
[Tc(NO)(Cp)(PPh3)Cl] and [Tc(NO)(Cp)(PPh3)(NCCH3)](PF6), and Their Reactions with Pyridine and Chalcogen Donors

 , and
, and
Abstract

1. Introduction
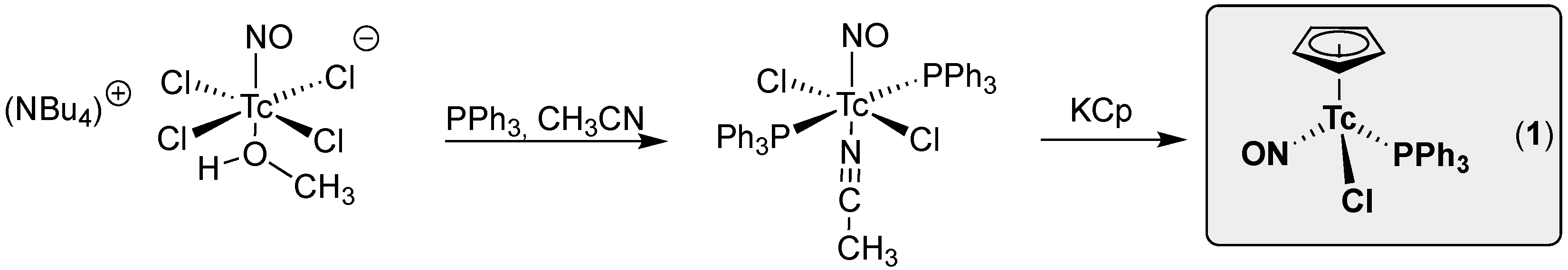
2. Results and Discussion
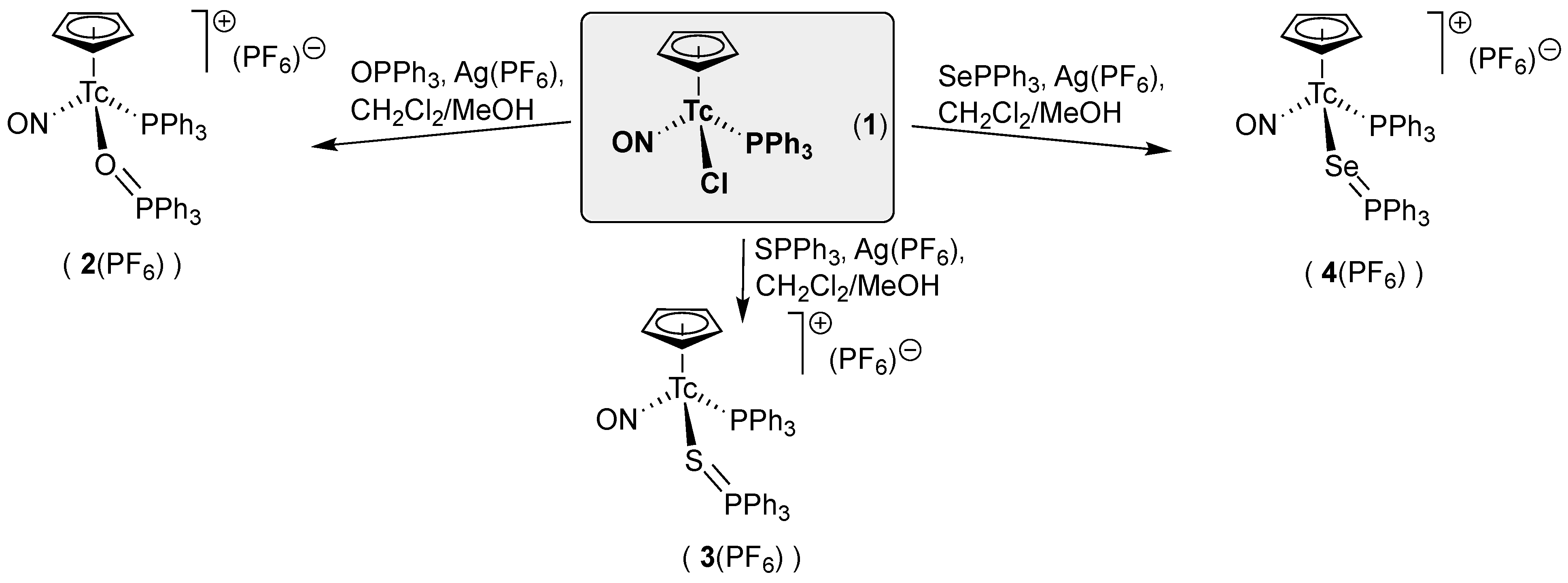
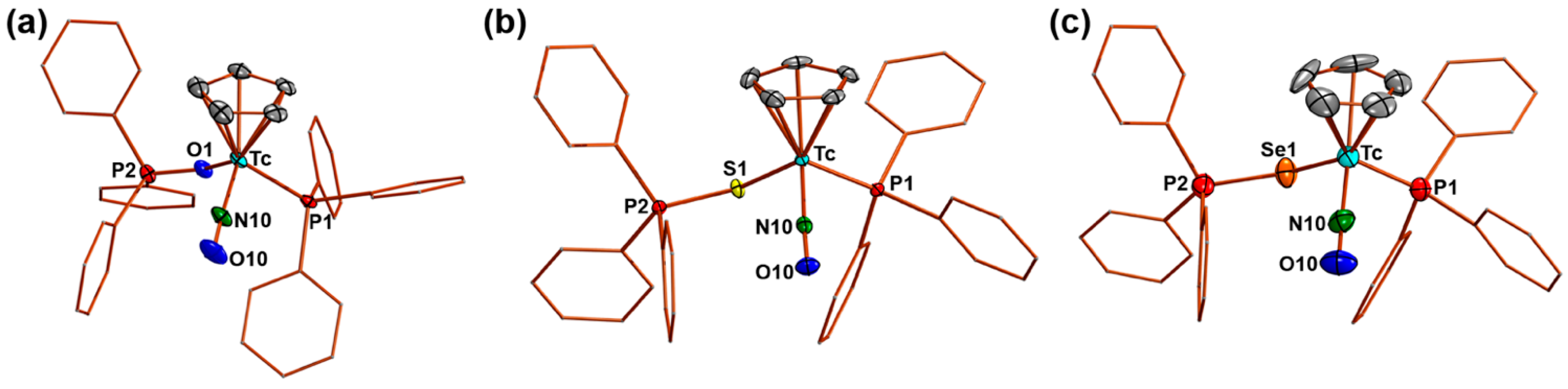
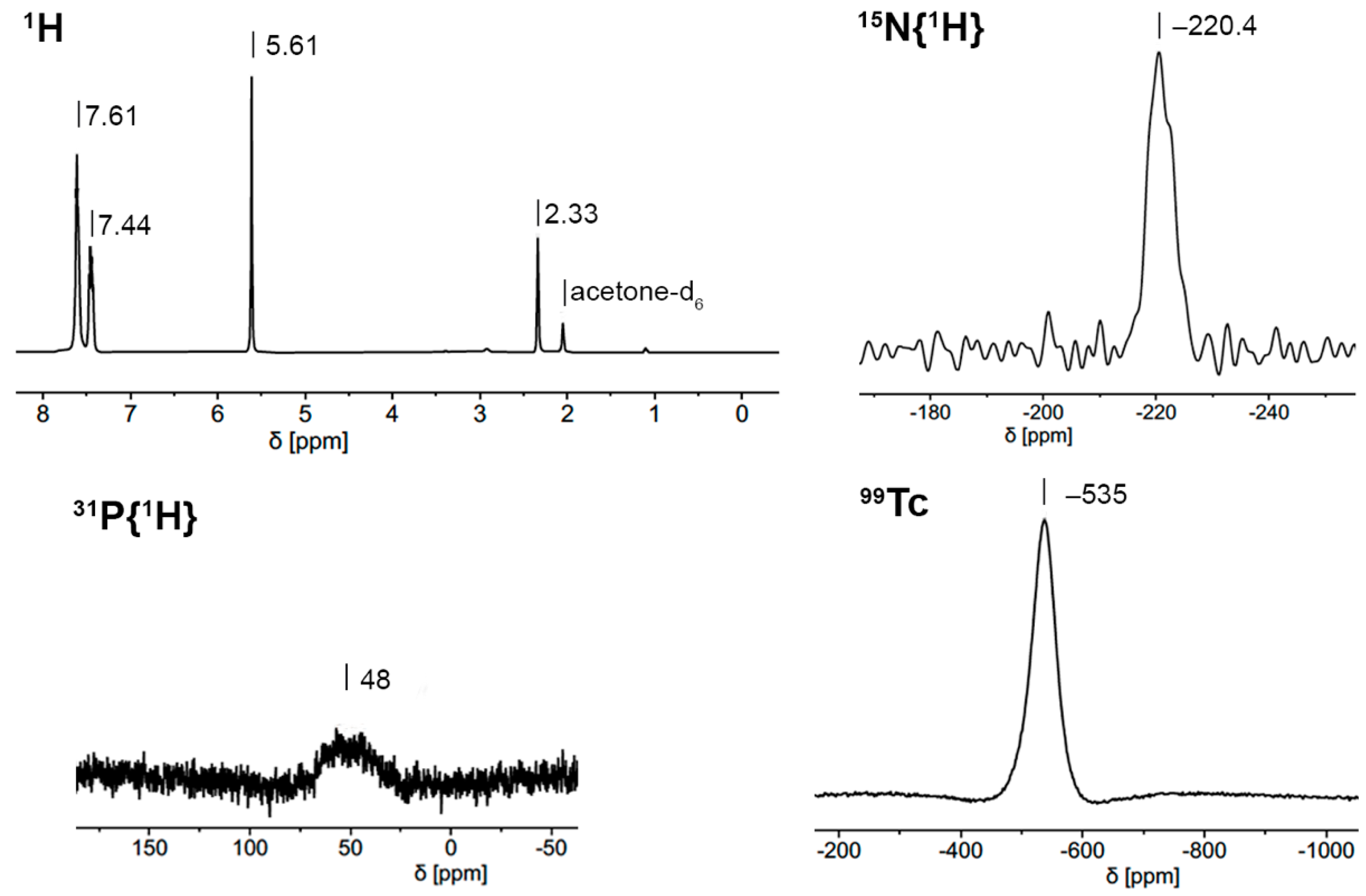
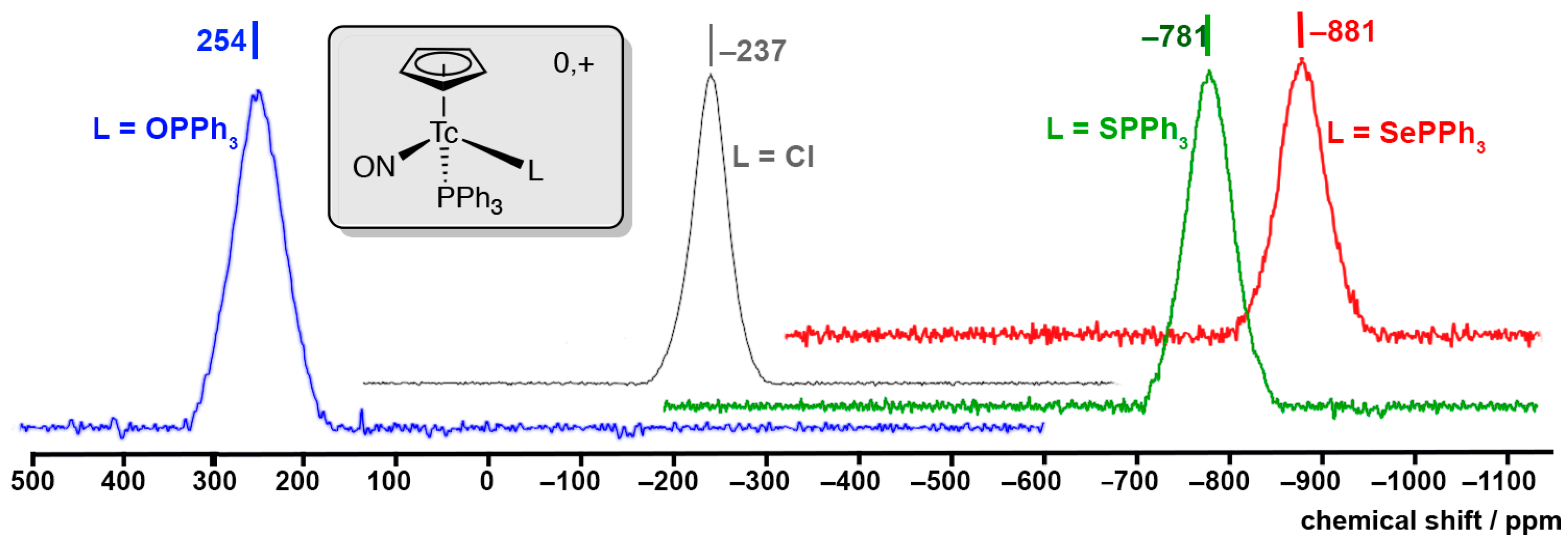
2.1. [Tc(NO)(Cp)(PPh3)(EPPh3)]+ (E = O, S, Se) Complexes
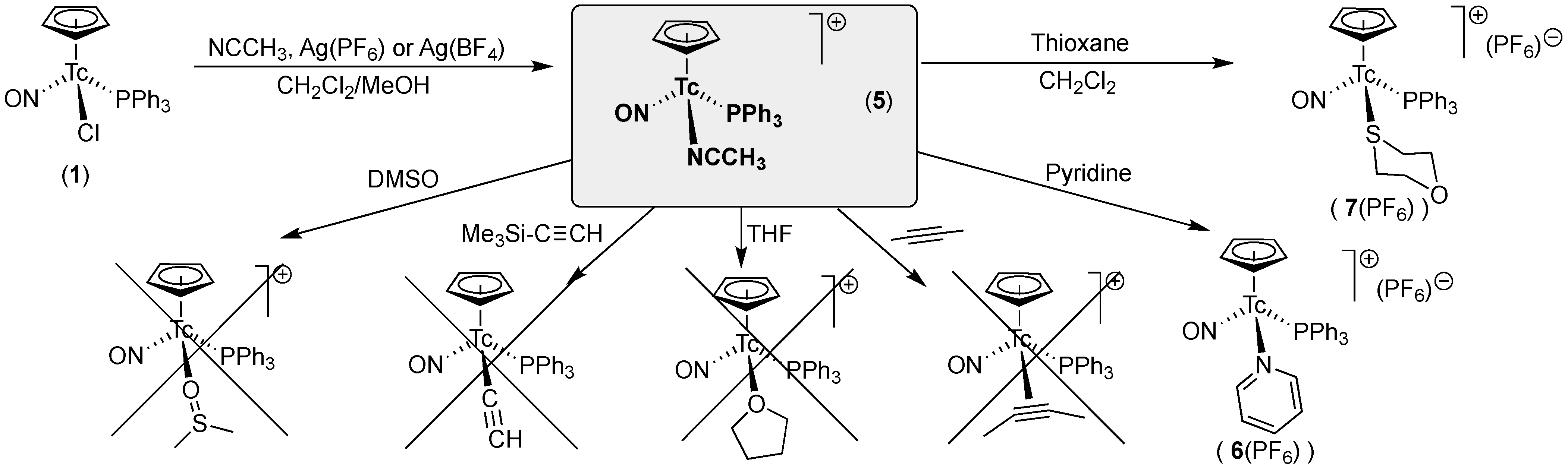
2.2. Synthesis and Reactions of [Tc(NO)(Cp)(PPh3)(NCCH3)]+ Salts
2.3. [Tc(NO)(CpR)(PPh3)(L)]0,+ Complexes
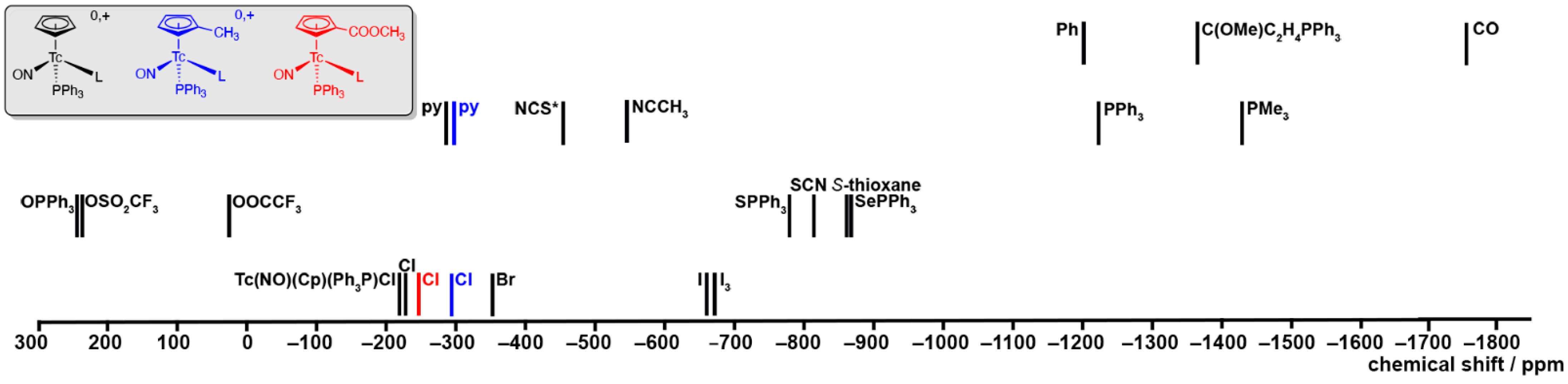
2.4. 99Tc NMR Spectra of Complexes with {Tc(NO)(Cp)(PPh3)}+ Units
3. Materials and Methods
3.1. Radiation Precaution
3.2. Syntheses
3.3. Spectroscopic Methods
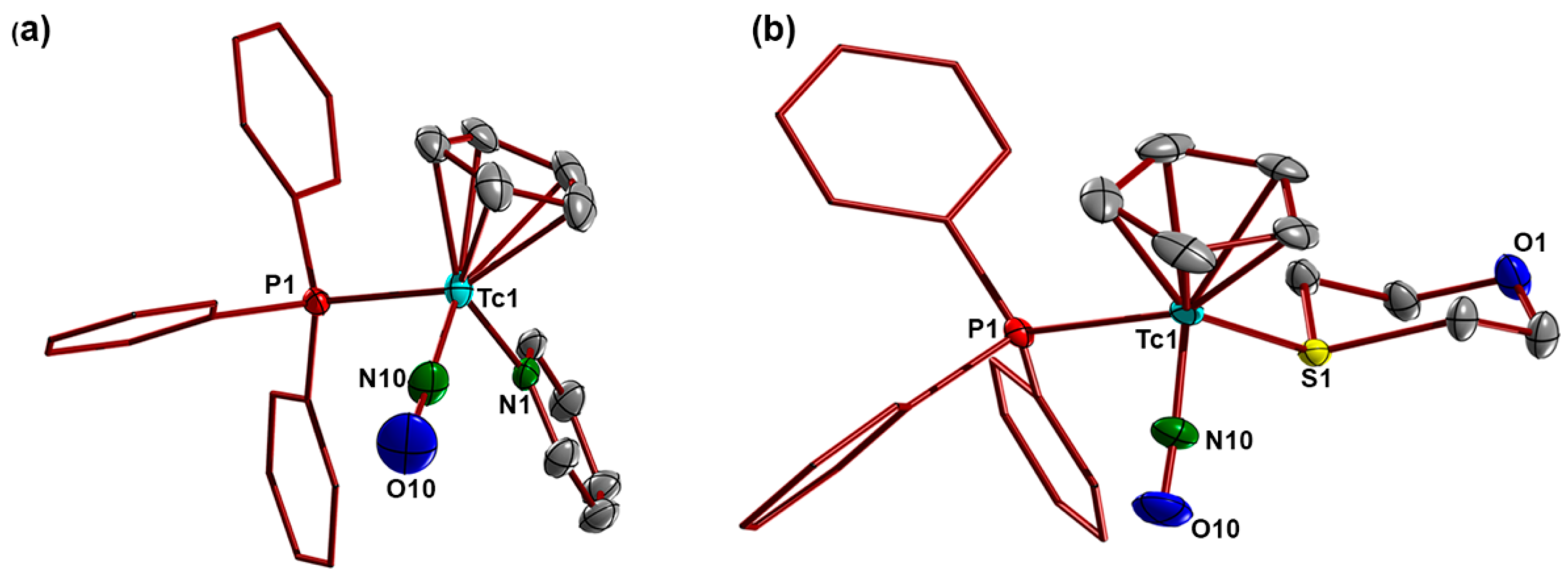
3.4. X-ray Crystallography
3.5. Computational Chemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tam, W.; Wong, W.K.; Gladysz, J.A. Neutral metal formyl complexes: Generation, reactivity, and models for Fischer−Tropsch catalyst intermediates. J. Am. Chem. Soc. 1979, 101, 1589–1591. [Google Scholar] [CrossRef]
