1. Introduction
Discovered in the 1980s, polymerase chain reaction (PCR) has become one of the most significant technological advancements in the fields of biology and clinical medicine. Technology that detects a broad range of bacteria or viruses based on PCR is widely applied for the screening of clinical, environmental, and manufacturing samples to diagnose infectious diseases or to detect bacterial contamination. Therefore, PCR technology plays a vital role in molecular biology, genetic engineering, and diagnostics [
1]. The
Taq DNA polymerase that is now widely used in PCR was first isolated from the extreme thermophilic bacteria
Thermus aquaticus YT-1 [
2].
Currently, the
Taq DNA polymerase is crucial for almost all PCR-based techniques, including RT-PCR and digital PCR. However, the
Taq DNA polymerase has the drawback of a relatively higher error frequency than other enzymes [
3]. Usually, product yields will decrease when the amplification fragment becomes longer than 1 kb due to the relatively low processivity and thermostability of the wild-type enzyme. Generally, the efficiency of amplification by the
Taq polymerase for targets shorter than 1 kb is approximately 80% [
4]. However,
Taq DNA polymerases become completely inhibited when the PCR mixture contains 0.2% blood. It seems that hemoglobin and lactoferrin have important roles in inhibiting the amplification process [
5]. Therefore, these drawbacks limit the applications of
Taq DNA polymerase.
Recently, a novel strategy was employed to overcome these limitations [
6]. Generating fusion proteins with
Taq DNA polymerases and a thermostable DNA-binding protein such as the Sso7d DNA-binding protein from
Sulfolobus solfataricus has been shown to enhance the processivity [
7], thermostability, and overall stability compared with the wild-type
Taq polymerase. The purified S-
Taq protein has shown acceptable limits of host genomic DNA levels without the use of DNases or other DNA precipitating agents, which highlights its potential for use in PCR-based diagnostics, in situ PCR, and forensic science [
7].
A previous study also found that adding 0.6% bovine serum albumin (BSA) to reaction mixtures containing the
Taq DNA polymerase reduced the inhibitory effect of blood and allowed for DNA amplification in the presence of 2% instead of 0.2% blood [
8]. Furthermore, BSA was found to be the most efficient amplification facilitator. As rapid and simple diagnostic methods are urgently required for blood analyses, modifications to
Taq DNA polymerase that enhance its tolerance to inhibition by blood are significant to medical diagnostics. Mutations to the
Taq DNA polymerase that render it resistant to inhibition by blood components are now commercially available [
9]. Additionally, there are
Taq polymerase mutants that are used in PCR-based tests of blood and soil samples that are widely used for diagnostics and forensic analyses and do not require pretreatment to purify the template DNA, and there are others that allow increased dye concentration to overcome fluorescence background and quenching in real-time PCR analyses of blood. Because of deficiencies in the 3′-5′ exonuclease domain, the
Taq DNA polymerase is widely applied to ARMS-PCR to detect single nucleotide polymorphisms (SNPs).
In recently reported studies, the
Taq polymerase was fused to DNA binding proteins to improve other properties [
10]. In this paper, we tried to apply the variant of
CE7 (Colicin E7 DNase) to achieve that effect. The
CE7 protein belongs to the category of highly toxic proteins because of its DNase domains (16 kDa). The wild
CE7 could bind with DNA and degrade it. Considering that the DNase activity of
CE7 is lethal to the cells, constructing the inactive variants of
CE7 is an appropriate option. Here, the active site histidine residues and DNA-binding residues were mutated to produce the
CL7 variant lacking the catalytic activities. That paper said that the
CL7 variant also lacked DNA-binding ability. However, we found that
CL7 could increase the stability of
Taq. By taking the features of
CL7, we fused
CL7 with
Taq to improve the properties of the
Taq polymerase. We demonstrated and functionally characterized the fusion enzymes and demonstrated that they showed improved processivity, sensitivity, amplification rates, and eliminated pre-PCR treatment steps. Finally, we demonstrated the practical benefits of PCR applications for enhancing the sensitivity of the polymerase.
