
Radiosynthesis and In Vitro Evaluation of [11C]tozadenant as Adenosine A2A Receptor Radioligand

 , , , and
, , , and
Abstract

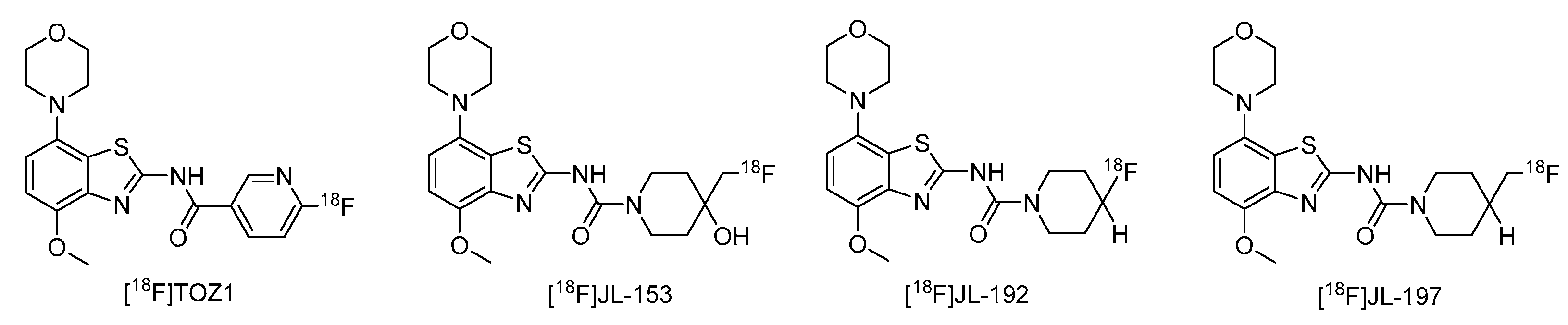
1. Introduction
2. Results and Discussion
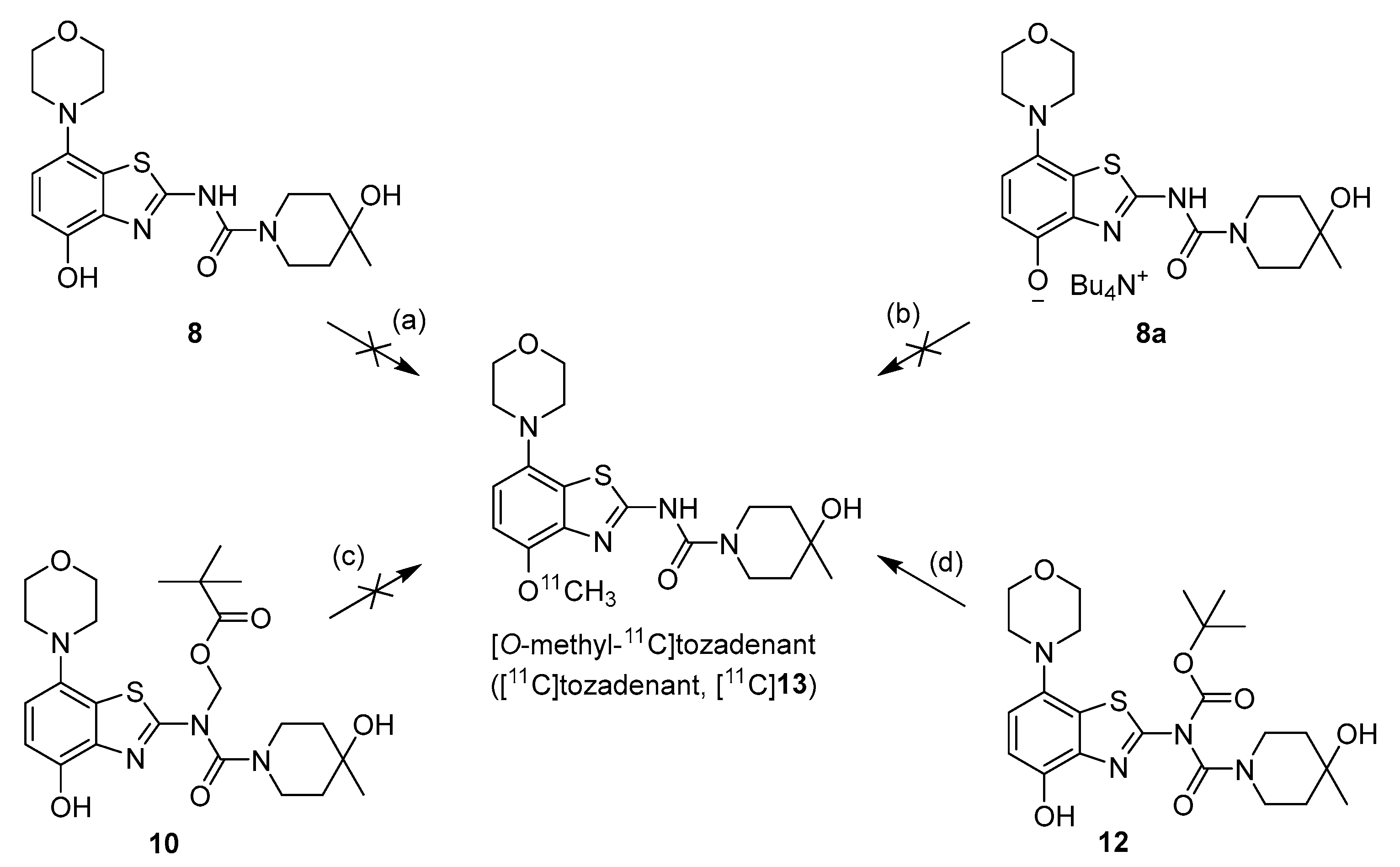
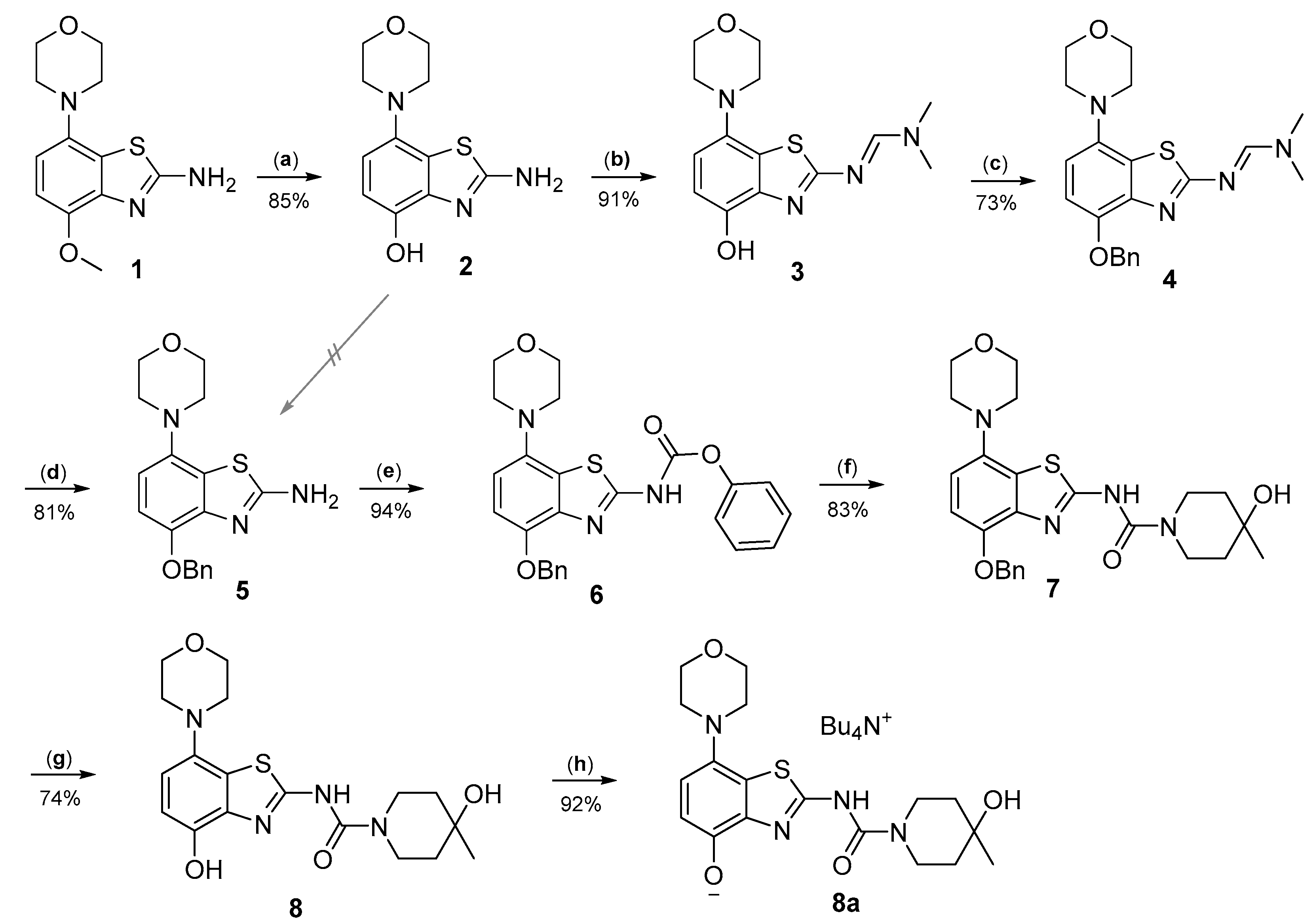
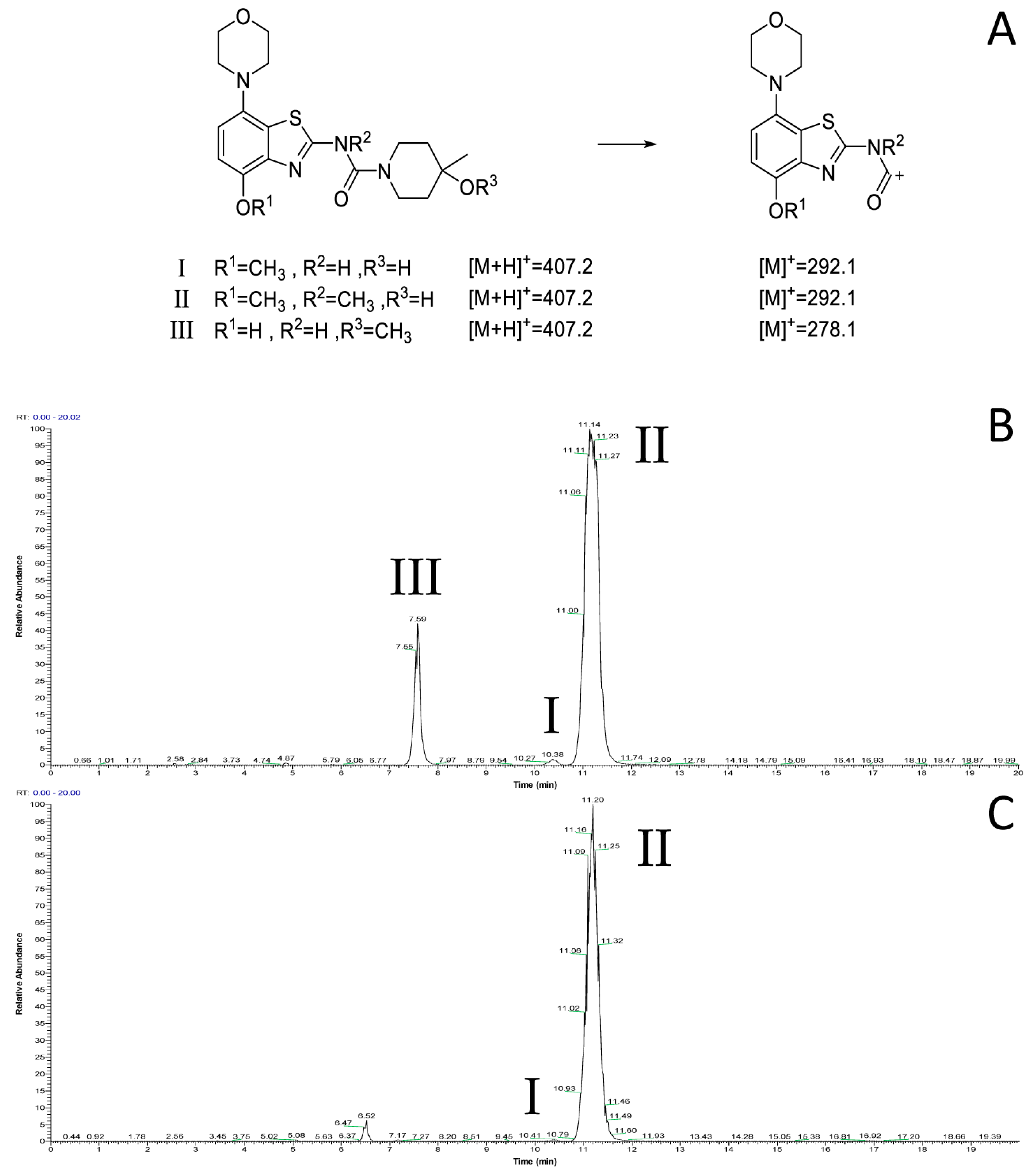
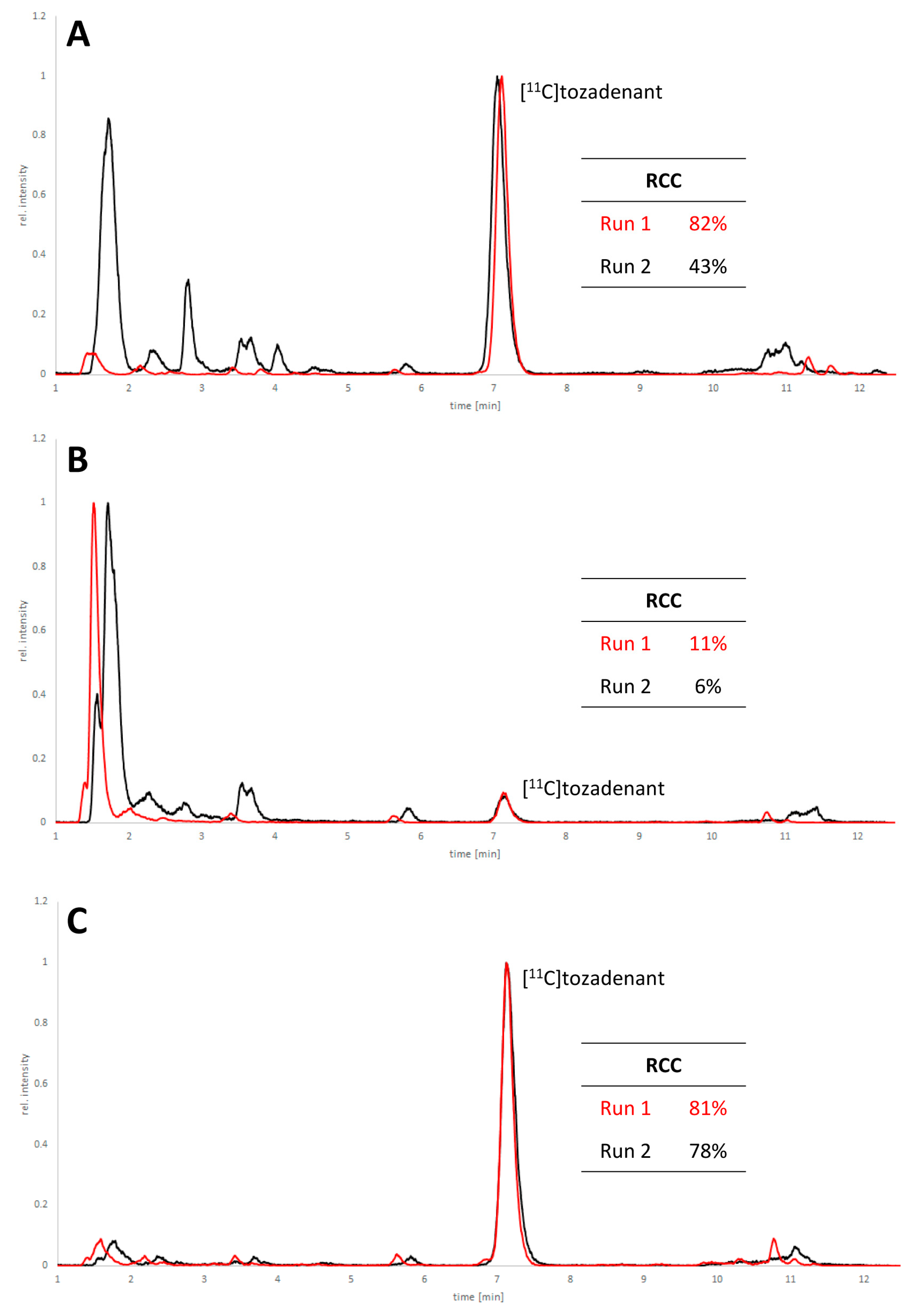
2.1. 11C-Methylation of Unprotected Desmethyl Precursors
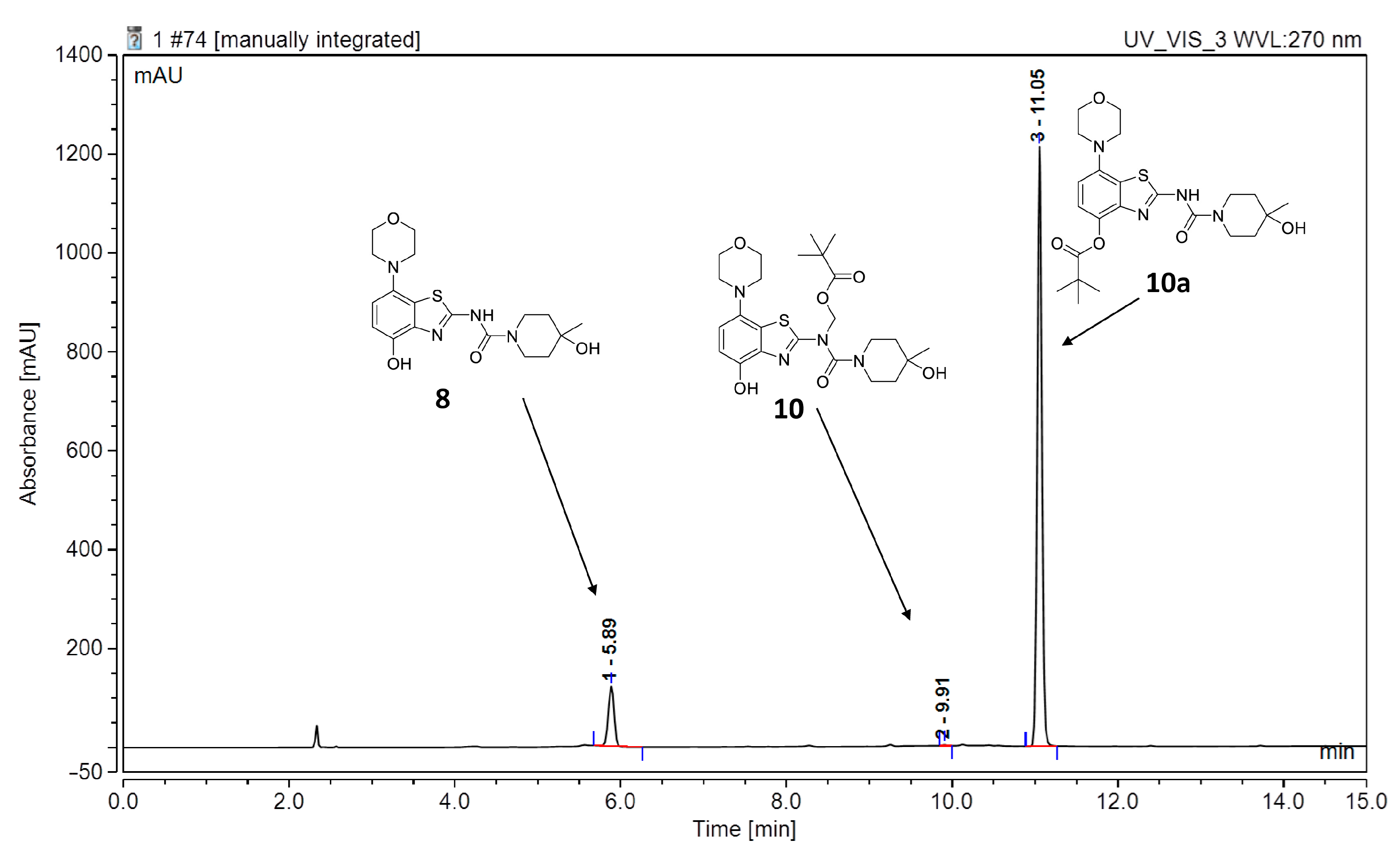
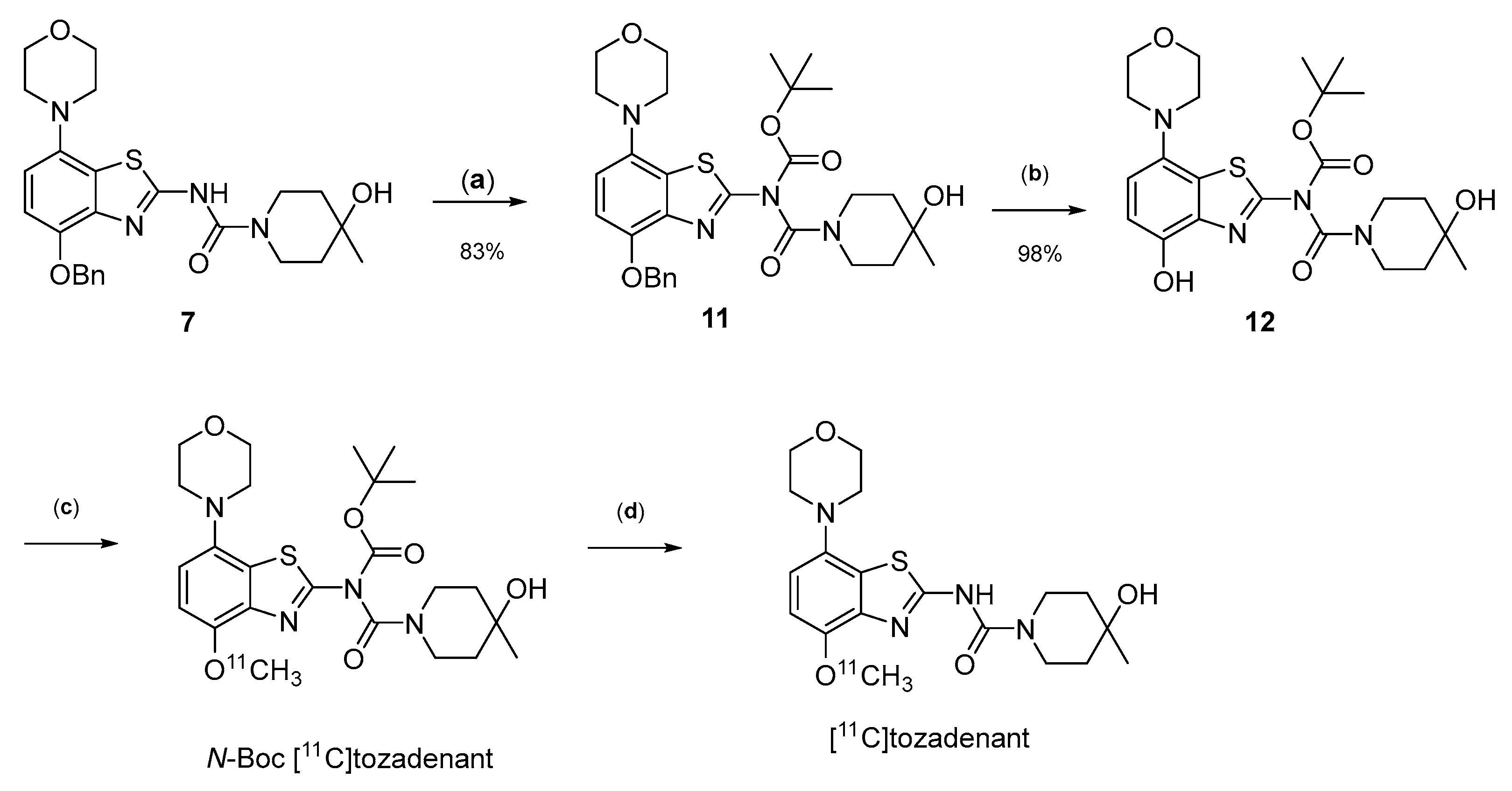
2.2. 11C-Methylation of a Base-Labile Protected Desmethyl Precursor
2.3. 11C-Methylation of an Acid-Labile Protected Desmethyl Precursor
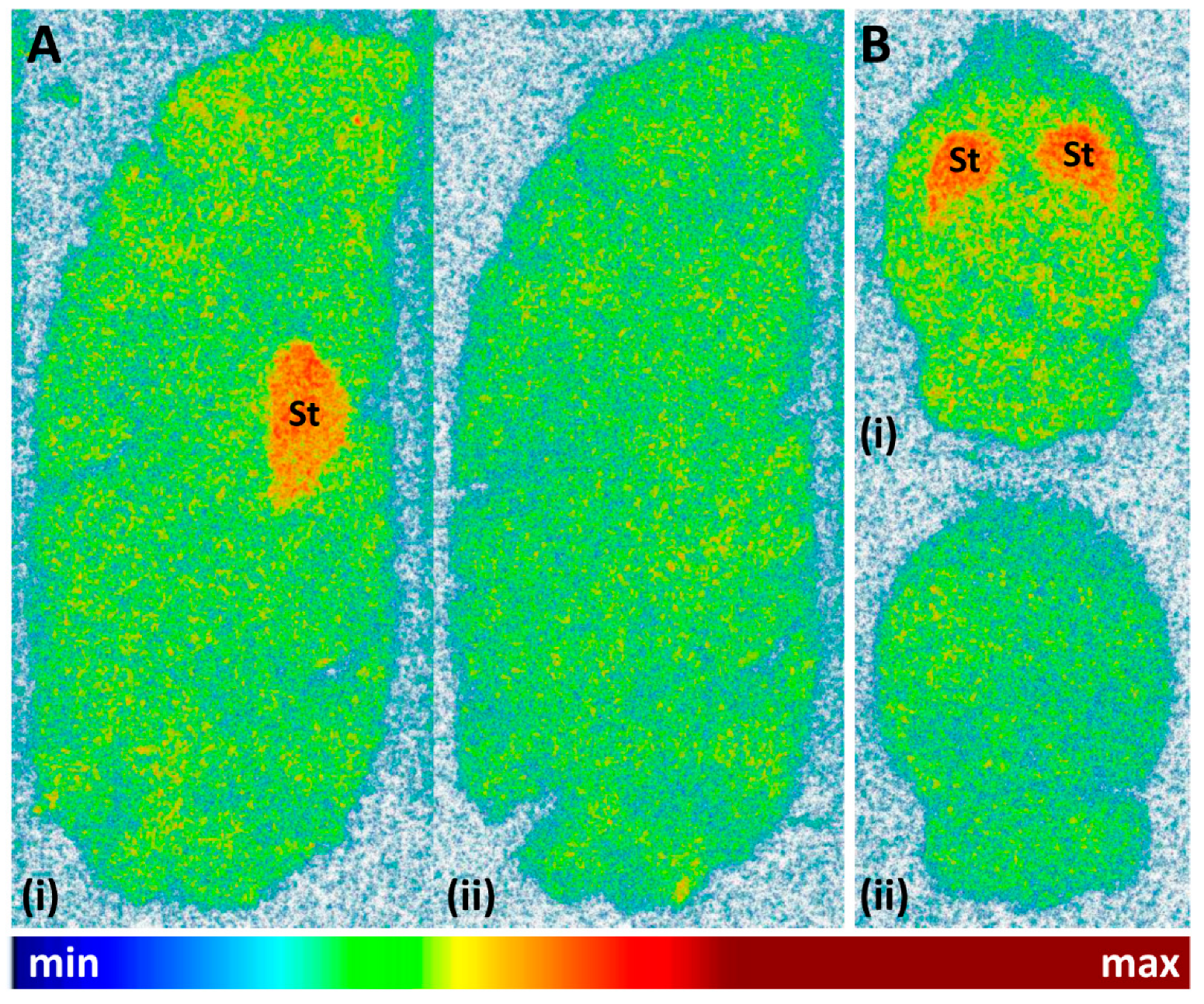
2.4. Preliminary In Vitro Evaluation of [11C]tozadenant
3. Materials and Methods
3.1. General Information
3.1.1. Solvents and Reagents
3.1.2. Spectroscopy
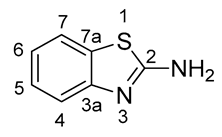
3.1.3. Chromatography
3.2. Chemical Syntheses
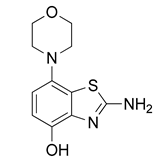
3.2.1. Preparation of 2-Amino-7-morpholinobenzo[d]thiazol-4-ol (2)
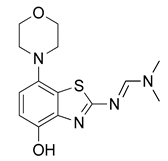
3.2.2. Preparation of (E)-N′-(4-Hydroxy-7-morpholinobenzo[d]thiazol-2-yl)-N,N-dimethylformamidine (3)
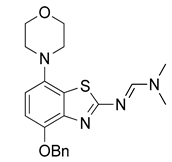
3.2.3. Preparation of (E)-N′-(4-Benzyloxy-7-morpholinobenzo[d]thiazol-2-yl)-N,N-dimethylformamidine (4)
3.2.4. Preparation of 4-Benzyloxy-7-morpholinobenzo[d]thiazol-2-amine (5)
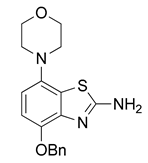
3.2.5. Preparation of Phenyl(4-benzyloxy)-7-morpholinobenzo[d]thiazol-2-yl)carbamate (6)
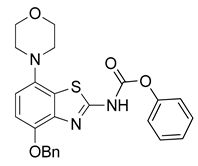
3.2.6. Preparation of N-(4-(Benzyloxy)-7-morpholinobenzo[d]thiazol-2-yl)-4-hydroxy-4-methylpiperidine-1-carboxamide (7) [25]