- Agbossou, F.; O’Connor, E.J.; Garner, C.M.; Quiros Mendez, N.; Fernandez, J.M.; Patton, A.T.; Ramsden, J.A.; Gladysz, J.A. Cyclopentadienyl Rhenium Complexes. Inorg. Synth. 1992, 29, 211–225. [Google Scholar]
- Gladysz, J.A.; Boone, B.J. Chiral Recognition in π Complexes of Alkynes, Aldehydes, and Ketones with Transition Metal Lewis Acids; Development of a General Model for Enantioface Binding Selectivities. Angew. Chem. Int. Ed. Engl. 1997, 36, 550–583. [Google Scholar] [CrossRef]
- Zhou, Y.; Dewey, M.A.; Gladysz, J.A. Synthesis and Reactivity of Chiral Rhenium Indenyl Complexes of the Formula [(η5-C9H7)Re(NO)(PPh3)(X)]n+. Organometallics 1993, 12, 3918–3923. [Google Scholar] [CrossRef]
- Seidel, S.N.; Prommesberger, M.; Eichenseher, S.; Meyer, O.; Hampel, F.; Gladysz, J.A. Syntheses and structural analyses of chiral rhenium containing amines of the formula (η5-C5H5)Re(NO)(PPh3)((CH2)nNRR′) (n = 0, 1). Inorg. Chim. Acta 2010, 363, 533–548. [Google Scholar] [CrossRef]
- Dembinski, R.; Lis, T.; Szafert, S.; Mayne, C.J.; Bartik, T.; Gladysz, J.A. Appreciably bent sp carbon chains: Synthesis, structure, and protonation of organometallic 1,3,5-triynes and 1,3,5,7-tetraynes of the formula (η5-C5H5)Re(NO)(PPh3) ((C≡C)n-p-C6H4Me). J. Organomet. Chem. 1999, 578, 229–246. [Google Scholar] [CrossRef]