2. Materials and Methods
2.1. Construction of Recombinant Plasmids
First, DNA coding sequences of the DNA polymerase from Thermus aquaticus (GenBank: P19821.1) and of
CL7, which is a mutant of
CE7 from
E. coli (GenBank: CP018986.1), were obtained [
11]. The two fragments were synthesized by GeneCreate (Wuhan, China). The designed primers for amplification are listed in
Table 1. The open reading frame synthesis procedure was performed as described previously. PCR amplification was then used to obtain two products: the
Taq DNA polymerase open reading frame (2493 bp) and the
CL7 gene (390 bp). The pET-30 vector was digested with the ndonucleasee Xba I (Takara, Shiga, Japan), and the product was purified using the DNA Gel Extraction Kit (Promega, Madison, WI, USA). Then, the PCR products were mixed together with the digested pET-30 vector. Thus, the open reading frame encoding a fragment of
Taq DNA polymerase was cloned into the pET-30 vector to generate a pET30/
Taq plasmid. This led to the expression of a
Taq DNA polymerase fusion protein with a C-terminal 6× histidine tag. Finally, T5 cloning (New England Biolabs, Ipswich, MA, USA) was performed, in which
CL7 was fused to the N-terminus of
Taq DNA polymerase with the 7-amino acid linker (ENLYFQG) and a 6× His tag to the C-terminus, resulting in plasmid pET30/
CL7-
Taq. This was necessary for the purification of a recombinant protein by metal affinity chromatography. Nucleotide sequences of the resulting recombinant plasmids, pET30/
Taq and pET30/
CL7-Taq, were confirmed by DNA sequencing (Sangon, Shanghai, China).
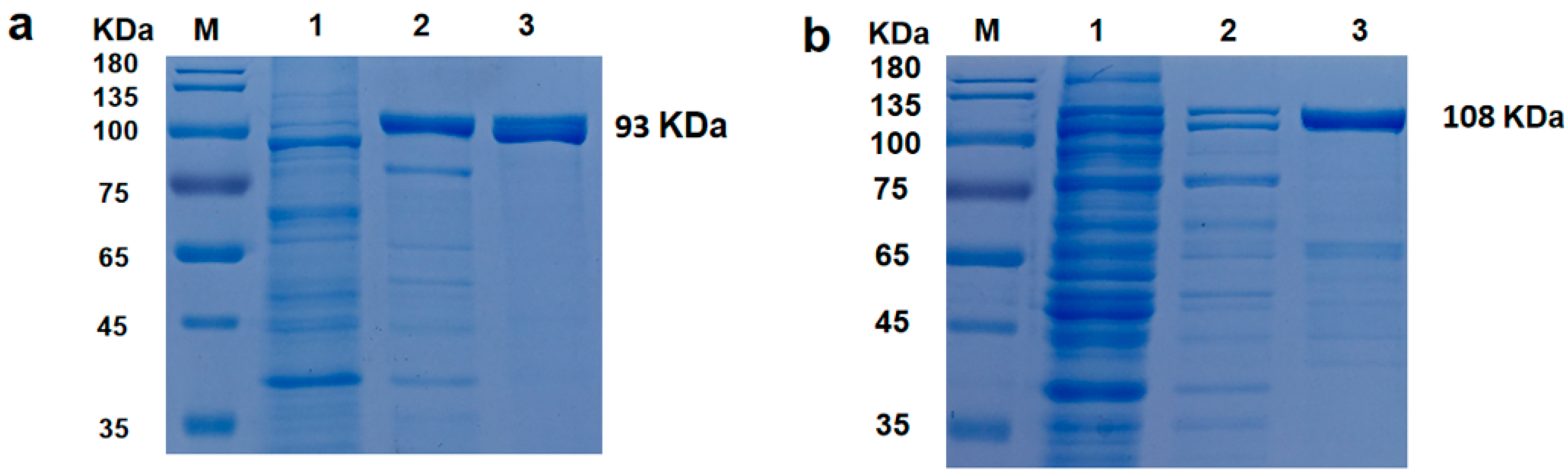
2.2. Protein Expression and Purification
Plasmids encoding Taq and CL7-Taq were used to transform the E. coli BL21 strain (DE3). Luria–Bertani (LB) medium was prepared. E. coli containing recombinant plasmid were grown to an A600 of 0.6 in LB containing 50 μg/mL kanamycin at 37 °C. Then, isopropyl-β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 1 mg/mL to induce Taq or CL7-Taq expression from the T7 promoter with shaking at 18 °C. After 12 h of incubation, the cells were centrifuged at 7000× g for 5 min, and the pellets were resuspended in 20 mL of lysis buffer (20 mM Tris-HCl [pH 9.0], 0.5 M NaCl, and 10 mM imidazole). Protein complexes were extracted by ultrasonic decomposition and the insoluble debris was removed by centrifugation at 12,000× g and 4 °C for 20 min.