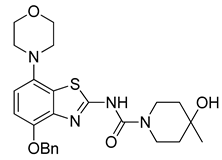
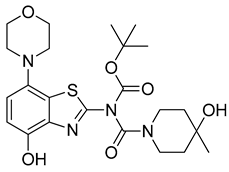
3.2.7. Preparation of 4-Hydroxy-N-(4-hydroxy-7-morpholinobenzo[d]thiazol-2-yl)-4-methylpiperidine-1-carboxamide (8) [34]
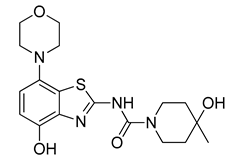
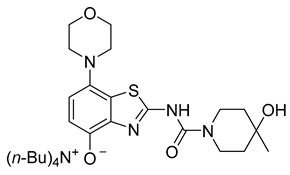
3.2.8. Preparation of Tetrabutylammonium 2-(4-Hydroxy-4-methylpiperidine-1-carboxamido)-7-morpholinobenzo[d]thiazol-4-olate (8a) (According to a Modification of the Procedure in [27])
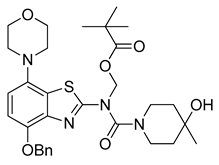
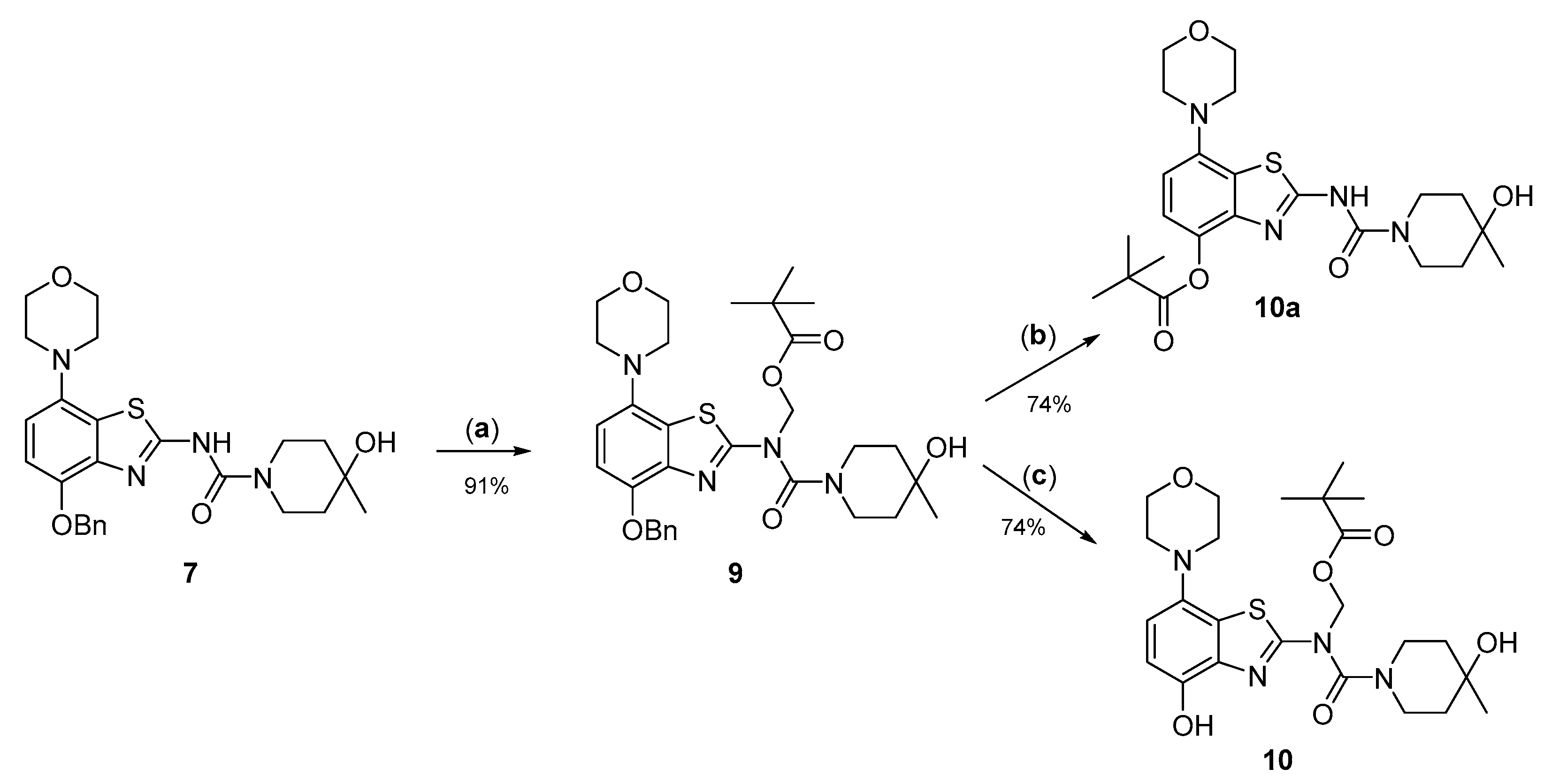
3.2.9. Preparation of (N-(4-(Benzyloxy)-7-morpholinobenzo[d]thiazol-2-yl)-4-hydroxy-4-methylpiperidine-1-carboxamido)methyl Pivalate (9)
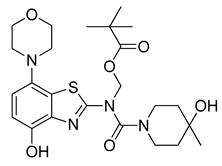
3.2.10. Preparation of (N-(4-(Hydroxy)-7-morpholinobenzo[d]thiazol-2-yl)-4-hydroxy-4-methylpiperidine-1-carboxamido)methyl Pivalate (10)
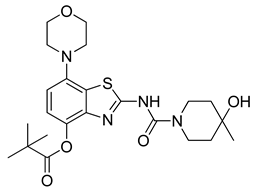
3.2.11. Preparation of 2-(4-Hydroxy-4-methylpiperidine-1-carboxamido)-7-morpholinobenzo[d]thiazol-4-yl Pivalate (10a)
3.2.12. Preparation of tert-Butyl (4-(benzyloxy)-7-morpholinobenzo[d]thiazol-2-yl)(4-hydroxy-4-methylpiperidine-1-carbonyl)carbamate (11)
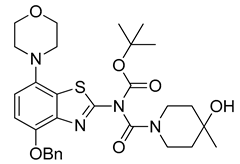
3.2.13. Preparation of tert-Butyl (4-hydroxy-4-methylpiperidine-1-carbonyl)(4-hydroxy-7-morpholinobenzo[d]thiazol-2-yl)carbamate (12)
3.3. Radiochemistry
3.3.1. General Radiochemistry
3.3.2. Manual Screening Reactions for the Preparation of [11C]tozadenant
3.3.3. Analytical HPLC
3.3.4. Automated Production of [11C]tozadenant ([11C]13)
3.3.5. Quality Control
