
Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges
Abstract
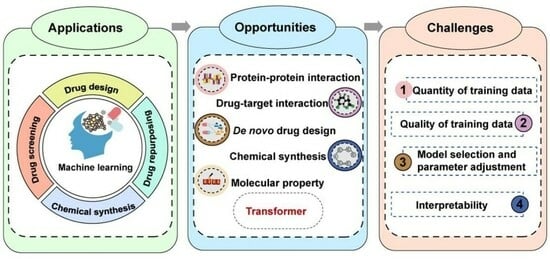
1. Introduction
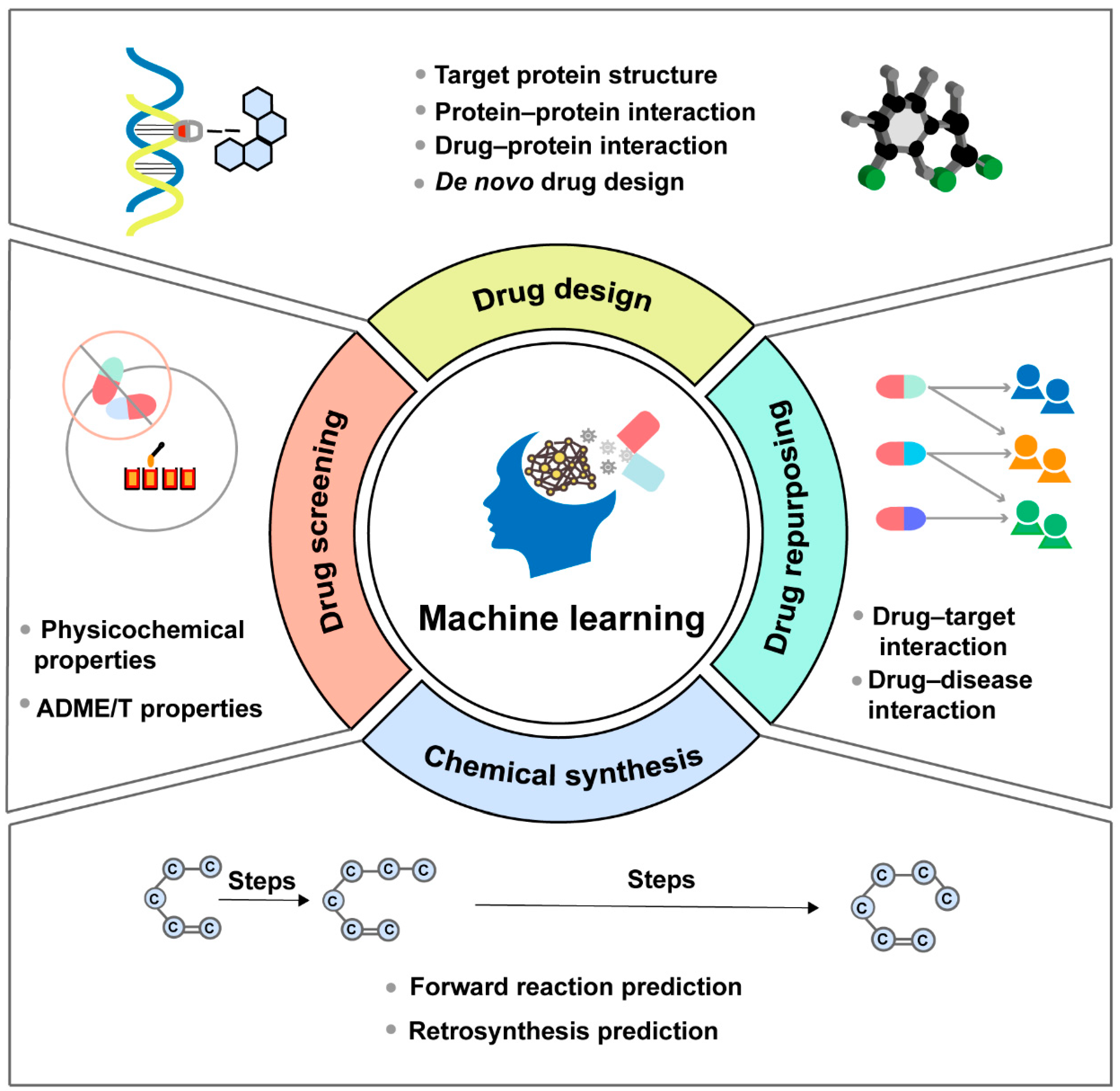
2. Applications of ML in Drug Discovery
2.1. Application of ML in Drug Design
2.1.1. Prediction of the Target Protein Structure
2.1.2. Prediction of PPIs
2.1.3. Prediction of DTIs
2.1.4. De Novo Drug Design
2.2. Application of ML in Drug Screening
2.2.1. Prediction of the Physicochemical Properties
2.2.2. Prediction of the ADME/T Properties
2.3. Application of ML in Drug Repurposing
2.4. Application of ML in Chemical Synthesis
2.4.1. Retrosynthesis Prediction
2.4.2. Forward Reaction Prediction
3. Opportunities for Transformer-Based ML Models in Empowering Drug Discovery
3.1. Opportunity 1: Transformer Models Empowering PPIs Identification
3.2. Opportunity 2: Transformer Models Empowering DTIs’ Identification
3.3. Opportunity 3: Transformer Models Empowering De Novo Drug Design
3.4. Opportunity 4: Transformer Models Empowering Molecular Property Prediction
3.5. Opportunity 5: Transformer Models Empowering Chemical Synthesis
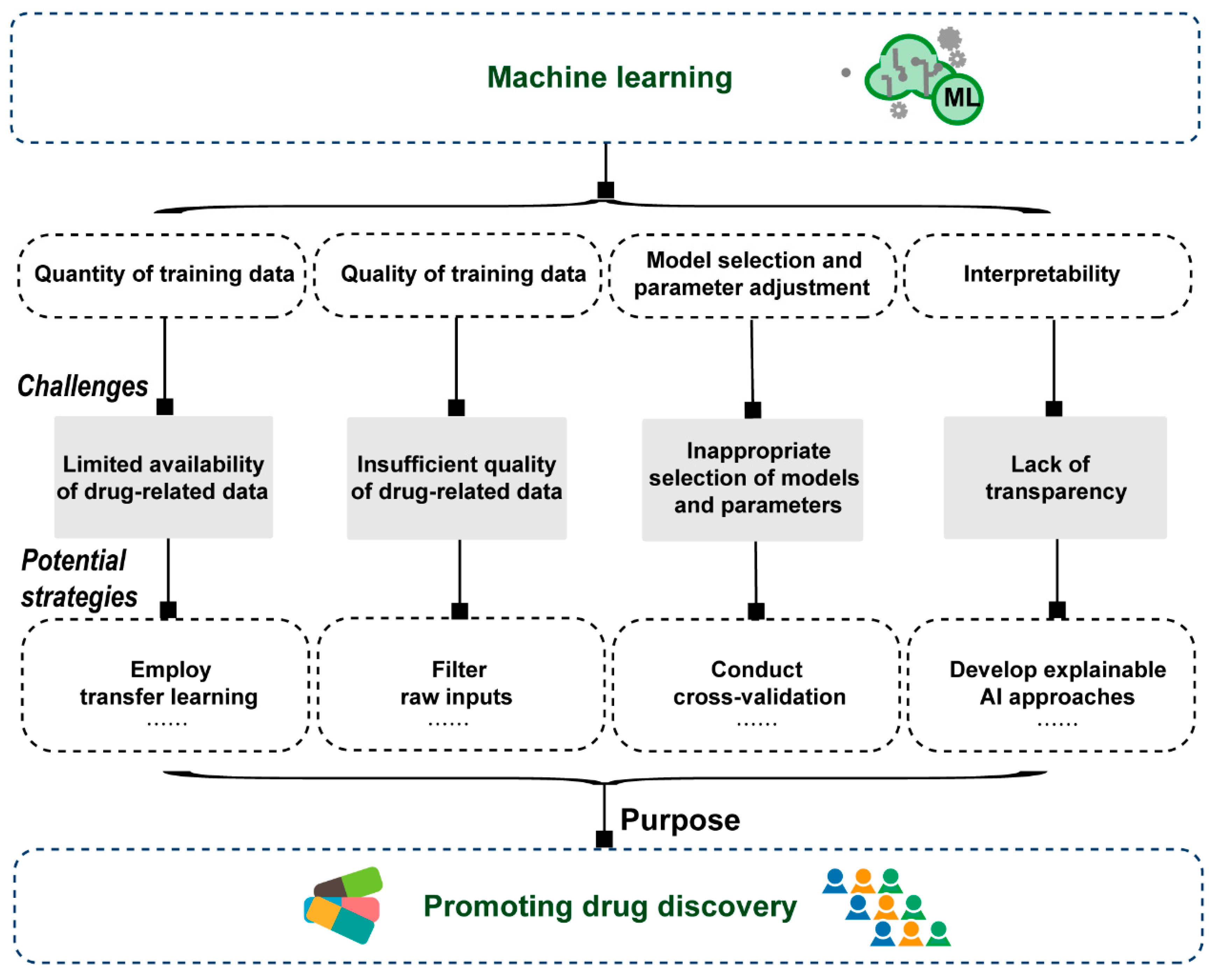
4. Challenges of ML-Based Models in Drug Discovery
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- DiMasi, J.A.; Grabowski, H.G.; Hansen, R.W. Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 2016, 47, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Yang, Z.; Ojima, I.; Samaras, D.; Wang, F. Artificial intelligence in drug discovery: Applications and techniques. Brief. Bioinform 2022, 23, bbab430. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Pichika, M.R. Artificial intelligence in drug development: Present status and future prospects. Drug Discov. Today 2019, 24, 773–780. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, R.; Li, Y.; Li, M. DeepDTAF: A deep learning method to predict protein-ligand binding affinity. Brief. Bioinform. 2021, 22, bbab072. [Google Scholar] [CrossRef]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, R.; Liu, Y.; Chen, S.; Liu, Q.; Zeng, W. Applications of transformer-based language models in bioinformatics: A survey. Bioinform. Adv. 2023, 3, vbad001. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 1–11. [Google Scholar]
- Kalakoti, Y.; Yadav, S.; Sundar, D. TransDTI: Transformer-Based Language Models for Estimating DTIs and Building a Drug Recommendation Workflow. ACS Omega 2022, 7, 2706–2717. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, S.; Hou, J.; Si, D.; Zhu, J.; Cao, R. ComplexQA: A deep graph learning approach for protein complex structure assessment. Brief. Bioinform. 2023, 24, bbad287. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A universal deep-learning model of protein sequence and function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Northey, T.C.; Barešić, A.; Martin, A.C.R. IntPred: A structure-based predictor of protein-protein interaction sites. Bioinformatics 2018, 34, 223–229. [Google Scholar] [CrossRef]
- Maheshwari, S.; Brylinski, M. Template-based identification of protein-protein interfaces using eFindSitePPI. Methods 2016, 93, 64–71. [Google Scholar] [CrossRef]
- Li, Y.; Golding, G.B.; Ilie, L. DELPHI: Accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 2021, 37, 896–904. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Yu, B.; Salhi, A.; Chen, R.; Wang, L.; Liu, Z. Prediction of protein-protein interaction sites through eXtreme gradient boosting with kernel principal component analysis. Comput. Biol. Med. 2021, 134, 104516. [Google Scholar] [CrossRef]