For heat treatment, the cleared lysate was immersed in a 75 °C orbital water shaker for 30 min, cooled on ice for 20 min, and then, the denatured host proteins were removed by centrifugation at 12,000×
g and 4 °C for 20 min. Following heat treatment, the exogenous proteins were purified in a one-step process using the Ni
2+-affinity chromatographic technique. A His-bind resin and His-bind buffer kit (GE Life Sciences, Chicago, IL, USA) were used to purify the His-tagged proteins according to the manufacturer’s instructions. The supernatant and produced enzyme were put into a column containing Ni–nitrilotriacetic acid (Ni-NTA) agarose (GE Life Sciences), which was previously prepared and equilibrated with lysis buffer. After binding to the Ni-NTA agarose column, the recombinant proteins were washed twice with washing buffer (20 mM Tris-HCl [pH 9.0], 0.5 M NaCl, and 50 mM imidazole), and then eluted with elution buffer (20 mM Tris-HCl [pH 9.0], 0.5 M NaCl, and 200 mM imidazole). An equal volume of each buffer (20 μL) was loaded onto a 12% (
w/
v) polyacrylamide gel for SDS-PAGE [
12], followed by staining with Coomassie Brilliant Blue G-250. Finally, the eluted fractions were dialyzed three times with storage buffer (20 mM Tris-HCl [pH 8.0], 100 mM KCl, and 0.2 mM EDTA). Protein concentrations were measured using the Bradford method.