3.4. In Vitro Autoradiography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Porkka-Heiskanen, T. Adenosine in Sleep and Wakefulness. Ann. Med. 1999, 31, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F. Adenosine Receptor Control of Cognition in Normal and Disease. Int. Rev. Neurobiol. 2014, 119, 257–307. [Google Scholar] [PubMed]
- Mori, A. How Do Adenosine A2A Receptors Regulate Motor Function? Park. Relat. Disord. 2020, 80, S13–S20. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R.; Stehle, J.H.; Rivkees, S.A. Molecular Cloning and Characterization of a Rat A1-Adenosine Receptor That Is Widely Expressed in Brain and Spinal Cord. Mol. Endocrinol. 1991, 5, 1037–1048. [Google Scholar] [CrossRef]
- Dixon, A.K.; Gubitz, A.K.; Sirinathsinghji, D.J.S.; Richardson, P.J.; Freeman, T.C. Tissue Distribution of Adenosine Receptor MRNAs in the Rat. Br. J. Pharmacol. 1996, 118, 1461–1468. [Google Scholar] [CrossRef]
- Martinez-Mir, M.I.; Probst, A.; Palacios, J.M. Adenosine A2 Receptors: Selective Localization in the Human Basal Ganglia and Alterations with Disease. Neuroscience 1991, 42, 697–706. [Google Scholar] [CrossRef]
- van Waarde, A.; Dierckx, R.A.J.O.; Zhou, X.; Khanapur, S.; Tsukada, H.; Ishiwata, K.; Luurtsema, G.; de Vries, E.F.J.; Elsinga, P.H. Potential Therapeutic Applications of Adenosine A2A Receptor Ligands and Opportunities for A2A Receptor Imaging. Med. Res. Rev. 2018, 38, 5–56. [Google Scholar] [CrossRef]
- Vuorimaa, A.; Rissanen, E.; Airas, L. In Vivo PET Imaging of Adenosine 2A Receptors in Neuroinflammatory and Neurodegenerative Disease. Contrast Media Mol. Imaging 2017, 2017, 6975841. [Google Scholar] [CrossRef]
- Domenici, M.R.; Ferrante, A.; Martire, A.; Chiodi, V.; Pepponi, R.; Tebano, M.T.; Popoli, P. Adenosine A2A Receptor as Potential Therapeutic Target in Neuropsychiatric Disorders. Pharmacol. Res. 2019, 147, 104338. [Google Scholar] [CrossRef]