- Quiros Mendez, N.; Arif, A.M.; Gladysz, J.A. Synthesis, structure, and dynamic behavior of rhenium sulfide and sulfoxide complexes of the formula [(η5-C5H5)Re(NO)(L)(XRR’)]+X− (X = S, SO). Organometallics 1991, 10, 2199–2209. [Google Scholar] [CrossRef]
- Eichenseher, S.; Delacroix, O.; Kromm, K.; Hampel, F.; Gladysz, J.A. Rhenium-Containing Phosphorus Donor Ligands for Palladium-Catalyzed Suzuki Cross-Coupling Reactions: A New Strategy for High-Activity Systems. Organometallics 2005, 24, 245–255. [Google Scholar] [CrossRef]
- Kromm, K.; Hampel, F.; Gladysz, J.A. A New Family of Chelating Diphosphines with Transition-Metal and Carbon Stereocenters in the Backbone: A Second-Generation Rhenium-Containing System. Organometallics 2002, 21, 4264–4274. [Google Scholar] [CrossRef]
- Dewey, M.A.; Bakke, J.M.; Gladysz, J.A. Synthesis and reactivity of chiral rhenium amine and amide complexes of the formulas [(η5-C5H5)Re(NO)(PPh3)(NHRR’)]+ TfO− and [(η5-C5H5)Re(NO)(PPh3)NRR’. Organometallics 1990, 9, 1349–1351. [Google Scholar] [CrossRef]
- Cambridge Structural Database, Release 5.40. November 2023.
