason, C.C.; Ahmann, J.M.; Senina, A.V.; Vellore, N.A.; Jones, C.L.; et al. SIRT5 Is a Druggable Metabolic Vulnerability in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 266–287. [Google Scholar] [CrossRef]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition? J. Biol. Chem. 2009, 284, 3823–3832. [Google Scholar] [CrossRef]
- Alhazzazi, T.Y.; Kamarajan, P.; Joo, N.; Huang, J.Y.; Verdin, E.; D’Silva, N.J.; Kapila, Y.L. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 2011, 117, 1670–1678. [Google Scholar] [CrossRef]
- Petrack, B.; Greengard, P.; Craston, A.; Kalinsky, H.J. Nicotinamide deamidase in rat liver and the biosynthesis of NAD. Biochem. Bioph. Res. Commun. 1963, 13, 472–477. [Google Scholar] [CrossRef]
- Suzuki, T.; Imai, K.; Nakagawa, H.; Miyata, N. 2-Anilinobenzamides as SIRT inhibitors. ChemMedChem 2006, 1, 1059–1062. [Google Scholar] [CrossRef]
- Suzuki, T.; Khan, M.N.; Sawada, H.; Imai, E.; Itoh, Y.; Yamatsuta, K.; Tokuda, N.; Takeuchi, J.; Seko, T.; Nakagawa, H.; et al. Design, synthesis, and biological activity of a novel series of human sirtuin-2-selective inhibitors. J. Med. Chem. 2012, 55, 5760–5773. [Google Scholar] [CrossRef] [PubMed]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Mesenzani, O.; Coppo, C.; Sorba, G.; Canonico, P.L.; Tron, G.C.; Genazzani, A.A. Identification of a sirtuin 3 inhibitor that displays selectivity over sirtuin 1 and 2. Eur. J. Med. Chem. 2012, 55, 58–66. [Google Scholar] [CrossRef]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, A.R.; Pirrie, L.; Hollick, J.J.; Ronseaux, S.; Campbell, J.; Higgins, M.; Staples, O.D.; Tran, F.; Slawin, A.M.; Lain, S.; et al. Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg. Med. Chem. 2012, 20, 1779–1793. [Google Scholar] [CrossRef]
- McCarthy, A.R.; Sachweh, M.C.; Higgins, M.; Campbell, J.; Drummond, C.J.; van Leeuwen, I.M.; Pirrie, L.; Ladds, M.J.; Westwood, N.J.; Laín, S. Tenovin-D3, a novel small-molecule inhibitor of sirtuin SirT2, increases p21 (CDKN1A) expression in a p53-independent manner. Mol. Cancer Ther. 2013, 12, 352–360. [Google Scholar] [CrossRef]
- Kallander, L.S.; Lu, Q.; Chen, W.; Tomaszek, T.; Yang, G.; Tew, D.; Meek, T.D.; Hofmann, G.A.; Schulz-Pritchard, C.K.; Smith, W.W.; et al. 4-Aryl-1,2,3-triazole: A Novel Template for a Reversible Methionine Aminopeptidase 2 Inhibitor, Optimized To Inhibit Angiogenesis in Vivo. J. Med. Chem. 2005, 48, 5644–5647. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, U.F.; Awad, L.; Grosdidier, A.; Larrieu, P.; Stroobant, V.; Colau, D.; Cerundolo, V.; Simpson, A.J.G.; Vogel, P.; Van den Eynde, B.J.; et al. Rational Design of Indoleamine 2,3-Dioxygenase Inhibitors. J. Med. Chem. 2010, 53, 1172–1189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-Gómez, G.; Díaz-Chávez, J.; Chávez-Blanco, A.; Gonzalez-Fierro, A.; Jiménez-Salazar, J.E.; Damián-Matsumura, P.; Gómez-Quiroz, L.E.; Dueñas-González, A. Nicotinamide sensitizes human breast cancer cells to the cytotoxic effects of radiation and cisplatin. Oncol. Rep. 2015, 33, 721–728. [Google Scholar] [CrossRef]