- Cieślak, M.; Komoszyński, M.; Wojtczak, A. Adenosine A2A Receptors in Parkinson’s Disease Treatment. Purinergic Signal. 2008, 4, 305–312. [Google Scholar] [CrossRef]
- Hauser, R.A.; Olanow, C.W.; Kieburtz, K.D.; Pourcher, E.; Docu-Axelerad, A.; Lew, M.; Kozyolkin, O.; Neale, A.; Resburg, C.; Meya, U.; et al. Tozadenant (SYN115) in Patients with Parkinson’s Disease Who Have Motor Fluctuations on Levodopa: A Phase 2b, Double-Blind, Randomised Trial. Lancet Neurol. 2014, 13, 767–776. [Google Scholar] [CrossRef]
- LeWitt, P.A.; Aradi, S.D.; Hauser, R.A.; Rascol, O. The Challenge of Developing Adenosine A2A Antagonists for Parkinson Disease: Istradefylline, Preladenant, and Tozadenant. Park. Relat. Disord. 2020, 80, S54–S63. [Google Scholar] [CrossRef]
- Renk, D.R.; Skraban, M.; Bier, D.; Schulze, A.; Wabbals, E.; Wedekind, F.; Neumaier, F.; Neumaier, B.; Holschbach, M. Design, Synthesis and Biological Evaluation of Tozadenant Analogues as Adenosine A2A Receptor Ligands. Eur. J. Med. Chem. 2021, 214, 113214. [Google Scholar] [CrossRef]
- Michel, A.; Downey, P.; Nicolas, J.-M.; Scheller, D. Unprecedented Therapeutic Potential with a Combination of A2A/NR2B Receptor Antagonists as Observed in the 6-OHDA Lesioned Rat Model of Parkinson’s Disease. PLoS ONE 2014, 9, e114086. [Google Scholar] [CrossRef] [PubMed]
- Mancel, V.; Mathy, F.-X.; Boulanger, P.; English, S.; Croft, M.; Kenney, C.; Knott, T.; Stockis, A.; Bani, M. Pharmacokinetics and Metabolism of [14C]-Tozadenant (SYN-115), a Novel A2a Receptor Antagonist Ligand, in Healthy Volunteers. Xenobiotica 2017, 47, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.I.; Park, M.-H.; Shin, S.