- Burzlaff, N.; Schenk, W.A. Chiral Rhenium Complexes of Functionalized Thioaldehydes. Eur. J. Inorg. Chem. 1999, 1999, 1435–1443. [Google Scholar] [CrossRef]
- Salzer, A.; Hosang, A.; Knuppertz, A.; Englert, U. Transition-Metal Complexes of the Optically Active Cyclopentadienyl Ligand PinCp*: Crystal Structure of (SRe)-(η5-PinCp*)Re(NO)(PPh3)[CONHCH(CH3)C10H7]. Eur. J. Inorg. Chem. 1999, 1999, 1497–1505. [Google Scholar] [CrossRef]
- Bernasconi, C.F.; Bhattacharya, S.; Wenzel, P.J.; Olmstead, M.M. Kinetic and Thermodynamic Acidity of [Cp(NO)(PPh3)Re(2,5-dimethyl-3-thienyl)carbene]+. Transition State Imbalance and Intrinsic Barriers. Organometallics 2006, 25, 4322–4330. [Google Scholar] [CrossRef]
- McCormick, F.B. Synthesis and structural characterization of a cationic rhenium selenoxoformaldehyde complex. Organometallics 1984, 3, 1924–1927. [Google Scholar] [CrossRef]
- Dilsky, S.; Schenk, W.A. Diastereomeric Halfsandwich Rhenium Complexes Containing Hemilabile Phosphane Ligands. Eur. J. Inorg. Chem. 2004, 2004, 4859–4870. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Rheingold, A.L. Bimetallic. mu.-malonyl compounds. Synthesis, characterization, and reactivity of (η5-C5Me5)Re(NO)(PPh3)-cyclo[(μ-,η1-,η2-COCH2CO)M(CO)4] (M = Re, Mn). Organometallics 1987, 6, 1987–1989. [Google Scholar] [CrossRef]
- Legoupy, S.; Crevisy, C.; Guillemin, J.-C.; Gree, R.; Toupet, L. Regio- and Stereoselective Nucleophilic Substitutions of Chiral Allylic Alcohol Rhenium Complexes. Chem.-Eur. J. 1998, 4, 2162–2172. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Rheingold, A.L. Thermodynamic control of stereochemistry in alkylation of chiral transition-metal .beta.-oxoacyl compounds: Enolization without epimerization. Organometallics 1988, 7, 2422–2424. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Chadha, R.K.; Tsuie, B.; Rheingold, A.L. Reactivity studies on bimetallic μ-malonyl complexes: Cleavage and alkylation chemistry of the malonyl ligand. J. Organomet. Chem. 1993, 455, 143–156. [Google Scholar] [CrossRef]
- Burzlaff, N.; Schenk, W.A. Synthesis of Chiral Rhenium Complexes Containing Functionalized Thiolate Ligands. Eur. J. Inorg. Chem. 1998, 1998, 2055–2061. [Google Scholar] [CrossRef]
- Stark, G.A.; Arif, M.; Gladysz, J.A. Diastereoselektive 1,2 additions of Nucleophiles to Quinoline Complexes of the Chiral Lewis Acid [(η5-C5H5)Re(NO)(PPh3)]+. Organometallics 1994, 13, 4523–4530. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Chadha, R.K. Conversion of a metallaenolate complex to a bimetallic μ-ketene complex: Molecular structure of (η5-C5Me5)(NO)(PPh3)Re[μ-(COCH2)-C1:C2]Re(CO)4(PPh3). Polyhedron 1993, 12, 527–532. [Google Scholar]
- Tetrick, S.M.; Cavanaugh, M.D.; Tha, F.S.; Cutler, A.R. Unusual Degradation of the Rhenium Silyl Ester Cp(NO)(PPh3)ReCO2SiMe2Ph to the Bimetallic μ-η1(C(Re)):η1(O,O′(Re)) Carbon Dioxide Complex Cp(NO)(PPh3)ReCO2Re(NO)(CO)(PPh3)OSiMe2Ph. Organometallics 1998, 17, 1925–1927. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Rheingold, A.L.; Staley, D.L.; Chadha, R.K. Synthesis and structural characterization of bimetallic. μ-malonyl complexes. J. Am. Chem. Soc. 1990, 112, 7585–7598. [Google Scholar] [CrossRef]
- Bosch, W.H.; Englert, U.; Pfister, B.; Stauber, R.; Salzer, A. Optically active transition-metal complexes II. Rhenium complexes with the optically active cyclopentadienyl ligand PCp: X-ray structures of the exo and endo isomers of PCpRe(CO)3 and of the derivative PCpReNO(CH3)PPh3. J. Organomet. Chem. 1996, 506, 273–285. [Google Scholar]
- O’Connor, J.M.; Uhrhammer, R.; Rheingold, A.L.; Staley, D.L. Synthesis and characterization of a novel bimetallic μ-malonyl complex. The first x-ray crystal structure of alkali metal chelation by a neutral malonyl compound. J. Am. Chem. Soc. 1989, 111, 7633–7634. [Google Scholar] [CrossRef]
- O’Connor, J.M.; Uhrhammer, R.; Rheingold, A.L.; Roddick, D.M. Keto-enol tautomerization in metal-acyl complexes: The enolization properties of bimetallic μ-malonyl compounds. J. Am. Chem. Soc. 1991, 113, 4530–4544. [Google Scholar] [CrossRef]
- White, C.J.; Angelici, R.J. Synthesis, Structure, and Reactivity of Thienyl-, Benzothienyl-, and Selenylcarbene Complexes of Rhenium: A New Mechanism for H/D Exchange During Hydrodesulfurization. Organometallics 1994, 13, 5132–5140. [Google Scholar]
- Pfister, B.; Englert, U.; Salzer, A. Optically Active Transition-Metal Complexes. 4. Rhenium Complexes with the Enantiopure Cyclopentadienyl Ligand PCp: X-ray Structure of the exo Isomer of SRe-(PCp)Re(NO)(PPh3)(CH3). Organometallics 1995, 14, 5561–5565. [Google Scholar] [CrossRef]
- Blanchard, S.S.; Nicholson, T.; Davison, A.; Davis, W.; Jones, A.G. The synthesis, characterization and substitution reactions of the mixed technetium(I) nitrosyl complex trans-trans-[(NO)(NCCH3)Cl2(PPh3)2Tc]. Inorg. Chim. Acta 1996, 244, 121–130. [Google Scholar] [CrossRef]
- Ackermann, J.; Nijki Noufele, C.; Hagenbach, A.; Abram, U. Nitrosyltechnetium(I) Complexes with 2-(Diphenylphosphanyl)aniline. Z. Anorg. Allg. Chem. 2019, 645, 8–13. [Google Scholar] [CrossRef]
- Ackermann, J.; Hagenbach, A.; Abram, U. {Tc(NO)(Cp)(PPh3)}+—A novel technetium(I) core. Chem. Commun. 2016, 52, 10285–10288. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, J.; Abdulkader, A.; Scholtysik, C.; Jungfer, M.R.; Hagenbach, A.; Abram, U. [TcI(NO)X(Cp)(PPh3)] Complexes (X− = I−, I3−, SCN−, CF3SO3−, or CF3COO−) and Their Reactions. Organometallics 2019, 38, 4471–4478. [Google Scholar] [CrossRef]
- Abdulkader, A.; Hagenbach, A.; Abram, U. [Tc(NO)Cl(Cp)(PPh3)]—A Technetium(I) Compound with an Unexpected Synthetic Potential, Eur. J. Inorg. Chem. 2021, 2021, 3812–3818. [Google Scholar]
- Fernandez, J.M.; Emerson, K.; Larsen, R.D.; Gladysz, J.A. The selective activation of one methyl ketone enantioface via σ-binding to a chiral metal template: Synthesis and reactivity of rhenium ketone complexes [(η5-C5H5)Re(NO)(PPh3)(η1-Me(R)CO)]+PF6−. J. Chem. Soc. Chem. Commun. 1988, 37–39. [Google Scholar] [CrossRef]
- Dalton, D.M.; Fernandez, J.M.; Emerson, K.; Larsen, R.D.; Arif, A.M.; Gladysz, J.A. Selective activation of one methyl ketone enantioface via sigma-binding to a chiral transition-metal template: Synthesis, structure, and reactivity of rhenium ketone complexes [(η5-C5H5)Re(NO)(PPh3)(η1-O=C(CH3)R)]+X−. J. Am. Chem. Soc. 1990, 112, 9198–9212. [Google Scholar] [CrossRef]
- Mendez, N.Q.; Arif, A.M.; Gladysz, J.A. π/σ Equilibria in Metal Complexes of Organic Carbonyl Compounds; Synthesis and Structure of Chiral Rhenium Complexes [(η5-C5H5)Re(NO)(PPh3)(O=CHAr)]X. Angew. Chem. Int. Ed. Engl. 1990, 29, 1473–1474. [Google Scholar] [CrossRef]