2.3. DNA Polymerase Activity Assay
The DNA polymerase activity of purified proteins was assayed using the EvaEZ Fluorometric Polymerase Activity Assay Kit (Biotium, Hayward, CA, USA). All assays were conducted in an isothermal reaction at 60 °C using a CFX Real-Time PCR instrument (Bio-Rad, Hercules, CA, USA) in accordance with the definition of one unit of enzyme activity (“One unit of DNA polymerase activity is usually defined as the amount of enzyme that will produce 10 nmol of nucleotides during a 30-min incubation”). Enzymatic activity was determined relative to a commercial
Taq DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) with an activity of 1 U/μL. When the DNA polymerase was active, the primer was extended to form a double-stranded product that bound the EvaGreen dye, resulting in increased fluorescence. The rate of increase is positively correlated with polymerase activity [
13].
2.4. Optimization of PCR Amplification
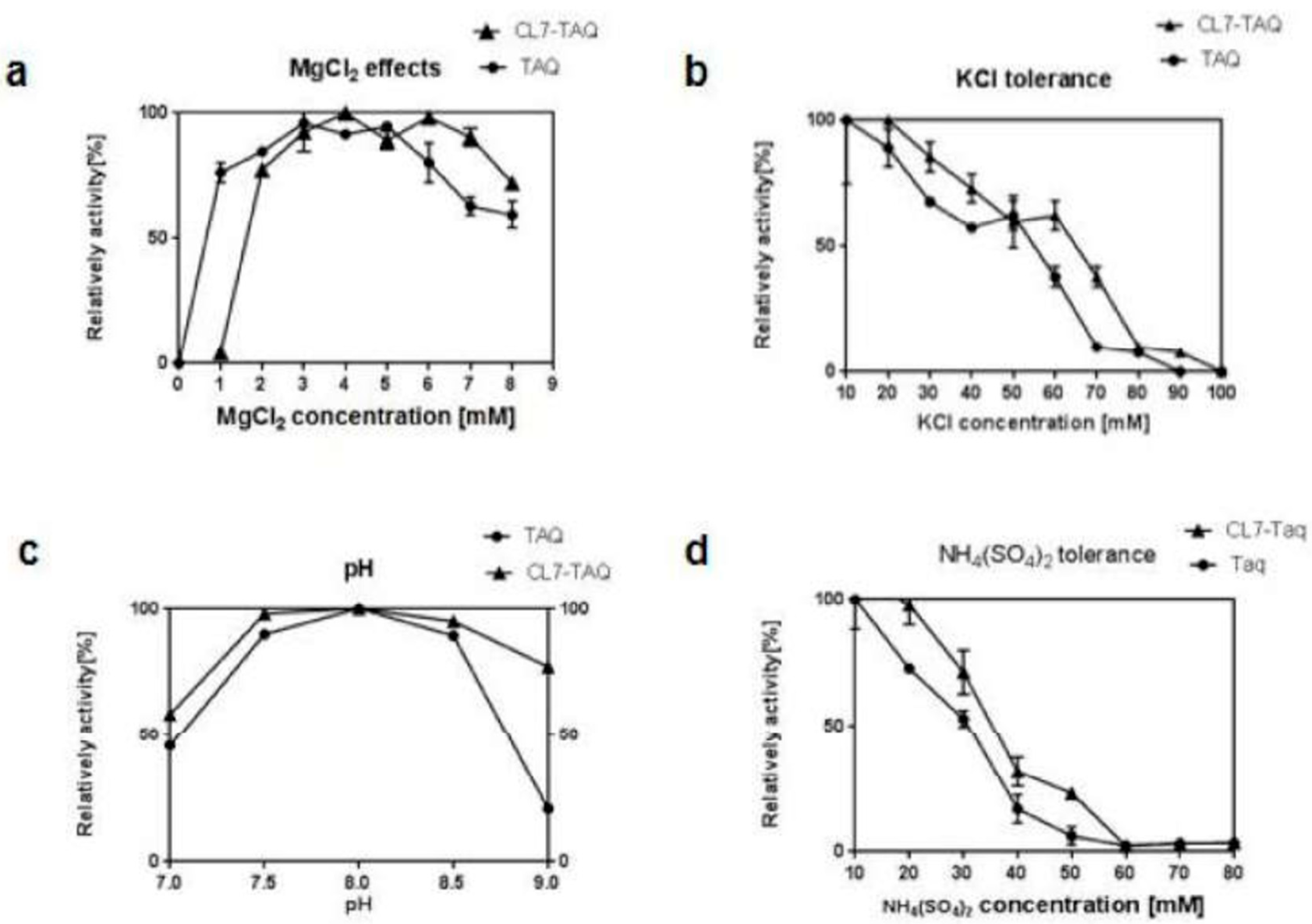
To optimize the amplification process, polymerase activity was measured using various concentrations of MgCl2, KCl, and (NH4)2SO4 in the buffer as well as various pHs. All PCR reactions were performed using 0.2 mM of each dNTP and 0.4 mM of each primer and of the pET30a-GFP plasmid DNA as template, containing