- Kang, Y.; Xu, Y.; Wang, X.; Pu, B.; Yang, X.; Rao, Y.; Chen, J. HN-PPISP: A hybrid network based on MLP-Mixer for protein-protein interaction site prediction. Brief. Bioinform. 2023, 24, bbac480. [Google Scholar] [CrossRef]
- Song, B.; Luo, X.; Luo, X.; Liu, Y.; Niu, Z.; Zeng, X. Learning spatial structures of proteins improves protein-protein interaction prediction. Brief. Bioinform. 2022, 23, bbab558. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, M.; Magner, A.; Saldinger, J.; Turali-Emre, E.S.; Elvati, P.; Kozarekar, S.; VanEpps, J.S.; Kotov, N.A.; Violi, A.; Hero, A.O. Struct2Graph: A graph attention network for structure based predictions of protein-protein interactions. BMC Bioinform. 2022, 23, 370. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Du, X.; Diao, Y.; Zhu, H. An integration of deep learning with feature embedding for protein-protein interaction prediction. PeerJ 2019, 7, e7126. [Google Scholar] [CrossRef]
- Huang, Y.; Wuchty, S.; Zhou, Y.; Zhang, Z. SGPPI: Structure-aware prediction of protein-protein interactions in rigorous conditions with graph convolutional network. Brief. Bioinform. 2023, 24, bbad020. [Google Scholar] [CrossRef]
- Du, X.; Sun, S.; Hu, C.; Yao, Y.; Yan, Y.; Zhang, Y. DeepPPI: Boosting Prediction of Protein-Protein Interactions with Deep Neural Networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, B.; Zhang, J.; Wang, Z.; Li, J. DL-PPI: A method on prediction of sequenced protein-protein interaction based on deep learning. BMC Bioinform. 2023, 24, 473. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.; Zhu, X.; Chen, X.; Lu, F.; Zhang, X. DeepSG2PPI: A Protein-Protein Interaction Prediction Method Based on Deep Learning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 2907–2919. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Mitra, P. MaTPIP: A deep-learning architecture with eXplainable AI for sequence-driven, feature mixed protein-protein interaction prediction. Comput. Methods Programs Biomed. 2024, 244, 107955. [Google Scholar] [CrossRef]
- Soleymani, F.; Paquet, E.; Viktor, H.L.; Michalowski, W.; Spinello, D. ProtInteract: A deep learning framework for predicting protein-protein interactions. Comput. Struct. Biotechnol. J. 2023, 21, 1324–1348. [Google Scholar] [CrossRef]
- Cui, Y.; Dong, Q.; Hong, D.; Wang, X. Predicting protein-ligand binding residues with deep convolutional neural networks. BMC Bioinform. 2019, 20, 93. [Google Scholar] [CrossRef]
- Mylonas, S.K.; Axenopoulos, A.; Daras, P. DeepSurf: A surface-based deep learning approach for the prediction of ligand binding sites on proteins. Bioinformatics 2021, 37, 1681–1690. [Google Scholar] [CrossRef]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019, 47, W345–w349. [Google Scholar] [CrossRef]
- Kandel, J.; Tayara, H.; Chong, K.T. PUResNet: Prediction of protein-ligand binding sites using deep residual neural network. J. Cheminform. 2021, 13, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jiang, Y.; Yang, Y. AGAT-PPIS: A novel protein-protein interaction site predictor based on augmented graph attention network with initial residual and identity mapping. Brief. Bioinform. 2023, 24, bbad122. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug-target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 24. [Google Scholar] [CrossRef]