-H.; Byeon, J.-J.; Park, Y.; Kim, N.; Choi, J.; Shin, Y.G. Quantitative Analysis of Tozadenant Using Liquid Chromatography-Mass Spectrometric Method in Rat Plasma and Its Human Pharmacokinetics Prediction Using Physiologically Based Pharmacokinetic Modeling. Molecules 2019, 24, 1295. [Google Scholar] [CrossRef] [PubMed]
- Zarrinmayeh, H.; Territo, P.R. Purinergic Receptors of the Central Nervous System: Biology, PET Ligands, and Their Applications. Mol. Imaging 2020, 19, 153601212092760. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.-D.; Stiles, G.L.; van Galen, P.J.M.; Jacobson, K.A. Characterization of Human Striatal A2-Adenosine Receptors Using Radioligand Binding and Photoaffinity Labeling. J. Recept. Res. 1992, 12, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Albasanz, J.L.; Rodríguez, A.; Ferrer, I.; Martín, M. Adenosine A2A Receptors Are Up-regulated in Pick’s Disease Frontal Cortex. Brain Pathol. 2006, 16, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Van de Bittner, G.C.; Ricq, E.L.; Hooker, J.M. A Philosophy for CNS Radiotracer Design. Acc. Chem. Res. 2014, 47, 3127–3134. [Google Scholar] [CrossRef]
- Lai, T.H.; Toussaint, M.; Teodoro, R.; Dukić-Stefanović, S.; Kranz, M.; Deuther-Conrad, W.; Moldovan, R.-P.; Brust, P. Synthesis and Biological Evaluation of a Novel 18F-Labeled Radiotracer for PET Imaging of the Adenosine A2A Receptor. Int. J. Mol. Sci. 2021, 22, 1182. [Google Scholar] [CrossRef]
- Wedekind, F.; Lang, M.; Humpert, S.; Schneider, D.; Drechsel, A.; Klein, S.; Mottaghy, F.; Tolba, R.; Neumaier, B.; Drzezga, A.; et al. Preclinical Evaluation of 18F-Labeled Tozadenant Analogs as Potential PET Tracers for Cerebral A2A Adenosine Receptors. In Proceedings of the World Molecular Imaging Congress, Prague, Czech Republic, 5–9 September 2023. [Google Scholar]