- Mendez, N.Q.; Seyler, J.W.; Arif, A.M.; Gladysz, J.A. Synthesis, structure, and spectroscopic properties of chiral rhenium aromatic aldehyde complexes [(η5-C5H5)Re(NO)(PPh3)(O=CHAr)]+X−: Equilibria between π and σ aldehyde binding modes. J. Am. Chem. Soc. 1993, 115, 2323–2334. [Google Scholar] [CrossRef]
- Bringmann, G.; Schupp, O.; Peters, K.; Walz, L.; von Schnering, H.G. Novel concepts in directed biaryl synthesis: XV. Chiral rhenium complexes [(η5-C5H5)Re(NO)(PPh3)(R,R-lactone)]+BF4− of lactone-bridged biaryls as ligands. J. Organomet. Chem. 1992, 438, 117–130. [Google Scholar] [CrossRef]
- Saura-Llamas, I.; Dalton, D.M.; Arif, A.M.; Gladysz, J.A. Synthesis, structure, and reactivity of chiral rhenium carboxylic and carbonic acid ester complexes of the formula [(η5-C5H5)Re(NO)(PPh3)(η1-O=C(X)X′)]+ X″−. Organometallics 1992, 11, 683–693. [Google Scholar] [CrossRef]
- Mendez, N.Q.; Arif, A.M.; Gladysz, J.A. Synthesis, Structure, and Dynamic Behavior of Rhenium Sulfide and Sulfoxide Complexes of the Formula [(η5-C5H5)Re(NO)(L)(XRR′)]+X′− (X = S, SO). Organometallics 1991, 10, 2199–2209. [Google Scholar] [CrossRef]
- Cagle, P.C.; Meyer, O.; Weickhardt, K.; Arif, A.M.; Gladysz, J.A. Enantioselective Synthesis of Organosulfur Compounds via [2,3] Rearrangements of Ylides Derived from Di(allyl) and Di(propargyl) Sulfide Complexes. Control of Carbon Configuration by an Easily Resolved and Recycled Chiral Transition Metal Auxiliary. J. Am. Chem. Soc. 1995, 117, 11730–11744. [Google Scholar] [CrossRef]
- Schenk, W.A.; Burzlaff, N.; Burzlaff, H.Z. Chiral Thioaldehyde Complexes of Rhenium, X-Ray Structure Determination of [Cp(NO)(Ph3P)Re(η2-S=CHPh)]PF6 [1]. Z. Naturforsch. 1994, B49, 1633–1639. [Google Scholar] [CrossRef]
- Schwochau, K. Technetium, Chemistry and Radiopharmaceutical Applications; Wiley: Weinheim, Germany, 2000. [Google Scholar]
- Alberto, R. Technetium. In Comprehensive Coordination Chemistry II; McCleverty, J.A., Meyer, T.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 5, p. 127. [Google Scholar]
- Besmer, M.L.; Braband, H.; Schneider, S.; Spingler, B.; Alberto, R. Exploring the Coordination Chemistry of N2 with Technetium PNP Pincer-Type Complexes. Inorg. Chem. 2021, 60, 6696–6701. [Google Scholar] [CrossRef] [PubMed]
- Besmer, M.L.; Braband, H.; Fox, T.; Spingler, B.; Sattelberger, A.P.; Alberto, R. Binding Small Molecules to a cis-Dicarbonyl 99TcI-PNP Complex via Metal-Ligand Cooperativity. Inorg. Chem. 2023, 62, 10727–10735. [Google Scholar] [CrossRef]
- Besmer, M.L.; Schwitter, F.; Battistin, F.; Braband, H.; Fox, T.; Spingler, B.; Alberto, R. Induced fac–mer rearrangements in {M(CO)3}+ complexes (M = Re, 99(m)Tc) by a PNP ligand. Dalton Trans. 2024, 53, 1434–1438. [Google Scholar] [CrossRef]
- Nadeem, Q.; Meola, G.; Braband, H.; Bolliger, R.; Blacque, O.; Hernandez-Valdes, D.; Alberto, A. To Sandwich Technetium: Highly Functionalized Bis-Arene Complexes [99mTc(η6-arene)2]+ Directly from Water and [99mTcO4]−. Angew. Chem. Int. Ed. 2020, 59, 1197–1200. [Google Scholar] [CrossRef]
- World Nuclear Association. Radioisotopes in Medicine. Available online: https://www.world-nuclear.org/information-library/non-power-nuclear-applications/radioisotopes-research/radioisotopes-in-medicine.aspx (accessed on 21 January 2024).
- Boschi, A.; Uccelli, L.; Martini, P. A Picture of Modern Tc-99m Radiopharmaceuticals: Production, Chemistry, and Applications in Molecular Imaging. Appl. Sci. 2019, 9, 2526. [Google Scholar] [CrossRef]
- Martini, P.; Pasquali, M.; Boschi, A.; Uccelli, L.; Giganti, M.; Duatti, A. Technetium Complexes and Radiopharmaceuticals with Scorpionate Ligands. Molecules 2018, 23, 2039. [Google Scholar] [CrossRef]
- Papagiannopoulou, D. Technetium-99m radiochemistry for pharmaceutical applications. J. Label. Compd. Radiopharm. 2017, 60, 502–520. [Google Scholar] [CrossRef] [PubMed]
- Alberto, R.; Braband, H. SPECT/PET Imaging with Technetium, Gallium, Copper, and Other Metallic Radionuclides. In Comprehensive Inorganic Chemistry II: From Elements to Applications, 2nd ed.; Reedjk, J., Poeppelmeier, K., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2013; pp. 785–817. [Google Scholar]
- Brunello, S.; Salvarese, N.; Carpanese, D.; Gobbi, C.; Melendez-Alafort, L.; Bolzati, C. A Review on the Current State and Future Perspectives of [99mTc]Tc-Housed PSMA-I in Prostate Cancer. Molecules 2022, 27, 2617. [Google Scholar] [CrossRef]
- Duatti, A. Review on 99mTc radiopharmaceuticals with emphasis on new advancements. Nucl. Med. Biol. 2021, 92, 202–216. [Google Scholar] [CrossRef]
- Fernandez, J.M.; Gladysz, J.A. Synthetic approaches to the chiral, pyramidal, transition-metal Lewis acid [(η5-C5H5)Re(NO)(PPh3)]+X−. Generation, characterization, and reactions of a dichloromethane adduct. Organometallics 1989, 8, 207–210. [Google Scholar] [CrossRef]
- Mikhalev, V.A. 99Tc NMR Spectroscopy. Radiochemistry 2005, 47, 319–333. [Google Scholar] [CrossRef]
- Balasekaran, S.M.; Hagenbach, A.; Drees, M.; Abram, U. [TcII(NO)(trifluoroacetate)4F]2−—Synthesis and reactions. Dalton Trans. 2017, 46, 13544–13552. [Google Scholar] [CrossRef]
- Roca Jungfer, M.; Elsholz, L.; Abram, U. Technetium Hydrides Revisited: Syntheses, Structures and Reactions of [TcH3(PPh3)4] and [TcH(CO)(PPh3)2]. Organometallics 2021, 40, 3095–3112. [Google Scholar] [CrossRef]
- Roca Jungfer, M.; Abram, U. [Tc(OH2)(CO)3(PPh3)2]+: A Synthon for Tc(I) Complexes and its Reactions with neutral ligands. Inorg. Chem. 2021, 60, 16734–16753. [Google Scholar] [CrossRef] [PubMed]
- Roca Jungfer, M.; Elsholz, L.; Abram, U. Technetium(I) Carbonyl Chemistry with Small Inorganic Ligands. Inorg. Chem. 2022, 61, 2980–2997. [Google Scholar] [CrossRef] [PubMed]
- Alberto, R.; Bergamaschi, G.; Braband, H.; Fox, T.; Amendola, V. 99TcO4−: Selective Recognition and Trapping in aqueous solution. Angew. Chem. Int. Ed. 2012, 51, 9772–9776. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.; Chun, E.; Mahmood, A.; Mueller, P.; Davison, A.; Jones, A.G. Synthesis, spectroscopy and structural analysis of Technetium and Rhenium nitrosyl complexes. Commun. Inorg. Synth. 2015, 3, 31–39. [Google Scholar]
- Hagenbach, A.; Yegen, E.; Abram, U. Technetium Tetrachloride as A Precursor for Small Technetium(IV) Complexes. Inorg. Chem. 2006, 45, 7331–7338. [Google Scholar] [CrossRef] [PubMed]
- Kaden, L.; Lorenz, B.; Kirmse, R.; Stach, J.; Behm, H.; Beurskens, P.T. Synthesis, characterization and x-ray molecular and crystal structure of Tc(NS)Cl3(Me2PhP)(Me2PhPO)-a first example of mixed phosphine/phosphine oxide coordination. Inorg. Chim. Acta 1990, 169, 43–48. [Google Scholar]