- Ahmed, A.; Mam, B.; Sowdhamini, R. DEELIG: A Deep Learning Approach to Predict Protein-Ligand Binding Affinity. Bioinform. Biol. Insights 2021, 15, 11779322211030364. [Google Scholar] [CrossRef]
- Karlov, D.S.; Sosnin, S.; Fedorov, M.V.; Popov, P. graphDelta: MPNN Scoring Function for the Affinity Prediction of Protein-Ligand Complexes. ACS Omega 2020, 5, 5150–5159. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Liyaqat, T.; Ahmad, T.; Saxena, C. TeM-DTBA: Time-efficient drug target binding affinity prediction using multiple modalities with Lasso feature selection. J. Comput. Aided Mol. Des. 2023, 37, 573–584. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Y.; Zhang, Y.; Li, K.; Lin, M.; Pan, F.; Wu, W.; Zhang, J. A reinforcement learning approach for protein-ligand binding pose prediction. BMC Bioinform. 2022, 23, 368. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Cang, Z.; Wu, K.; Wang, M.; Cao, Y.; Wei, G.W. Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges. J. Comput. Aided Mol. Des. 2019, 33, 71–82. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, Y.; Chen, Q. AMMVF-DTI: A Novel Model Predicting Drug-Target Interactions Based on Attention Mechanism and Multi-View Fusion. Int. J. Mol. Sci. 2023, 24, 14142. [Google Scholar] [CrossRef]
- Popova, M.; Isayev, O.; Tropsha, A. Deep reinforcement learning for de novo drug design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Liu, Z. Multi-objective de novo drug design with conditional graph generative model. J. Cheminform. 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Born, J.; Manica, M.; Oskooei, A.; Cadow, J.; Markert, G.; Rodríguez Martínez, M. PaccMann(RL): De novo generation of hit-like anticancer molecules from transcriptomic data via reinforcement learning. iScience 2021, 24, 102269. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef]
- Coley, C.W.; Rogers, L.; Green, W.H.; Jensen, K.F. SCScore: Synthetic Complexity Learned from a Reaction Corpus. J. Chem. Inf. Model. 2018, 58, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Schoenmaker, L.; Béquignon, O.J.M.; Jespers, W.; van Westen, G.J.P. UnCorrupt SMILES: A novel approach to de novo design. J. Cheminform. 2023, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, C.; Han, P.; Li, X.; Chen, W.; Rodríguez Patón, A.; Wang, S.; Zheng, P. PETrans: De Novo Drug Design with Protein-Specific Encoding Based on Transfer Learning. Int. J. Mol. Sci. 2023, 24, 1146. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, N.R.C.; Pereira, T.O.; Machado, A.C.D.; Oliveira, J.L.; Abbasi, M.; Arrais, J.P. FSM-DDTR: End-to-end feedback strategy for multi-objective De Novo drug design using transformers. Comput. Biol. Med. 2023, 164, 107285. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Ren, Y.; Wang, S.; Han, P.; Wang, L.; Li, X.; Rodriguez-Patón, A. DNMG: Deep molecular generative model by fusion of 3D information for de novo drug design. Methods 2023, 211, 10–22. [Google Scholar] [CrossRef]
- Macedo, B.; Ribeiro Vaz, I.; Taveira Gomes, T. MedGAN: Optimized generative adversarial network with graph convolutional networks for novel molecule design. Sci. Rep. 2024, 14, 1212. [Google Scholar] [CrossRef]
- Panapitiya, G.; Girard, M.; Hollas, A.; Sepulveda, J.; Murugesan, V.; Wang, W.; Saldanha, E. Evaluation of Deep Learning Architectures for Aqueous Solubility Prediction. ACS Omega 2022, 7, 15695–15710. [Google Scholar] [CrossRef]