- Merighi, S.; Borea, P.A.; Varani, K.; Vincenzi, F.; Travagli, A.; Nigro, M.; Pasquini, S.; Suresh, R.R.; Kim, S.W.; Volkow, N.D.; et al. Pathophysiological Role and Medicinal Chemistry of A2A Adenosine Receptor Antagonists in Alzheimer’s Disease. Molecules 2022, 27, 2680. [Google Scholar] [CrossRef] [PubMed]
- Thavonekham, B. A Practical Synthesis of Ureas from Phenyl Carbamates. Synthesis 1997, 1997, 1189–1194. [Google Scholar] [CrossRef]
- Anwer, M.K.; Spatola, A.F. An Advantageous Method for the Rapid Removal of Hydrogenolysable Protecting Groups under Ambient Conditions; Synthesis of Leucine-Enkephalin. Synthesis 1980, 1980, 929–932. [Google Scholar] [CrossRef]
- Mayer, R.J.; Breugst, M.; Hampel, N.; Ofial, A.R.; Mayr, H. Ambident Reactivity of Phenolate Anions Revisited: A Quantitative Approach to Phenolate Reactivities. J. Org. Chem. 2019, 84, 8837–8858. [Google Scholar] [CrossRef]
- Holschbach, M.; Schüller, M. An On-Line Method for the Preparation of n.c.a. [11CH3]Trifluoromethanesulfonic Acid Methyl Ester. Appl. Radiat. Isot. 1993, 44, 897–898. [Google Scholar] [CrossRef]
- Rasmussen, M.; Leonard, N.J. Synthesis of 3-(2′-Deoxy-D-Erythro-Pentofuranosyl)Adenine. Application of a New Protecting Group, Pivaloyloxymethyl(Pom). J. Am. Chem. Soc. 1967, 89, 5439–5445. [Google Scholar] [CrossRef]
- Dobrovolná, Z.; Červený, L. Ammonium Formate Decomposition Using Palladium Catalyst. Res. Chem. Intermed. 2000, 26, 489–497. [Google Scholar] [CrossRef]