a known target sequence (PCR product of 445 bp). PCR assays were performed using 1 U of purified CL7-Taq or Taq DNA polymerase in a 20 μL reaction mixture containing 1 ng of DNA template. The PCR conditions were as follows: 3 min at 95 °C, and then 25 cycles of 15 s at 95 °C, 15 s at 60 °C, and 30 s at 72 °C. To determine the optimum MgCl2 concentration, PCR was performed at various pHs (7.0–9.0) with the use of the Tris–HCl buffer. Then, we used the Tris-HCl buffer (pH 8.0) containing increasing concentrations of MgCl2 (0–9 mM) to detect the enzyme activity. Furthermore, PCR was performed with various concentrations of KCl (10–90 mM) and (NH4)2SO4 (10–90 mM).
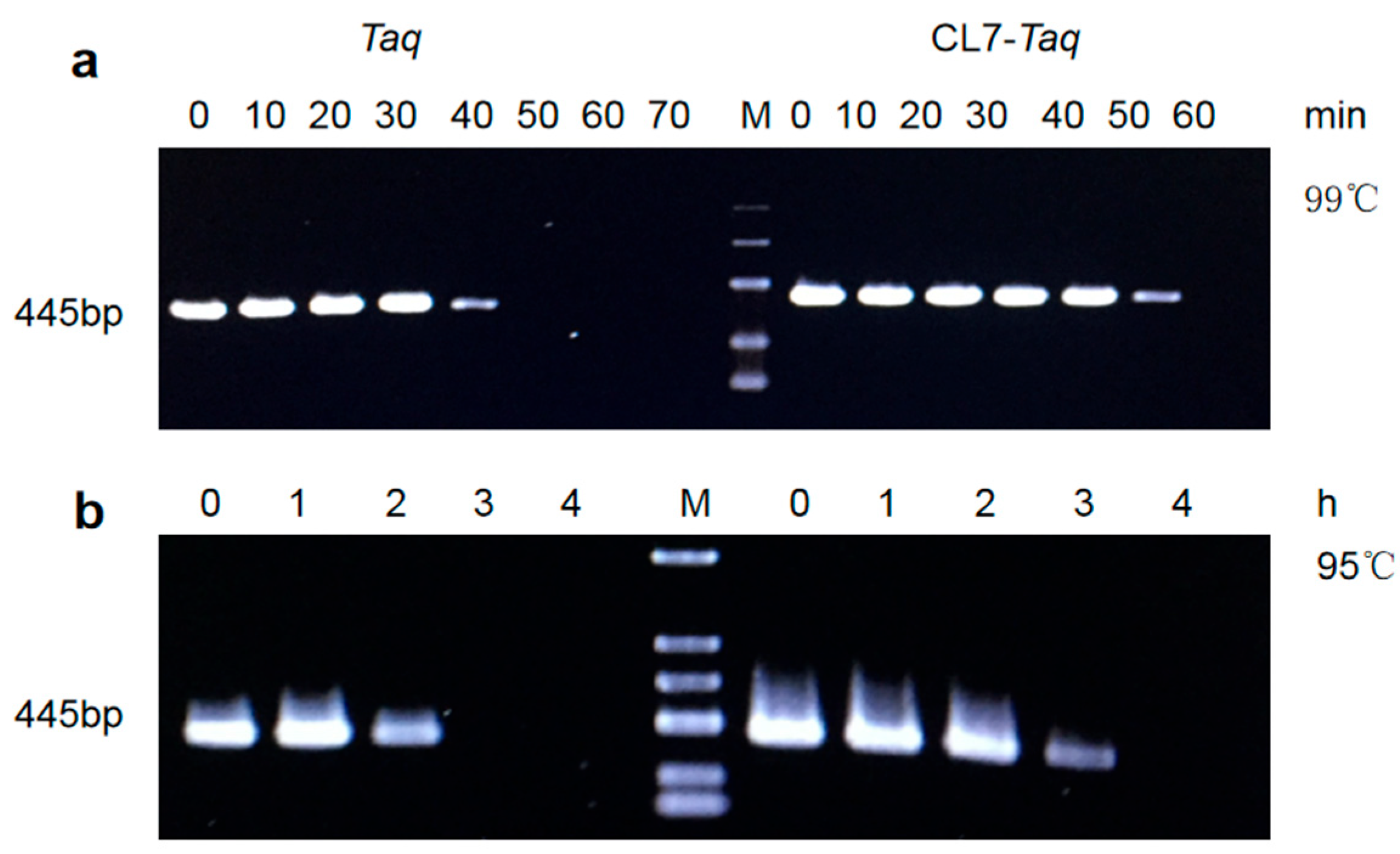
Temperature stability was also assayed [
14]. One unit of purified
CL7-
Taq and
Taq DNA polymerases was heated at 99 °C for 10, 20, 30, 40, 50, and 60 min, and at 95 °C for 1, 2, 3, 4, and 5 h. Then, the same amount of enzyme was used to amplify a 445 bp target fragment under the optimal reaction buffer (in the same PCR conditions determined in the optimization process): 20 mM Tris-HCl (pH 8.0), 4 mM MgCl
2, 10 mM (NH
4)
2SO
4, and 20 mM KCl.
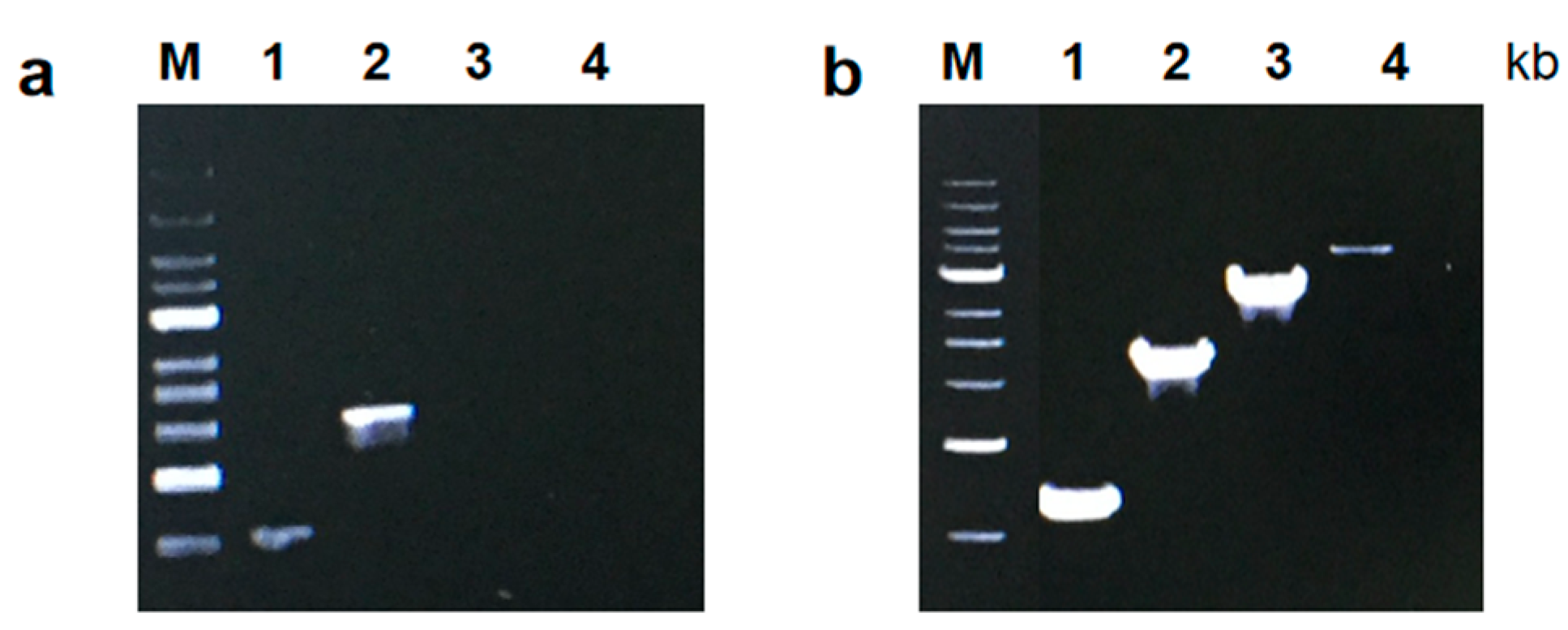
2.5. Measuring the PCR Amplification Rate
PCR amplification rates were measured using the protocol described by [
15].