- Francoeur, P.G.; Koes, D.R. SolTranNet-A Machine Learning Tool for Fast Aqueous Solubility Prediction. J. Chem. Inf. Model. 2021, 61, 2530–2536. [Google Scholar] [CrossRef]
- Zang, Q.; Mansouri, K.; Williams, A.J.; Judson, R.S.; Allen, D.G.; Casey, W.M.; Kleinstreuer, N.C. In Silico Prediction of Physicochemical Properties of Environmental Chemicals Using Molecular Fingerprints and Machine Learning. J. Chem. Inf. Model. 2017, 57, 36–49. [Google Scholar] [CrossRef]
- Tian, H.; Ketkar, R.; Tao, P. ADMETboost: A web server for accurate ADMET prediction. J. Mol. Model. 2022, 28, 408. [Google Scholar] [CrossRef]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN Web Server for ADMET Predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, S.; Li, Z.; Wan, Z.; Lin, J. Interpretable-ADMET: A web service for ADMET prediction and optimization based on deep neural representation. Bioinformatics 2022, 38, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Chen, X.; Zhang, R.; Lei, Z.; Wang, X.; Zhou, F. XGraphBoost: Extracting Graph Neural Network-Based Features for a Better Prediction of Molecular Properties. J. Chem. Inf. Model. 2021, 61, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity prediction using deep learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Lai, L.; Pei, J. Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 2018, 15, 4336–4345. [Google Scholar] [CrossRef]
- Shaker, B.; Yu, M.S.; Song, J.S.; Ahn, S.; Ryu, J.Y.; Oh, K.S.; Na, D. LightBBB: Computational prediction model of blood-brain-barrier penetration based on LightGBM. Bioinformatics 2021, 37, 1135–1139. [Google Scholar] [CrossRef]
- Tang, Q.; Nie, F.; Zhao, Q.; Chen, W. A merged molecular representation deep learning method for blood-brain barrier permeability prediction. Brief. Bioinform. 2022, 23, bbac357. [Google Scholar] [CrossRef]
- Jang, W.D.; Jang, J.; Song, J.S.; Ahn, S.; Oh, K.S. PredPS: Attention-based graph neural network for predicting stability of compounds in human plasma. Comput. Struct. Biotechnol. J. 2023, 21, 3532–3539. [Google Scholar] [CrossRef]
- Khaouane, A.; Khaouane, L.; Ferhat, S.; Hanini, S. Deep Learning for Drug Development: Using CNNs in MIA-QSAR to Predict Plasma Protein Binding of Drugs. AAPS PharmSciTech 2023, 24, 232. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhu, S.; Lu, W.; Liu, Z.; Huang, J.; Zhou, Y.; Fang, J.; Huang, Y.; Guo, H.; Li, L.; et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem. Sci. 2020, 11, 1775–1797. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Hong, L.; Xiao, A.; Jiang, T.; Zeng, J. NeoDTI: Neural integration of neighbor information from a heterogeneous network for discovering new drug-target interactions. Bioinformatics 2019, 35, 104–111. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef]
- Luo, H.; Wang, J.; Li, M.; Luo, J.; Peng, X.; Wu, F.X.; Pan, Y. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics 2016, 32, 2664–2671. [Google Scholar] [CrossRef]
- Doshi, S.; Chepuri, S.P. A computational approach to drug repurposing using graph neural networks. Comput. Biol. Med. 2022, 150, 105992. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Zhu, S.; Liu, X.; Zhou, Y.; Nussinov, R.; Cheng, F. deepDR: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019, 35, 5191–5198. [Google Scholar] [CrossRef]