- Flörke, U. Cambridge Structural Database. Private Communication. 2015; Entry RUCNEK. [Google Scholar]
- Flörke, U. Cambridge Structural Database. Private Communication. 2015; Entry VUCGUX. [Google Scholar]
- Balasekaran, S.M.; Hagenbach, A.; Spandl, J.; Abram, U. The Reaction of Cs2[Tc(NO)F5] with BF3 in Acetonitrile: Formation and Structure of [{Tc(NO)(CH3CN)4}2(μ-F)](BF4)3. Z. Anorg. Allg. Chem. 2017, 643, 1146–1149. [Google Scholar] [CrossRef]
- Linder, K.E.; Davison, A.; Dewan, J.C.; Costello, C.E.; Maleknia, S. Nitrosyl complexes of technetium: Synthesis and characterization of [TcI(NO)(CNCMe3)5](PF6)2 and Tc(NO)Br2(CNCMe3)3 and the crystal structure of Tc(NO)Br2(CNCMe3)3. Inorg. Chem. 1986, 25, 2085–2089. [Google Scholar] [CrossRef]
- Ernst, M.J.; Roca Jungfer, M.; Abram, U. Reactions of TcI(NO) and TcVN Complexes with Alkynes and Alkynides. Organometallics 2022, 41, 2011–2021. [Google Scholar] [CrossRef]
- Ackermann, J.; Hagenbach, A.; Abram, U. Nitrosyltechnetium complexes with (2-aminomethylphenyl)diphenylphosphine. Inorg. Chim. Acta 2014, 419, 59–65. [Google Scholar] [CrossRef]
- Schibli, R.; Mati, N.; Spingler, B.; Lehaire, M.-L.; Gramlich, V.; Barnes, C.L. Syntheses and Characterization of Dicarbonyl−Nitrosyl Complexes of Technetium(I) and Rhenium(I) in Aqueous Media: Spectroscopic, Structural, and DFT Analyses. Inorg. Chem. 2005, 44, 683–690. [Google Scholar] [CrossRef]
- Brown, D.S.; Newman, J.L.; Thornback, J.R.; Davison, A. Structure of the tetra-n-butylammonium salt of the tetrachloro(methanol)nitrosyltechnetium(II) anion. Acta Crystallogr. Struct. Commun. 1987, 43, 1692–1694. [Google Scholar] [CrossRef]
- Brown, D.S.; Newman, J.L.; Thornback, J.R. The structure of the tetraphenylarsonium salt of the trichloro(pentane-2,4-dionato)nitrosyltechnetium(II) anion. Acta Crystallogr. Struct. Commun. 1988, 44, 973–975. [Google Scholar] [CrossRef]
- Brown, D.S.; Newman, J.L.; Thornback, J.R.; Pearlstein, P.M.; Davison, A.; Lawson, A. The synthesis and characterisation of the trichloronitrosyl(acetylacetonato)technetium(II) anion, a novel technetium(II) complex. Inorg. Chim. Acta 1988, 150, 169–193. [Google Scholar] [CrossRef]
- Claude, G.; Salsi, F.; Hagenbach, A.; Gembicky, M.; Neville, M.; Chan, C.; Figueroa, J.S.; Abram, U. Structural and Redox Variations in Technetium Complexes Supported by m-Terphenyl Isocyanides. Organometallics 2020, 39, 2287–2294. [Google Scholar] [CrossRef]
- Banberry, H.J.; Hamor, T.A. Chloronitrosylbis[o-phenylenebis(dimethylarsine)]technetium(I) chloride-tetrabutylammonium chloride (1/1). Acta Crystalogr. Sect. C Cryst. Struct. Commun. 1994, 50, 44–46. [Google Scholar] [CrossRef]
- Lu, J.; Clarke, M.J. Modulation of Tc–NX (X = O or S) bonds by π-acceptor ligands. J. Chem. Soc. Dalton Trans. 1992, 1243–1248. [Google Scholar] [CrossRef]
- Roca Jungfer, M.; Ernst, M.J.; Hagenbach, A.; Abram, U. [{TcI(NO)(LOMe)(PPh3)Cl}2Ag](PF6) and [TcII(NO)(LOMe)(PPh3)Cl](PF6): Two Unusual Technetium Complexes with a “Kläui-type” Ligand. Z. Anorg. Allg. Chem. 2022, 648, e2021003. [Google Scholar] [CrossRef]
- Balasekaran, S.M.; Spandl, J.; Hagenbach, A.; Köhler, K.; Drees, M.; Abram, U. Fluoridonitrosyl Complexes of Technetium(I) and Technetium(II). Synthesis, Characterization, Reactions, and DFT Calculations. Inorg. Chem. 2014, 53, 5117–5128. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, T.; Hirsch-Kuchma, M.; Shellenberger-Jones, A.; Davison, A.; Jones, A.G. The synthesis and characterization of a technetium nitrosyl complex with cis-{2-pyridyl,diphenylphosphine} coligands. The X-ray crystal structure of [TcCl2(NO)(pyPPh2-P,N)(pyPPh2-P)]. Inorg. Chim. Acta 1998, 267, 319–322. [Google Scholar] [CrossRef]
- Grunwald, A.C.; Scholysik, C.; Hagenbach, A.; Abram, U. One Ligand, One Metal, Seven Oxidation States: Stable Technetium Complexes with the “Kläui Ligand”. Inorg. Chem. 2020, 59, 9396–9405. [Google Scholar] [CrossRef]
- Nicholson, T.; Müller, P.; Davison, A.; Jones, A.G. The synthesis and characterization of a cationic technetium nitrosyl complex: The X-ray crystal structure of [TcCl(NO)(DPPE)2](PF6) · CH2Cl2. Inorg. Chim. Acta 2006, 359, 1296–1298. [Google Scholar] [CrossRef]
- Kirmse, R.; Lorenz, B. EPR on trichloro-nitrosyl-bis(dimethylphenylphosphine)technetium(II) TcCl3(NO)(PMe2Ph)2. Polyhedron 1983, 2, 935–939. [Google Scholar] [CrossRef]
- White, C.; Thompson, S.J.; Maitlis, P.M. Pentamethylcyclopentadienyl-rhodium and-iridium complexes XIV. The solvolysis of coordinated acetone solvent species to tris (μ-difluorophosphato) bis [η5-pentamethylcyclopentadienylrhodium (III)] hexafluorophosphate, to the η5-(2, 4-dimethyl-1-oxapenta-1, 3-dienyl)(pentamethylcyclopentadienyl) iridium cation, or to the η5-(2-hydroxy-4-methylpentadienyl)(η5-pentamethylcyclopentadienyl) iridium cation. J. Organomet. Chem. 1977, 134, 319–325. [Google Scholar]
- Thompson, S.J.; Bailey, P.M.; White, C.; Maitlis, P.M. Solvolysis of the Hexafluorophosphate Ion and the Structure of [Tris (μ-difluorophosphato) bis (penta-methylcyclopentadienylrhodium)] Hexafluorophosphate. Angew. Chem. Int. Ed. 1976, 15, 490–491. [Google Scholar] [CrossRef]
- Keyzer, E.N.; Matthews, P.D.; Liu, Z.; Bond, A.D.; Grey, C.P.; Wright, D.S. Synthesis of Ca(PF6)2, formed via nitrosonium oxidation of calcium. Chem. Commun. 2017, 53, 4573–4576. [Google Scholar] [CrossRef]
- Cotton, F.A.; Haefner, S.C.; Sattelberger, A.P. Metal-metal multiply-bonded complexes of technetium. 6. A μ, η1, η2-CH3CN complex prepared via reductive cleavage of the electron-rich Tc-Tc triple bond in decakis-acetonitrile ditechnetium tetrafluoroborate. Inorg. Chim. Acta 1997, 266, 55–63. [Google Scholar] [CrossRef]
- Abram, U.; Abram, S.; Schibli, R.; Alberto, R.; Dilworth, J.R. Synthesis and structures of technetium(I) and rhenium(I) tricarbonyl complexes with bis(diphenylthiophosphoryl)amide, [M(CO)3[(Ph2PS)2N](CH3CN)] (M = Tc, Re). Polyhedron 1998, 17, 1303–1309. [Google Scholar] [CrossRef]
- Miroslavov, A.E.; Sidorenko, G.V.; Lumpov, A.A.; Suglobov, D.N.; Sizova, O.V.; Maltsev, D.A.; Gurzhiy, V.V.; Polotskii, Y.S. Reaction of technetium hexacarbonyl cation with acetonitrile: Kinetics, product structure, DFT calculations. J. Organomet. Chem. 2012, 720, 1–6. [Google Scholar] [CrossRef]
- Freiberg, E.; Davies, W.M.; Davison, A.; Jones, A.G. Synthesis and Characterization of Technetiumtetrakis(acetonitrile)bis(triphenylphosphine) Cationic Complexes. Inorg.Chem. 2002, 41, 3337–3339. [Google Scholar] [CrossRef]