CL7-
Taq and
Taq DNA polymerases were used to amplify PCR products of 1, 2, 3, and 4 kb under the conditions determined in the optimization process and using pET23a/dcas9 plasmid DNA as a template, which was recombined in our laboratory. PCR amplification started with an initial denaturation at 95 °C for 3 min and included 25 cycles of 30 s at 95 °C, 30 s at 60 °C, and 60 s at 72 °C.
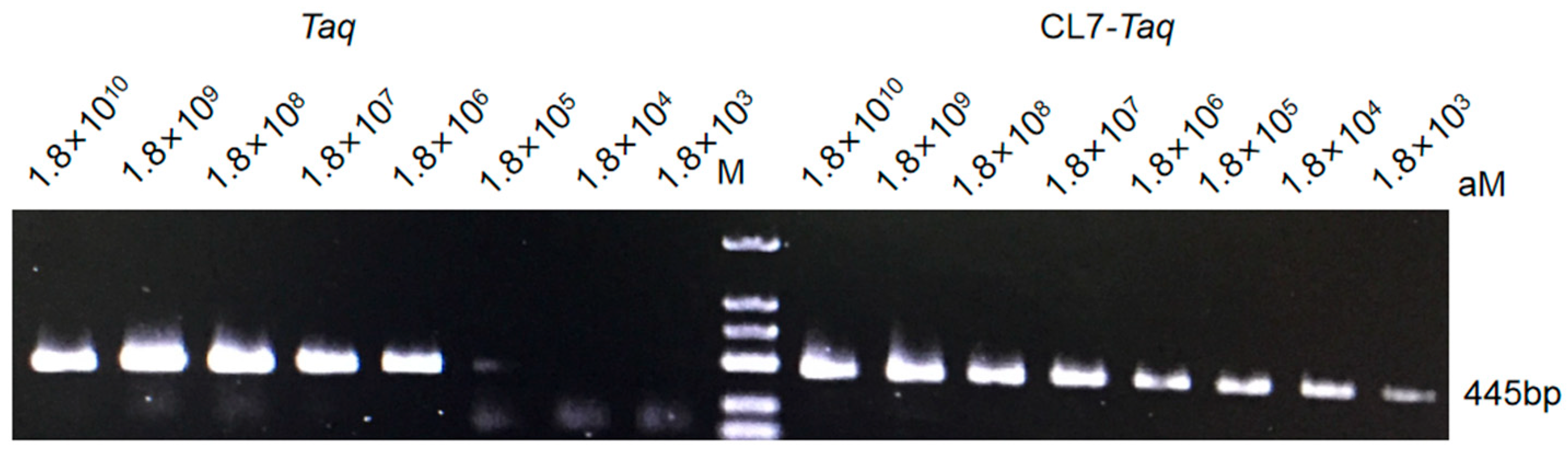
2.6. Sensitivity
Following sufficient modifications, DNA polymerase sensitivity (affinity for template) was measured using the protocol by [
16]. PCR was conducted under conditions optimized for the fusion polymerases
CL7-
Taq and
Taq. We used pET23a-GFP plasmid as a template along with the primers 5′-TGGTCTTCAATGCTTTGCGAGATAA-3′ (forward) and 5′-CTTTTCGTTGGGATCTTTCG-3′ (reverse). The product of the reaction was 445 bp. The reaction was conducted at decreasing template concentrations (serial 10-fold dilutions of the template) and included an initial denaturation at 95 °C for 3 min followed by 35 cycles of 30 s at 95 °C, 30 s at 60 °C, and 90 s at 72 °C. The amplified fragments were analyzed in a 1.5% agarose gel stained with ethidium bromide.