- Jiang, H.J.; Huang, Y.A.; You, Z.H. Predicting Drug-Disease Associations via Using Gaussian Interaction Profile and Kernel-Based Autoencoder. BioMed Res. Int. 2019, 2019, 2426958. [Google Scholar] [CrossRef]
- Ghorbanali, Z.; Zare-Mirakabad, F.; Salehi, N.; Akbari, M.; Masoudi-Nejad, A. DrugRep-HeSiaGraph: When heterogenous siamese neural network meets knowledge graphs for drug repurposing. BMC Bioinform. 2023, 24, 374. [Google Scholar] [CrossRef] [PubMed]
- Suviriyapaisal, N.; Wichadakul, D. iEdgeDTA: Integrated edge information and 1D graph convolutional neural networks for binding affinity prediction. RSC Adv. 2023, 13, 25218–25228. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Luu Nguyen, Q.; Ho, S.; Sloane, J.; Wender, P.; Pande, V. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Cent. Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef]
- Thakkar, A.; Chadimová, V.; Bjerrum, E.J.; Engkvist, O.; Reymond, J.L. Retrosynthetic accessibility score (RAscore)—Rapid machine learned synthesizability classification from AI driven retrosynthetic planning. Chem. Sci. 2021, 12, 3339–3349. [Google Scholar] [CrossRef]
- Wei, J.N.; Duvenaud, D.; Aspuru-Guzik, A. Neural Networks for the Prediction of Organic Chemistry Reactions. ACS Cent. Sci. 2016, 2, 725–732. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Using Machine Learning To Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar] [CrossRef]
- Marcou, G.; Aires de Sousa, J.; Latino, D.A.; de Luca, A.; Horvath, D.; Rietsch, V.; Varnek, A. Expert system for predicting reaction conditions: The Michael reaction case. J. Chem. Inf. Model. 2015, 55, 239–250. [Google Scholar] [CrossRef]
- You, Z.H.; Li, S.; Gao, X.; Luo, X.; Ji, Z. Large-scale protein-protein interactions detection by integrating big biosensing data with computational model. BioMed Res. Int. 2014, 2014, 598129. [Google Scholar] [CrossRef]
- Chan, H.C.S.; Shan, H.; Dahoun, T.; Vogel, H.; Yuan, S. Advancing Drug Discovery via Artificial Intelligence. Trends Pharmacol. Sci. 2019, 40, 592–604. [Google Scholar] [CrossRef]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. The protein structure prediction problem could be solved using the current PDB library. Proc. Natl. Acad. Sci. USA 2005, 102, 1029–1034. [Google Scholar] [CrossRef]
- Tang, T.; Zhang, X.; Liu, Y.; Peng, H.; Zheng, B.; Yin, Y.; Zeng, X. Machine learning on protein-protein interaction prediction: Models, challenges and trends. Brief. Bioinform. 2023, 24, bbad076. [Google Scholar] [CrossRef]
- Soleymani, F.; Paquet, E.; Viktor, H.; Michalowski, W.; Spinello, D. Protein-protein interaction prediction with deep learning: A comprehensive review. Comput. Struct. Biotechnol. J. 2022, 20, 5316–5341. [Google Scholar] [CrossRef]
- Li, S.; Wu, S.; Wang, L.; Li, F.; Jiang, H.; Bai, F. Recent advances in predicting protein-protein interactions with the aid of artificial intelligence algorithms. Curr. Opin. Struct. Biol. 2022, 73, 102344. [Google Scholar] [CrossRef]
- Tripathi, N.; Goshisht, M.K.; Sahu, S.K.; Arora, C. Applications of artificial intelligence to drug design and discovery in the big data era: A comprehensive review. Mol. Divers. 2021, 25, 1643–1664. [Google Scholar] [CrossRef]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J. Artificial intelligence in the prediction of protein-ligand interactions: Recent advances and future directions. Brief. Bioinform. 2022, 23, bbab476. [Google Scholar] [CrossRef]