- Garcia, J.; Sorrentino, J.; Diller, E.J.; Chapman, D.; Woydziak, Z.R. General Method for Nucleophilic Aromatic Substitution of Aryl Fluorides and Chlorides with Dimethylamine Using Hydroxide-Assisted Decomposition of N,N-Dimethylforamide. Synth. Commun. 2016, 46, 475–481. [Google Scholar] [CrossRef]
- Gündel, D.; Toussaint, M.; Lai, T.H.; Deuther-Conrad, W.; Cumming, P.; Schröder, S.; Teodoro, R.; Moldovan, R.-P.; Pan-Montojo, F.; Sattler, B.; et al. Quantitation of the A2A Adenosine Receptor Density in the Striatum of Mice and Pigs with [18F]FLUDA by Positron Emission Tomography. Pharmaceuticals 2022, 15, 516. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, MA, USA; Elsevier Inc.: Burlington, MA, USA, 2009; ISBN 9780080878249. [Google Scholar]
- Anwer, M.K.; Spatola, A.F. A Rapid Non-Acidolytic Method for Deprotection and Removal of Peptides from Solid Phase Resins—Applications of Ammonium Formate Catalytic Transfer Hydrogenation. Tetrahedron Lett. 1981, 22, 4369–4372. [Google Scholar] [CrossRef]
- Holschbach, M.; Schüller, M. A New and Simple On-Line Method for the Preparation of n.c.a. [11C]Methyl Iodide. Appl. Radiat. Isot. 1993, 44, 779–780. [Google Scholar] [CrossRef]
- Humpert, S.; Hoffmann, C.; Neumaier, F.; Zlatopolskiy, B.D.; Neumaier, B. Validation of Analytical HPLC with Post-Column Injection as a Method for Rapid and Precise Quantification of Radiochemical Yields. J. Chromatogr. B 2023, 1228, 123847. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Method | Unit | Specification | Result |
|---|---|---|---|---|
| Appearance | visually | - | clear, no particles | conform |
| Activity concentration at EOS | calculated | MBq/mL | ≥30 | 172 ± 95 |
| Radiochemical identity | HPLC | % | ±5 | 0 ± 2 |
| Radiochemical purity | HPLC | % | ≥90 | 95.0 ± 2.8 |
| Carrier identity | HPLC | % | ±5 | 0 ± 2 |