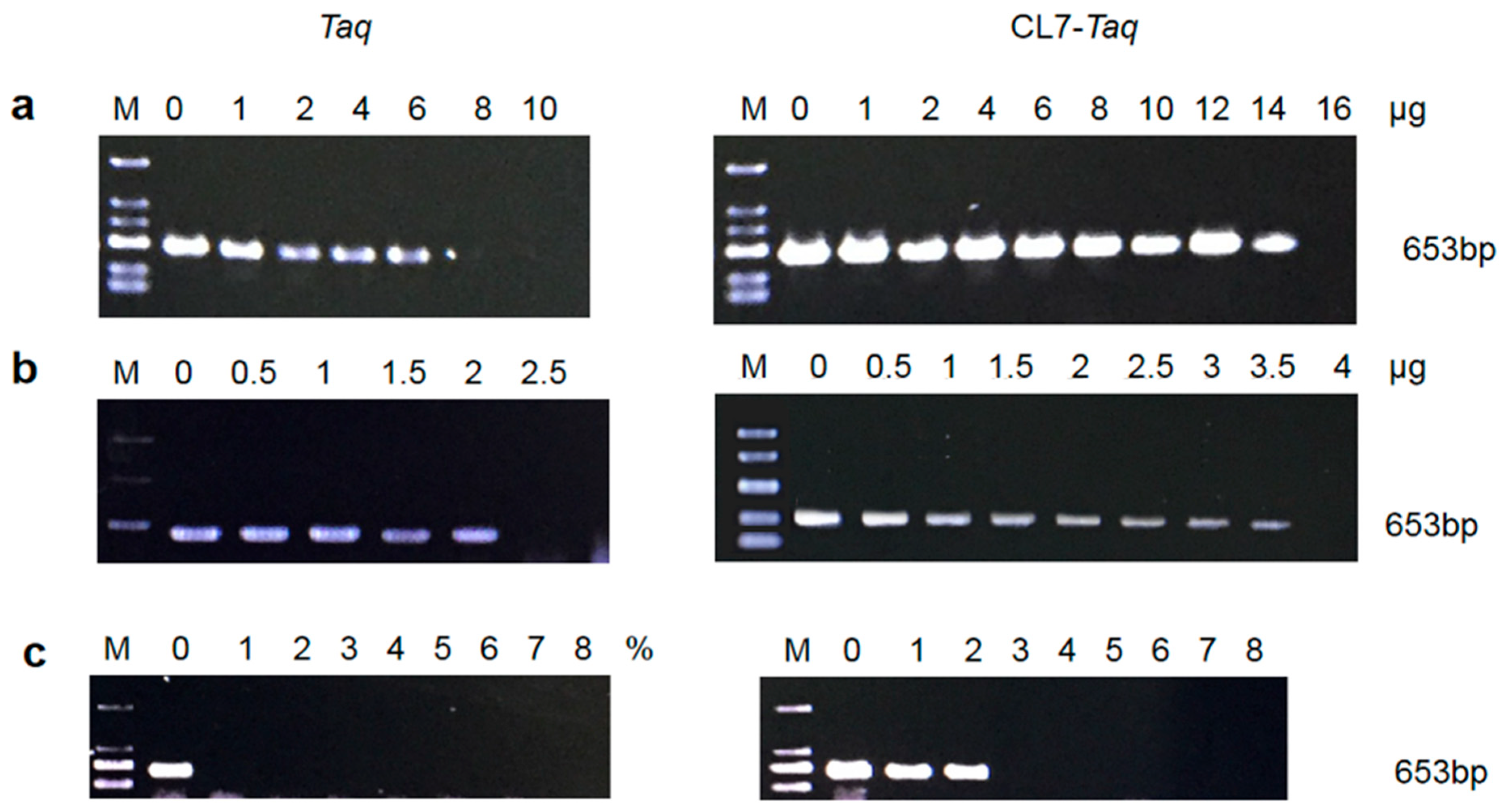
2.7. Resistance to Inhibitors
The effect of PCR inhibitors such as heparin (Sigma-Aldrich, St. Louis, MO, USA) at a range from 16 to 1 μg, lactoferrin (Sigma-Aldrich) at a range from 4 to 0.5 μg, and blood at a range from 8% to 1% on the catalytic activity of the
CL7-
Taq and
Taq DNA polymerases was assessed by a PCR reaction using human genomic DNA as a template and the specific β-actin primers 5′-AGAGATGGCCACGGCTGCTT-3′ (forward) and 5′-ATTTGCGGTGGACGATGGAG-3′ (reverse) [
16]. The amplified fragments were analyzed in a 1.5% agarose gel stained with ethidium bromide.
2.8. Fidelity of CL7-Taq
A PCR-based forward mutation assay was performed by Wang Fei [
15]. The mannanase activity was applied to measure the error rate of the
CL7-
Taq DNA polymerase. PCR was carried out by
CL7-
Taq and
Taq polymerases under optimized conditions. The resulting PCR products were treated with
DpnI, subjected to agarose gel DNA purification, and transformed into
E. coli XL10-Gold. These clones were transferred to LB plates containing 100 mM arabinose and 0.03% (
m/
v) trypan blue (which gives the plates a blue appearance) and 0.5% (
m/
v) Konjac Flour as the substrate for the mannanase. A clear zone was visible around mannanase-expressing colonies capable of hydrolyzing the Konjac glucomannan. A single colony of
E. coli XL10-Gold that contained the plasmid pBAD-man was used as a positive control, and a single colony of
E. coli XL10-Gold that contained the plasmid pBAD-His-GFP was used as a negative control. Hydrolysis holes with different sizes were observed and measured after 12 h of culture. Error rates were calculated based on the sizes of the hydrolysis holes. Clones that produced much larger or smaller hydrolysis holes were considered mutants.