- Nicolaou, C.A.; Kannas, C.; Loizidou, E. Multi-objective optimization methods in de novo drug design. Mini Rev. Med. Chem. 2012, 12, 979–987. [Google Scholar] [CrossRef]
- Zhong, F.; Xing, J.; Li, X.; Liu, X.; Fu, Z.; Xiong, Z.; Lu, D.; Wu, X.; Zhao, J.; Tan, X.; et al. Artificial intelligence in drug design. Sci. China. Life Sci. 2018, 61, 1191–1204. [Google Scholar] [CrossRef]
- Hessler, G.; Baringhaus, K.H. Artificial Intelligence in Drug Design. Molecules 2018, 23, 2520. [Google Scholar] [CrossRef]
- Schneider, G.; Funatsu, K.; Okuno, Y.; Winkler, D. De novo Drug Design—Ye olde Scoring Problem Revisited. Mol. Inform. 2017, 36, 1681031. [Google Scholar] [CrossRef]
- Wang, L.; Ding, J.; Pan, L.; Cao, D.; Jiang, H.; Ding, X. Artificial intelligence facilitates drug design in the big data era. Chemom. Intell. Lab. Syst. 2019, 194, 103850. [Google Scholar] [CrossRef]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef]
- Yu, E.; Xu, Y.; Shi, Y.; Yu, Q.; Liu, J.; Xu, L. Discovery of novel natural compound inhibitors targeting estrogen receptor α by an integrated virtual screening strategy. J. Mol. Model. 2019, 25, 278. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, L.; Yang, Z.; Yu-Chian Chen, C. Graph convolutional network approach to investigate potential selective Limk1 inhibitors. J. Mol. Graph. Model. 2021, 107, 107965. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, F.; Tang, J.; Nussinov, R.; Cheng, F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit. Health 2020, 2, e667–e676. [Google Scholar] [CrossRef]
- Pan, X.; Lin, X.; Cao, D.; Zeng, X.; Yu, P.S.; He, L.; Nussinov, R.; Cheng, F. Deep learning for drug repurposing: Methods, databases, and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1597. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, M.; Liu, Y.; Su, Y.; Zeng, X. Deep learning in retrosynthesis planning: Datasets, models and tools. Brief. Bioinform. 2022, 23, bbab391. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.A.; Yang, Q.; Sresht, V.; Bolgar, P.; Hou, X.; Klug-McLeod, J.L.; Butler, C.R. Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space. Chem. Commun. 2019, 55, 12152–12155. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Y. Graph-sequence attention and transformer for predicting drug-target affinity. RSC Adv. 2022, 12, 29525–29534. [Google Scholar] [CrossRef]
- Lee, M. Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review. Molecules 2023, 28, 5169. [Google Scholar] [CrossRef]
- Lin, P.; Yan, Y.; Huang, S.Y. DeepHomo2.0: Improved protein-protein contact prediction of homodimers by transformer-enhanced deep learning. Brief. Bioinform. 2023, 24, bbac499. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Elofsson, A.; Jiang, Y.; Huang, W.; Yu, M.; Li, Z. AFTGAN: Prediction of multi-type PPI based on attention free transformer and graph attention network. Bioinformatics 2023, 39, btad052. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Z.; Che, C.; Jin, B. DeepMGT-DTI: Transformer network incorporating multilayer graph information for Drug-Target interaction prediction. Comput. Biol. Med. 2022, 142, 105214. [Google Scholar] [CrossRef]
- Qian, H.; Lin, C.; Zhao, D.; Tu, S.; Xu, L. AlphaDrug: Protein target specific de novo molecular generation. PNAS Nexus 2022, 1, pgac227. [Google Scholar] [CrossRef]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training. 2018. Available online: https://www.mikecaptain.com/resources/pdf/GPT-1.pdf (accessed on 14 January 2024).