- Berthelier, V.; Tixier, J.M.; Muller-Steffner, H.; Schuber, F.; Deterre, P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem. J. 1998, 330 Pt 3, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Burgos, E.S.; Schramm, V.L. Weak Coupling of ATP Hydrolysis to the Chemical Equilibrium of Human Nicotinamide Phosphoribosyltransferase. Biochemistry 2008, 47, 11086–11096. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.; Berman, J.; Culp-Hill, R.; Reisz, J.; Ling, T.; Rondeau, V.; Li, M.; Yang, M.; Hong, J.Y.; Lin, H.; et al. Sirtuin 3 Inhibition Targets AML Stem Cells through Perturbation of Fatty Acid Oxidation. Blood 2021, 138, 2240. [Google Scholar] [CrossRef]
- Hong, J.Y.; Fernandez, I.; Anmangandla, A.; Lu, X.; Bai, J.J.; Lin, H. Pharmacological Advantage of SIRT2-Selective versus pan-SIRT1–3 Inhibitors. ACS Chem. Biol. 2021, 16, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Hu, J.; Zhang, X.; Lin, H. Thiomyristoyl peptides as cell-permeable Sirt6 inhibitors. Org. Biomol. Chem. 2014, 12, 7498–7502. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liu, Z.-M.; Zhu, K.-R.; Cui, G.-L.; Liu, L.-X.; Yan, Y.-H.; Ning, X.-L.; Yu, Z.-J.; Li, G.-B.; Qi, Q.-R. New ε-N-thioglutaryl-lysine derivatives as SIRT5 inhibitors: Chemical synthesis, kinetic and crystallographic studies. Bioorg. Chem. 2023, 135, 106487. [Google Scholar] [CrossRef]
- Neal, R.A.; Halpert, J. Toxicology of Thiono-Sulfur Compounds. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Ruse, M.J.; Waring, R.H. The effect of methimazole on thioamide bioactivation and toxicity. Toxicol. Lett. 1991, 58, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.N.; Davidson, V.P.; Judd, C.E.; Stodgell, C.; Traiger, G.J. The effect of partial hepatectomy on the metabolism, distribution, and nephrotoxicity of para-methylthiobenzamide in the rat. Toxicol. Appl. Pharmacol. 1992, 113, 246–252. [Google Scholar] [CrossRef]
- Leblond, J.; Mignet, N.; Largeau, C.; Seguin, J.; Scherman, D.; Herscovici, J. Lipopolythiourea transfecting agents: Lysine thiourea derivatives. Bioconjug. Chem. 2008, 19, 306–314. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; He, B.; Du, J.; Lin, H.; Cerione, R.A.; Hao, Q. The bicyclic intermediate structure provides insights into the desuccinylation mechanism of human sirtuin 5 (SIRT5). J. Biol. Chem. 2012, 287, 28307–28314. [Google Scholar] [CrossRef]
- Rajabi, N.; Hansen, T.N.; Nielsen, A.L.; Nguyen, H.T.; Bæk, M.; Bolding, J.E.; Bahlke, O.Ø.; Petersen, S.E.G.; Bartling, C.R.O.; Strømgaard, K.; et al. Investigation of Carboxylic Acid Isosteres and Prodrugs for Inhibition of the Human SIRT5 Lysine Deacylase Enzyme. Angew. Chem. Int. Ed. 2022, 61, e202115805. [Google Scholar] [CrossRef]
- Bolding, J.E.; Martín-Gago, P.; Rajabi, N.; Gamon, L.F.; Hansen, T.N.; Bartling, C.R.O.; Strømgaard, K.; Davies, M.J.; Olsen, C.A. Aryl Fluorosulfate Based Inhibitors That Covalently Target the SIRT5 Lysine Deacylase. Angew. Chem. Int. Ed. 2022, 61, e202204565. [Google Scholar] [CrossRef]