- Kowalczyk, J.J.; Arif, A.M.; Gladysz, J.A. Synthesis, Structure, and Reactivity of Chiral Rhenium Alkyne Complexes of the Formula [(η5-C5H5)Re(NO)(PPh3)(RC=CR’)](BF4)]. Organometallics 1991, 10, 1079–1088. [Google Scholar] [CrossRef]
- Kowalczyk, J.J.; Agbossou, K.S.; Gladysz, A.J. Generation and reactivity of the chiral rhenium chlorobenzene complex [(η5-C5H5)Re(NO)(PPh3)(CIC6H5)]+BF4−: An improved functional equivalent of the chiral Lewis acid [(η5-C5H5)Re(NO)(PPh3)]+BF4−. J. Organomet. Chem. 1990, 397, 333–346. [Google Scholar] [CrossRef]
- Otto, M.; Boone, J.B.; Arif, M.A.; Gladysz, A.J. Synthesis, structure, and interconversion of chiral rhenium oxygenand sulfur-bound sulfoxide complexes of formula [(η5-C5H5)Re(NO)(PPh3)(OS(Me)R)]+ X−; diastereoselective oxidations of coordinated methyl alkyl sulfides. J. Chem. Soc. Dalton Trans. 2001, 1218–1229. [Google Scholar] [CrossRef]
- Abram, U.; Lorenz, B.; Kaden, L.; Scheller, D. Nitrido Complexes of Technetium with tertiary phosphines and Arsines. Polyhedron 1988, 7, 285–289. [Google Scholar] [CrossRef]
- O’Connell, L.A.; Pearlstein, R.M.; Davison, A.; Thornback, J.R.; Kronauge, J.F.; Jones, A.G. Technetium-99 NMR spectroscopy: Chemical shift trends and long range coupling effects. Inorg. Chim. Acta 1989, 161, 39–43. [Google Scholar] [CrossRef]
- Claude, G.; Zeh, L.; Roca Jungfer, M.; Hagenbach, A.; Figueroa, J.S.; Abram, U. The Chemistry of Phenyltechnetium(V) Complexes with Isocyanides: Steric and Electronic Factors. Molecules 2022, 27, 8546. [Google Scholar] [CrossRef]
- Miroslavov, A.E.; Sidorenko, G.V.; Suglobov, D.N.; Lumpov, A.A.; Gurzhiy, V.V.; Grigorev, M.S.; Mikhalev, V.A. Technetium(I) Dithiocarbamates and Xanthates. Inorg. Chem. 2011, 50, 1098–1104. [Google Scholar] [CrossRef]
- Zuhayra, M.; Lützen, U.; Lützen, A.; Papp, L.; Henze, E.; Friedrichs, G.; Oberdorfer, F. C-H Bond Activation of Coordinated Pyridine: Ortho-Pyridyl-Ditechnetiumhydridocarbonyl Metal Cyclus. Crystal Structure and Dynamic Behavior in Solution. Inorg. Chem. 2008, 47, 10177–10182. [Google Scholar] [CrossRef] [PubMed]
- Leibnitz, P.; Reck, G.; Pietzsch, H.-J.; Spies, H. Cambridge Structural Database. 2001; entry AMUBIT.
- Leibnitz, P.; Reck, G.; Pietzsch, H.-J.; Spies, H. Cambridge Structural Database. 2001; entry AMUBUF.
- Schibli, R.; Alberto, R.; Abram, U.; Abram, S.; Egli, A.; Schubiger, P.A.; Kaden, T.A. Structural and 99Tc NMR Investigations of Complexes with fac-[Tc(CO)3]+ Moieties and Macrocyclic Thioethers of Various Ring Sizes: Synthesis and X-ray Structure of the Complexes fac-[Tc(9-ane-S3)(CO)3]Br, fac-[Tc2(tosylate)2(18-ane-S6)(CO)6], and fac-[Tc2(20-ane-S6-OH)(CO)6][tosylate]2. Inorg. Chem. 1998, 37, 3509–3516. [Google Scholar] [PubMed]
- Pietzsch, H.-J.; Tisato, F.; Refosco, F.; Leibnitz, P.; Seifert, S.; Spies, H. Synthesis and Characterization of Novel Trigonal Bipyramidal Technetium(III) Mixed-Ligand Complexes with SES/S/P Coordination (E = O, N(CH3), S). Inorg. Chem. 2001, 40, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Oehlke, E.; Alberto, R.; Abram, U. Synthesis, Characterization, and Structures of R3EOTcO3 Complexes (E = C, Si, Ge, Sn, Pb) and Related Compounds. Inorg. Chem. 2010, 49, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Alberto, R. Role of Pure Technetium Chemistry: Are There Still Links to Applications in Imaging? Inorg. Chem. 2023, 62, 20539–20548. [Google Scholar] [CrossRef]
- Wald, J.; Alberto, R.; Ortner, K.; Candreia, L. Aqueous One-Pot Synthesis of Derivatized Cyclopentadienyl–Tricarbonyl Complexes of 99mTc with an In Situ CO Source: Application to a Serotonergic Receptor Ligand. Angew. Chem. Int. Ed. 2001, 40, 3062–3066. [Google Scholar] [CrossRef]
- Masi, S.; Top, S.; Boubekeur, L.; Jaouen, G.; Mundwiler, S.; Spingler, B.; Alberto, R. Direct Synthesis of Tricarbonyl(cyclopentadienyl)rhenium and Tricarbonyl(cyclopentadienyl)technetium Units from Ferrocenyl Moieties—Preparation of 17α-Ethynylestradiol Derivatives Bearing a Tricarbonyl(cyclopentadienyl)technetium Group. Eur. J. Inorg. Chem. 2004, 2004, 2013–2017. [Google Scholar] [CrossRef]
- Zobi, F.; Spingler, B.; Alberto, R. Syntheses, Structures and Reactivities of [CpTc(CO)3X]+ and [CpRe(CO)3X]. Eur. J. Inorg. Chem. 2008, 4205–4214. [Google Scholar] [CrossRef]
- Benz, M.; Braband, H.; Schmutz, P.; Halter, J.; Alberto, R. From TcVII to TcI; facile syntheses of bis-arene complexes [99(m)Tc(arene)2]+ from pertechnetate. Chem. Sci. 2015, 6, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Braband, H.; Jordi, S.; Fox, T.; Blacque, O.; Spingler, B.; Alberto, R. Structure and reactivities of rhenium and technetium bis-arene sandwich complexes [M(η6-arene)2]+. Dalton Trans. 2017, 46, 14631–14637. [Google Scholar] [CrossRef]
- Meola, G.; Braband, H.; Hernandez-Valdes, G.; Gotzmann, C.; Fox, T.; Spingler, B.; Alberto, R. A Mixed-Ring Sandwich Complex from Unexpected Ring Contraction in [Re(η6-C6H5Br)(η6-C6R6)](PF6). Inorg. Chem. 2017, 56, 6297–6301. [Google Scholar] [CrossRef]
- Mindt, T.L.; Struthers, H.; Brans, L.; Anguelov, T.; Schweinsberg, C.; Maes, V.; Tourwe, D.; Schibli, R. “Click to Chelate”: Synthesis and Installation of Metal Chelates into Biomolecules in a Single Step. J. Am. Chem. Soc. 2006, 128, 15096–15097. [Google Scholar] [CrossRef]
- Spang, P.; Herrmann, C.; Roesch, F. Bifunctional Gallium-68 Chelators: Past, Present, and Future. Sem. Nucl. Chem. 2016, 46, 373–394. [Google Scholar] [CrossRef]
- Claude, G.; Puccio, D.; Roca Jungfer, M.; Hagenbach, A.; Spreckelmeyer, S.; Abram, U. Technetium Complexes with an Isocyano-alkyne Ligand and its Reaction Products. Inorg. Chem. 2023, 62, 12445–12452. [Google Scholar] [CrossRef]
- Johnston, P.; Loonat, S.M.; Ingham, L.W.; Carlton, L.; Coville, J.N. Substituted Cyclopentadienyl Complexes. 1. The Proton Nuclear Magnetic Resonance Spectra off [(η5-C5H4Me)Fe(CO)(LI] and [(η5-C9H7)Fe(CO)(L)I]. Organometallics 1987, 6, 2121–2127. [Google Scholar] [CrossRef]
- Carlton, L.; Johnston, P.; Coville, J.N. Substituted cyclopentadienyl complexes II. 13C NMR spectra of some [(η5-C5H4Me)Fe(CO)(L)I] complexes. J. Organomet. Chem. 1988, 339, 339–343. [Google Scholar] [CrossRef]
- Winter, C.H.; Veal, W.R.; Garner, C.N.; Arif, A.M.; Gladysz, J.A. Synthesis, structure, and reactivity of stable alkyl and aryl iodide complexes of the formula [(η5-C5H5(Re(NO)(PPh3)(IR)]+BF4−. J. Am. Chem. Soc. 1989, 111, 4766–4776. [Google Scholar] [CrossRef]
- Kowalczyk, J.J.; Arif, A.M.; Gladysz, J.A. Synthesis, Structure, and Reactivity of Chiral Rhenium Cycloalkene Complexes of the Formula [(η5-C5H5)Re(NO)(PPh3)(CH=CH(CH2)n−2]+(BF4)− Facile Vinylic Deprotonation of a Coordinated Alkene. Chem. Ber. 1991, 124, 729–742. [Google Scholar] [CrossRef]