- Wang, Y.; Zhao, H.; Sciabola, S.; Wang, W. cMolGPT: A Conditional Generative Pre-Trained Transformer for Target-Specific De Novo Molecular Generation. Molecules 2023, 28, 4430. [Google Scholar] [CrossRef]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Liu, Z.; Roberts, R.A.; Lal-Nag, M.; Chen, X.; Huang, R.; Tong, W. AI-based language models powering drug discovery and development. Drug Discov. Today 2021, 26, 2593–2607. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jiang, D.; Wang, J.; Zhang, X.; Du, H.; Pan, L.; Hsieh, C.Y.; Cao, D.; Hou, T. Knowledge-based BERT: A method to extract molecular features like computational chemists. Brief. Bioinform. 2022, 23, bbac131. [Google Scholar] [CrossRef]
- Wang, S.; Guo, Y.; Wang, Y.; Sun, H.; Huang, J. Smiles-bert: Large scale unsupervised pre-training for molecular property prediction. In Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Niagara Falls, NY, USA, 7–10 September 2019; pp. 429–436. [Google Scholar]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef]
- Andronov, M.; Voinarovska, V.; Andronova, N.; Wand, M.; Clevert, D.-A.; Schmidhuber, J. Reagent prediction with a molecular transformer improves reaction data quality. Chem. Sci. 2023, 14, 3235–3246. [Google Scholar] [CrossRef]
- Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V.H.; Haeuselmann, R.A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. [Google Scholar] [CrossRef]
- Mamoshina, P.; Vieira, A.; Putin, E.; Zhavoronkov, A. Applications of Deep Learning in Biomedicine. Mol. Pharm. 2016, 13, 1445–1454. [Google Scholar] [CrossRef]
- Lu, J.; Wang, C.; Zhang, Y. Predicting Molecular Energy Using Force-Field Optimized Geometries and Atomic Vector Representations Learned from an Improved Deep Tensor Neural Network. J. Chem. Theory Comput. 2019, 15, 4113–4121. [Google Scholar] [CrossRef]
- Cai, C.; Wang, S.; Xu, Y.; Zhang, W.; Tang, K.; Ouyang, Q.; Lai, L.; Pei, J. Transfer Learning for Drug Discovery. J. Med. Chem. 2020, 63, 8683–8694. [Google Scholar] [CrossRef]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Commun. ACM 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Chang, M. AI for Drug Development and Well-Being. 2020. Available online: http://ctrisoft.net/StatisticiansOrg/AI/AIforWellbingebook5.5x8.5in.pdf (accessed on 14 January 2024).
- Erhan, D.; Bengio, Y.; Courville, A.C.; Vincent, P. Visualizing Higher-Layer Features of a Deep Network; University of Montreal: Montreal, QC, USA, 2009. [Google Scholar]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Algorithm | Specific Function | PMID |
|---|---|---|---|
| Prediction of the target protein structure | |||
| TrRosetta server | DNN | Predict 3D structures of proteins | [13] |
| AlphaFold | DNN | Predict 3D structures of proteins | [14] |
| ComplexQA | GNN | Predict protein complex structure | [15] |
| ProteinBERT | Transformer | Predict secondary structure | [16] |
| ESMfold | Transformer | Predict structure of proteins | [17] |
| Predicting protein–protein interactions | |||
| IntPred | RF | Predict PPI interface sites | [18] |
| eFindSite | SVM; NBC | Predict PPI interfaces | [19] |
| DELPHI | RNN; CNN | Predict PPI sites | [20] |
| PPISP-XGBoost | XGBoost | Predict PPI sites | [21] |
| HN-PPISP | CNN | Predict PPI sites | [22] |
| TAGPPI | GCN | Predict PPIs | [23] |
| Struct2Graph | GAT | Predict PPIs | [24] |
| DeepFE-PPI | DNN | Predict PPIs | [25] |
| SGPPI | GCN | Predict PPIs | [26] |
| DeepPPI | DNN | Predict PPIs | [27] |
| DL-PPI | GNN | Predict PPIs | [28] |
| DeepSG2PPI | CNN | Predict PPIs | [29] |
| MaTPIP | Transformer; CNN | Predict PPIs | [30] |
| ProtInteract | Autoencoder; CNN | Predict PPIs | [31] |
| Predicting drug–target interactions | |||
| DeepC-SeqSite | CNN | Predict DTI binding sites | [32] |
| DeepSurf | CNN; ResNet | Predict DTI binding sites | [33] |
| PrankWeb | RF | Predict DTI binding sites | [34] |
| PUResNet | ResNet | Predict DTI binding sites | [35] |
| AGAT-PPIS | GNN | Predict DTI binding sites | [36] |
| DeepDTA | CNN | Predict DTI binding affinity | [37] |
| SimBoost | GBM | Predict DTI binding affinity | [38] |
| DEELIG | CNN | Predict DTI binding affinity | [39] |
| DeepDTAF | CNN | Predict DTI binding affinity | [8] |