- Asaba, T.; Suzuki, T.; Ueda, R.; Tsumoto, H.; Nakagawa, H.; Miyata, N. Inhibition of Human Sirtuins by in Situ Generation of an Acetylated Lysine−ADP−Ribose Conjugate. J. Am. Chem. Soc. 2009, 131, 6989–6996. [Google Scholar] [CrossRef]
- Hirsch, B.M.; Du, Z.; Li, X.; Sylvester, J.A.; Wesdemiotis, C.; Wang, Z.; Zheng, W. Potent sirtuin inhibition bestowed by l-2-amino-7-carboxamidoheptanoic acid (l-ACAH), a Nε-acetyl-lysine analog. MedChemComm 2011, 2, 291–299. [Google Scholar] [CrossRef]
- He, Y.; Yan, L.; Zang, W.; Zheng, W. Novel sirtuin inhibitory warheads derived from the Nε-acetyl-lysine analog l-2-amino-7-carboxamidoheptanoic acid. Org. Biomol. Chem. 2015, 13, 10442–10450. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.; Sousa, E.; Fernandes, C. Cyclic Peptides in Pipeline: What Future for These Great Molecules? Pharmaceuticals 2023, 16, 996. [Google Scholar] [CrossRef]
- Jiang, Y.; Zheng, W. Cyclic Tripeptide-based Potent and Selective Human SIRT5 Inhibitors. Med. Chem. 2020, 16, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, B.; Zheng, W. Cyclic tripeptide-based potent human SIRT7 inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 461–465. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, W. Cyclic peptide-based potent human SIRT6 inhibitors. Org. Biomol. Chem. 2016, 14, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, J.; Hayashi, Y.; Suga, H. Discovery of Macrocyclic Peptides Armed with a Mechanism-Based Warhead: Isoform-Selective Inhibition of Human Deacetylase SIRT2. Angew. Chem. Int. Ed. 2012, 51, 3423–3427. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Sir2 Deacetylases Exhibit Nucleophilic Participation of Acetyl-Lysine in NAD+ Cleavage. J. Am. Chem. Soc. 2007, 129, 5802–5803. [Google Scholar] [CrossRef]
- Bolding, J.E.; Nielsen, A.L.; Jensen, I.; Hansen, T.N.; Ryberg, L.A.; Jameson, S.T.; Harris, P.; Peters, G.H.J.; Denu, J.M.; Rogers, J.M.; et al. Substrates and Cyclic Peptide Inhibitors of the Oligonucleotide-Activated Sirtuin 7. Angew. Chem. Int. Ed. 2023, 62, e202314597. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Structure | Targeted Sirtuin Isoform | Potency (EC50), μM | Fold Activation | Cell Active | Citations |
|---|---|---|---|---|---|---|
| Resveratrol |  | Sirt1 | ~100 | 13.4 | Yes | [58,59] |
| SRT1720 |  | Sirt1 | ~0.10 | ~8 | Yes | [59] |
| SRT2104 |  | Sirt1 | 0.43 a | ~2 | Yes | [60] |
| Honokiol |  | Sirt3 | N.D. | ~2 | Yes | [61] |
| ADTL-SA1215 |  | Sirt3 | 0.21 | 2 | Yes | [62] |
| Compound 31 |  | Sirt3 | ~100–200 | ~5–1000 | Yes | [63] |
| Compound 30 |  | Sirt5 | ~40 | ~3–5 | Yes | [63] |
| UBCS039 |  | Sirt6 | 38 | 2 | Yes | [64] |
| LPA |  | Sirt6 | 25 | 48 | Yes | [65] |
| MDL-811 |  | Sirt6 | 7.1 | 44 | Yes | [66] |
| Compound 12q |  | Sirt6 | 8.9 * 5.4 ** | ~12–18 * ~40 ** | Yes | [67] |
| Compound Name | Structure | Targeted Sirtuin Isoform | Potency (IC50), μM | Selectivity | Cell Active | Citations |
|---|---|---|---|---|---|---|