2.9. PCR Amplification under Standard Buffer Conditions
CL7-Taq and Taq DNA polymerases (Takara, Shiga, Japan) were used to amplify PCR products of 1, 2, 3, and 4 kb under standard buffer conditions and using pET23a/dcas9 plasmid DNA as a template. The reactions performed here used the provided Taq enzyme buffer (Takara, Shiga, Japan). The optimum condition was obtained as above: 20 mM Tris-HCl (pH 8.0), 4 mM MgCl2, 10 mM (NH4)2SO4, and 20 mM KCl.
4. Discussion
PCR technology has been widely applied in the fields of molecular biology, genetic engineering, and diagnostics [
8]. PCR amplification efficiency is strongly dependent on the properties of the DNA polymerase and reaction conditions. Wild-type DNA polymerase has some drawbacks; therefore, modern diagnostic methods and genetic engineering techniques require modified DNA polymerases with better properties, such as those that possess higher sensitivity and/or amplification rates. For this reason, we engineered fusions of the DNA polymerase to enhance its ability. In this study, the N-terminus of the
Taq DNA polymerase was fused with the thermoduric
CL7 protein using a seven-amino-acid linker (Gly-Asn-Leu-Tyr-Phe-Gln-Cys).
The most common challenges during the amplification of environmental and blood samples are inhibitors present in the tested material [
17]. Our observations of successful amplification of the
β-actin open reading frame directly from human blood using
CL7-
Taq is very promising and opens up a new avenue for this polymerase as a valuable tool for medical diagnostics and forensic science, where sample availability is minimal. We found that the enhanced salt tolerance of
CL7-
Taq is responsible for its successful use in direct PCR of the human genome without preprocessing. Previous studies have shown that PCR inhibitors alter DNA or block enzymes though these pathways, such as inhibiting the active site or blocking access to the active site for cofactors such as Mg
2+ ions [
8,
18]. Therefore, inhibitors either weaken the efficiency of PCR amplification or block it completely. However, commercially available native enzymes are not always able to deal with these PCR issues. Our data showed that the fusion
CL7-
Taq DNA polymerase exhibited a higher tolerance to PCR inhibitors (blood, lactoferrin, and heparin) compared with the
Taq DNA polymerase. Therefore, the
Taq DNA polymerase containing
CL7 had better performance in the amplification of complicated templates.
Our data showed that the fusion of CL7 to the Taq DNA polymerase improved enzymatic properties such as thermostability, amplification rate, and template sensitivity. In thermostable tests, we found that the thermostability of the CL7-Taq DNA polymerase was remarkably higher than that of the Taq DNA polymerase. The thermostability of CL7-Taq was over 1 h longer than that of the Taq DNA polymerase at 95 °C. Mg2+ and other salts were critical components of PCR reactions; thus, optimizing their concentrations was essential for native DNA polymerase. Unlike the Taq DNA polymerase, the CL7-Taq DNA polymerase exhibited sufficient amplification efficiency within a wide range of Mg2+ concentrations. Furthermore, the CL7-Taq DNA polymerase had a satisfactory amplification efficiency at various concentrations of KCl and (NH4)2SO4. Thus, the salt tolerance of CL7-Taq was remarkably enhanced.
Our observation that CL7-Taq had higher sensitivity than Taq makes it an attractive enzyme for PCR-based diagnostics. Therefore, it is tempting to speculate that one could explore using CL7-Taq for ARMS-PCR to detect SNPs or common DNA viruses such as HPV, Herpesvirus, and Parvoviruses.
5. Conclusions
In this study, we aimed to obtain and characterize a new CL7-Taq fusion DNA polymerase, in which the DNA coding sequence of the Taq DNA polymerase was fused with that of CL7, a variant of CE7 (Colicin E7 DNase) from Escherichia coli. Our results showed that the sensitivity of the CL7-Taq DNA polymerase was 100-fold higher than that of the wild-type Taq, which required a template concentration of at least 1.8 × 105 nM. Moreover, the extension rate of CL7-Taq was 4 kb/min, which remarkably exceeded the rate of the Taq DNA polymerase (2 kb/min). The recombinant CL7-Taq protein exhibited excellent thermostability, extension rate, sensitivity, and resistance to PCR inhibitors. Thus, this is a novel enzyme that improved the properties of the Taq DNA polymerase and thus may have wide application in molecular biology and diagnostics.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}