| Carrier concentration | HPLC | µg/mL | ≤5.0 | 1.12 ± 0.48 |
| Total concentration of unknown impurities a | HPLC | µg/mL | ≤5.0 | 0.16 ± 0.16 |
| Methanol concentration | GC | mg/mL | ≤1.0 | 0.06 ± 0.03 |
| Ethanol concentration | GC | %vol | ≤10 | 9.10 ± 0.49 |
| Acetone concentration | GC | mg/mL | ≤1.0 | n.d. |
| Acetonitrile concentration | GC | mg/mL | ≤0.41 | n.d. |
| DMSO concentration | GC | mg/mL | ≤1.0 | n.d. |
| DMA concentration | GC | mg/mL | ≤1.0 | n.d. |
| pH | indicator | - | 4.5–8.5 | 5.3 ± 0.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humpert, S.; Schneider, D.; Lang, M.; Schulze, A.; Neumaier, F.; Holschbach, M.; Bier, D.; Neumaier, B. Radiosynthesis and In Vitro Evaluation of [11C]tozadenant as Adenosine A2A Receptor Radioligand. Molecules 2024, 29, 1089. https://doi.org/10.3390/molecules29051089
Humpert S, Schneider D, Lang M, Schulze A, Neumaier F, Holschbach M, Bier D, Neumaier B. Radiosynthesis and In Vitro Evaluation of [11C]tozadenant as Adenosine A2A Receptor Radioligand. Molecules. 2024; 29(5):1089. https://doi.org/10.3390/molecules29051089
Chicago/Turabian StyleHumpert, Swen, Daniela Schneider, Markus Lang, Annette Schulze, Felix Neumaier, Marcus Holschbach, Dirk Bier, and Bernd Neumaier. 2024. "Radiosynthesis and In Vitro Evaluation of [11C]tozadenant as Adenosine A2A Receptor Radioligand" Molecules 29, no. 5: 1089. https://doi.org/10.3390/molecules29051089
APA StyleHumpert, S., Schneider, D., Lang, M., Schulze, A., Neumaier, F., Holschbach, M., Bier, D., & Neumaier, B. (2024). Radiosynthesis and In Vitro Evaluation of [11C]tozadenant as Adenosine A2A Receptor Radioligand. Molecules, 29(5), 1089. https://doi.org/10.3390/molecules29051089