| ELT-31 |  | Sirt1/2/3 | 0.001–0.033 | Nonselective | N.D. | [126] |
| EX-527 |  | Sirt1 | 0.12 | ~20–800 fold over Sirt2/3 | Yes | [127] |
| Compound 86 |  | Sirt2 | 0.02 | ~100–400 fold over Sirt1/3 | Yes | [128] |
| AGK2 |  | Sirt2 | 3.5 | >14 fold over Sirt1/3 | Yes | [129] |
| AK7 |  | Sirt2 | 15.5 | >4 fold over Sirt1/3 | Yes | [130] |
| SirReal2 |  | Sirt2 | 0.14 | >1000 fold over Sirt1/3/4/5/6 | Yes | [124] |
| MIND4 |  | Sirt2 | 3.5 | Selective over Sirt1/3 | Yes | [131] |
| Compound 60 |  | Sirt4 | 0.9 | Non-selective over Sirt2; ~3.5–5.5 fold over Sirt1/3/5/6 | Yes | [132] |
| Compound 69 |  | Sirt4 | 16 | ~2–3 fold over Sirt1/2/3/5/6 | Yes | [132] |
| Compound 47 |  | Sirt5 | 0.21 | >3800 fold Sirt5 over Sirt1/2/3/6 | N.D. | [133] |
| Lysine Derivative | Warhead | Targeted Sirtuin Isoform(s) | Potency (IC50), μM | Selectivity | Cell Active | Citations |
|---|---|---|---|---|---|---|
| Thioacetyl |  | Sirt1/2/3 | 0.18–29.4 | Largely non-selective | Yes | [175,176,177,178,179,180,181,182,183] |
| Thiocarbamoyl |  | Sirt1/2/3 | 1.7–159.1 | Non-selective | Yes | [184,185] |
| Thiomyristoyl (TM) |  | Sirt2 | 0.03–0.09 * 0.04–3.3 ** | Up to ~3500–7100 fold over Sirt1/3/5/6/7 | Yes | [186,187,188] |
| Soluble TM |  | Sirt2 | 0.03 | ~72–94 fold over Sirt1/3; >1500 fold over Sirt6 | Yes | [189] |
| Fatty acyl thiocarbamoyl |  | Sirt2 | 0.06–0.15 | ~180–2500 fold over Sirt1/3 | Yes | [190] |
| Cyclic thioacetyl |  | Sirt1 Sirt2 | 0.002 0.01 | 19–62 fold over Sirt2/3; 7–8 fold over Sirt1/3 | Yes | [191,192,193] |
| Mitochondrially-targeted TM |  | Sirt3 | 0.53 | N.D.C. | Yes | [194] |
| Mitochondrially-targeted thiocarbamoyl |  | Sirt3 | 1.1 | N.D.C. | Yes | [195] |
| Targeted Sirtuin Isoform | Activators | Inhibitors |
|---|---|---|
| Sirt1 | SRT1720 [59], SRT2104 [60] | EX-527 [127] |
| Sirt2 | N/A * | TM [186], AF8/10 [189,190], SirReal2 [124] |
| Sirt3 | ADTL-SA1215 [62] | Mitochondrially targeted myristoyl-thiocarbamoyl pseudopeptides [194,195] |
| Sirt4 | N/A * | Compound 60/69 [132] |
| Sirt5 | 1,4-dihydropyridine derivative Compound 31 [63] | Succinyl-thiocarbamoyl pseudopeptides [201,202] |
| Sirt6 | LPA [65], MDL-811 [107] | N/A * |
| Sirt7 | N/A * | Peptide lariat Compound 41 [240] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bursch, K.L.; Goetz, C.J.; Smith, B.C. Current Trends in Sirtuin Activator and Inhibitor Development. Molecules 2024, 29, 1185. https://doi.org/10.3390/molecules29051185
Bursch KL, Goetz CJ, Smith BC. Current Trends in Sirtuin Activator and Inhibitor Development. Molecules. 2024; 29(5):1185. https://doi.org/10.3390/molecules29051185
Chicago/Turabian StyleBursch, Karina L., Christopher J. Goetz, and Brian C. Smith. 2024. "Current Trends in Sirtuin Activator and Inhibitor Development" Molecules 29, no. 5: 1185. https://doi.org/10.3390/molecules29051185
APA StyleBursch, K. L., Goetz, C. J., & Smith, B. C. (2024). Current Trends in Sirtuin Activator and Inhibitor Development. Molecules, 29(5), 1185. https://doi.org/10.3390/molecules29051185