| GraphDelta | CNN | Predict DTI binding affinity | [40] |
| PotentialNet | CNN | Predict DTI binding affinity | [41] |
| DeepAffinity | RNN, CNN | Predict DTI binding affinity | [9] |
| TeM-DTBA | CNN | Predict DTI binding affinity | [42] |
| Wang et al.’s method | RL | Predict DTI binding pose | [43] |
| Nguyen et al.’s method | RF; CNN | Predict DTI binding pose | [44] |
| AMMVF-DTI | GAT; NTN | Predict drug–target interactions | [45] |
| De novo drug design | |||
| ReLeaSE | RNN; RL | Conduct de novo drug design | [46] |
| ChemVAE | CNN; GRU | Conduct de novo drug design | [47] |
| MolRNN | RNN | Conduct multi-objective de novo drug design | [48] |
| PaccMann(RL) | VAE | Generate compounds with anti-cancer drug properties | [49] |
| druGAN | AAE | Conduct de novo drug design | [50] |
| SCScore | CNN | Evaluate the molecular accessibility | [51] |
| UnCorrupt SMILES | Transformer | Conduct de novo drug design | [52] |
| PETrans | Transfer learning | Conduct de novo drug design | [53] |
| FSM-DDTR | Transformer | Conduct de novo drug design | [54] |
| DNMG | GAN | Conduct de novo drug design | [55] |
| MedGAN | GAN | Design novel molecule | [56] |
| Prediction of the physicochemical properties | |||
| Panapitiya et al.’s method | GNN | Predict aqueous solubility | [57] |
| SolTranNet | Transformer | Predict aqueous solubility | [58] |
| Zang et al.’s method | SVM | Predict multiple physicochemical properties | [59] |
| Prediction of the ADME/T properties | |||
| ADMETboost | XGBoost | Predict ADME/T properties | [60] |
| vNN | k-NN | Predict ADME/T properties | [61] |
| Interpretable-ADMET | CNN; GAT | Predict ADME/T properties | [62] |
| XGraphBoost | GNN | Predict ADME/T properties | [63] |
| DeepTox | DNN | Predict toxicity of compounds | [64] |
| Li et al.’s method | DNN | Predict human Cytochrome P450 inhibition | [65] |
| LightBBB | LightGBM | Predict blood–brain barrier | [66] |
| Deep-B3 | CNN | Predict blood–brain barrier | [67] |
| PredPS | GNN | Predict stability of compounds in human plasma | [68] |
| Khaouane et al.’s method | CNN | Predict plasma protein binding | [69] |
| Application of AI in drug repurposing | |||
| deepDTnet | Autoencoder | Predict new targets of known drugs | [70] |
| NeoDTI | GCN | Predict new targets of known drugs | [71] |
| DTINet | Network diffusion algorithm and the dimensionality reduction | Predict new targets of known drugs | [72] |
| MBiRW | Birandom walk algorithm | Predict new indications of known drugs | [73] |
| GDRnet | GNN | Predict new indications of known drugs | [74] |
| deepDR | VAE | Predict new indications of known drugs | [75] |
| GIPAE | VAE | Predict new indications of known drugs | [76] |
| DrugRep-HeSiaGraph | Heterogeneous siamese neural network | Predict new indications of known drugs | [77] |
| iEdgeDTA | GCNN | Predict DTI binding affinity | [78] |
| Retrosynthesis prediction | |||
| Segler et al.’s method | MCTS, DNN | Predict retrosynthetic analysis | [79] |
| Liu et al.’s method | RNN | Predict retrosynthetic analysis | [80] |
| RAscore | RF | Predict retrosynthetic accessibility score | [81] |
| Reaction prediction | |||
| Wei et al.’s method | Neural network | Predict reaction classes | [82] |
| Coley et al.’s method | Neural network | Predict products of chemical reactions | [83] |
| Gao et al.’s method | Neural network | Predict optimal reaction conditions | [84] |
| Marcou et al.’s method | RF | Evaluate reaction feasibility | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, X.; Zhao, Y.; Qi, Z.; Hou, S.; Chen, J. Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges. Molecules 2024, 29, 903. https://doi.org/10.3390/molecules29040903
Qi X, Zhao Y, Qi Z, Hou S, Chen J. Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges. Molecules. 2024; 29(4):903. https://doi.org/10.3390/molecules29040903
Chicago/Turabian StyleQi, Xin, Yuanchun Zhao, Zhuang Qi, Siyu Hou, and Jiajia Chen. 2024. "Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges" Molecules 29, no. 4: 903. https://doi.org/10.3390/molecules29040903
APA StyleQi, X., Zhao, Y., Qi, Z., Hou, S., & Chen, J. (2024). Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges. Molecules, 29(4), 903. https://doi.org/10.3390/molecules29040903