- Friedlein, F.K.; Kromm, K.; Hampel, F.; Gladysz, J.A. Synthesis, Structure, and Reactivity of Palladacycles That Contain a Chiral Rhenium Fragment in the Backbone: New Cyclometalation and Catalyst Design Strategies. Chem.-Eur. J. 2006, 12, 5267–5281. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR nomenclature. nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Wendland, D.; Bauche, J.; Luc, P. Hyperfine structure in Tc I: Experiment and theory. J. Phys. Ser. B 1977, 10, 1989–2002. [Google Scholar] [CrossRef]
- Claude, G.; Genz, J.; Weh, D.; Roca Jungfer, M.; Hagenbach, A.; Gembicky, M.; Figueroa, J.S.; Abram, U. Mixed-Isocyanide Complexes of Technetium under Steric and Electronic Control. Inorg. Chem. 2022, 61, 16163–16176. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Panda, T.K.; Gamer, M.T.; Roesky, P.W.; Yoo, H.; Berry, H.H. Sodium and Potassium Cyclopentadienide. Inorg. Synth. 2014, 36, 35–37. [Google Scholar]
- Jana, R.; Kumar, M.S.; Singh, N.; Elias, A.J. Synthesis, reactivity and structural studies of (η5-methylcyclopentadienyl)(η4-tetraphenylcyclopentadiene)cobalt and its derivatives. J. Organomet. Chem. 2008, 693, 3780–3786. [Google Scholar] [CrossRef]
- Hart, P.W.; Shihua, D.; Rausch, D.M. The formation and reactions of (η5-carboxycyclopentadienyl)dicarbonylcobalt. J. Organomet. Chem. 1985, 282, 111–121. [Google Scholar] [CrossRef]
- Sheldrick, G. SADABS; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Coppens, P. The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis; Crystallographic Computing: Copenhagen, Muksgaard, 1979. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Putz, H.; Brandenburg, K. Diamond-Crystal and Molecular Structure Visualization Crystal Impact; Version 5.1; GbR: Bonn, Germany, 2023. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree−Fock and local density functional theories. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Andrae, D.; Haußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustable initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart−Dresden−Bonn relativistic effective core potentials: The atoms Ga−Kr and In−Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Gaussian basis sets for molecular calculations. In Methods of Electronic Structure Theory; Modern Theoretical Chemistry, Schaefer, H.F., Eds.; Springer: Boston, MA, USA, 1977; Volume 3, pp. 1–27. [Google Scholar]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Clark, T.H.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B. 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Franzke, Y.J.; Treß, R.; Pazdera, T.M.; Weigend, F. Error-consistent segmented contracted all-electron relativistic basis sets of double- and triple-zeta quality for NMR shielding constants. Phys. Chem. Chem. Phys. 2019, 21, 16658–16664. [Google Scholar] [CrossRef] [PubMed]
- Bühl, M.; Golubnychiy, V. Density-functional computation of 99Tc NMR chemical shifts. Magn. Reson. Chem. 2008, 46, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Holfeltz, V.E.; Hall, G.B.; Johnson, I.E.; Walter, E.D.; Lee, S.; Reinhart, B.; Lukens, W.W.; Machara, N.P.; Levitskaia, T.G. Identification and Quantification of Technetium Species in Hanford Waste Tank AN-102. Anal. Chem. 2020, 92, 13961–13970. [Google Scholar] [CrossRef] [PubMed]
- de Andrade, T.F.C.B.; Dos Santos, H.F.; Fonseca Guerra, C.; Paschoal, D.F.S. Computational Prediction of Tc-99 NMR Chemical Shifts in Technetium Complexes with Radiopharmaceutical Applications. J. Phys. Chem. A 2022, 126, 5434–5448. [Google Scholar] [CrossRef]
- Chatterjee, S.D.; Andersen, A.; Du, Y.; Engelhard, M.H.; Hall, G.B.; Levitskaia, T.G.; Lukens, W.W.; Shutthanandan; Walter, E.D.; Washton, N.M. Tech. Report PNNL-26265; Pacific Northwest National Laboratory: Columbus, OH, USA, 2017. [Google Scholar]
- Hall, G.B.; Andersen, A.; Washton, N.M.; Chatterjee, S.; Levitskaia, T.G. Theoretical modeling of 99Tc NMR chemical shifts. Inorg. Chem. 2016, 55, 8341–8347. [Google Scholar] [CrossRef] [PubMed]
- Luksic, S.A.; Kim, D.; Levitskaia, T.; Chatterjee, S.; Lukens, W.; Kruger, A.A. Redox and volatility of Tc(CO)3+ compounds in waste glass melting. J. Nucl. Mat. 2019, 515, 199–205. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Mayer, I.; Salvador, P. Overlap populations, bond orders and valences for ‘fuzzy’ atoms. Chem. Phys. Lett. 2004, 383, 368–375. [Google Scholar] [CrossRef]
- Matito, E.; Poater, J.; Solà, M.; Duran, M.; Salvador, P. Comparison of the AIM Delocalization Index and the Mayer and Fuzzy Atom Bond Orders. J. Phys. Chem. A 2005, 109, 9904–9910. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tc-N10 | N10-O10 | Tc-E | Tc-P1 | E-P2 | Tc-N10-O10 | Tc-E-P2 | N10-Tc-E | N10-Tc-P1 | |
|---|---|---|---|---|---|---|---|---|---|
| 2(PF6) | 1.755(4) | 1.184(6) | 2.149(3) | 2.399(1) | 1.519(3) | 170.4(4) | 133.3(2) | 102.4(2) | 91.4(2) |
| 3(PF6) | 1.768(2) | 1.177(3) | 2.424(1) | 2.373(1) | 2.024(1) | 174.0(2) | 109.22(3) | 98.65(6) | 91.45(6) |
| 4(PF6) | 1.757(7) | 1.186(8) | 2.530(1) | 2.381(2) | 2.183(2) | 174.8(6) | 107.36(7) | 98.9(2) | 91.5(2) |
| Tc-N10 | N10-O10 | Tc-N1/S1 | Tc-P1 | Tc-N10-O10 | N10-Tc-N1/S1 | N10-Tc-P1 | |
|---|---|---|---|---|---|---|---|
| 6(PF6) | 1.762(2) | 1.175(3) | 2.161(2) | 2.3925(7) | 169.3(2) | 98.10(8) | 90.22(6) |
| 7(PF6) | 1.771(2) | 1.174/3) | 2.4047(6) | 2.3805(6) | 172.7(2) | 93.20(7) | 90.03(7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ernst, M.J.; Abdulkader, A.; Hagenbach, A.; Claude, G.; Roca Jungfer, M.; Abram, U. [Tc(NO)(Cp)(PPh3)Cl] and [Tc(NO)(Cp)(PPh3)(NCCH3)](PF6), and Their Reactions with Pyridine and Chalcogen Donors. Molecules 2024, 29, 1114. https://doi.org/10.3390/molecules29051114
Ernst MJ, Abdulkader A, Hagenbach A, Claude G, Roca Jungfer M, Abram U. [Tc(NO)(Cp)(PPh3)Cl] and [Tc(NO)(Cp)(PPh3)(NCCH3)](PF6), and Their Reactions with Pyridine and Chalcogen Donors. Molecules. 2024; 29(5):1114. https://doi.org/10.3390/molecules29051114
Chicago/Turabian StyleErnst, Moritz Johannes, Abdullah Abdulkader, Adelheid Hagenbach, Guilhem Claude, Maximilian Roca Jungfer, and Ulrich Abram. 2024. "[Tc(NO)(Cp)(PPh3)Cl] and [Tc(NO)(Cp)(PPh3)(NCCH3)](PF6), and Their Reactions with Pyridine and Chalcogen Donors" Molecules 29, no. 5: 1114. https://doi.org/10.3390/molecules29051114
APA StyleErnst, M. J., Abdulkader, A., Hagenbach, A., Claude, G., Roca Jungfer, M., & Abram, U. (2024). [Tc(NO)(Cp)(PPh3)Cl] and [Tc(NO)(Cp)(PPh3)(NCCH3)](PF6), and Their Reactions with Pyridine and Chalcogen Donors. Molecules, 29(5), 1114. https://doi.org/10.3390/molecules29051114