
Metal Complexes Containing Homoleptic Diorganoselenium(II) Ligands: Synthesis, Characterization and Investigation of Optical Properties



Abstract
1. Introduction
2. Results and Discussion
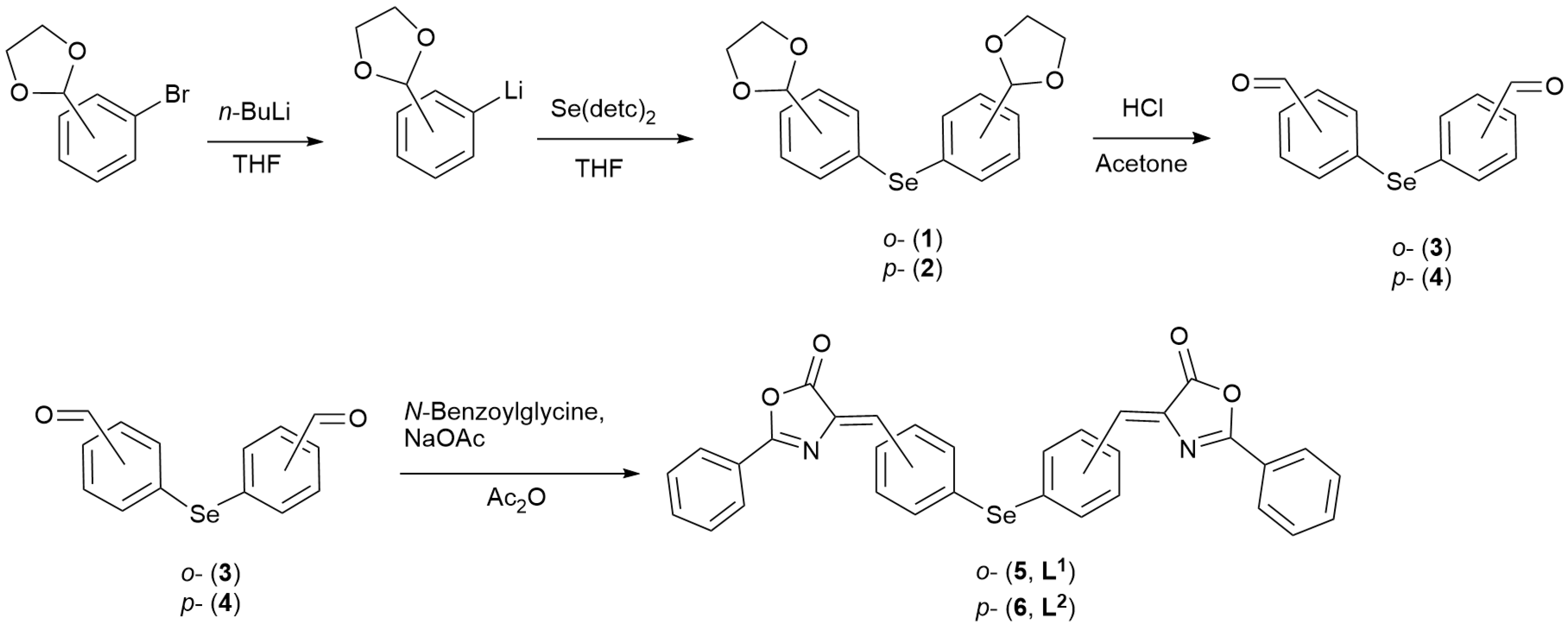
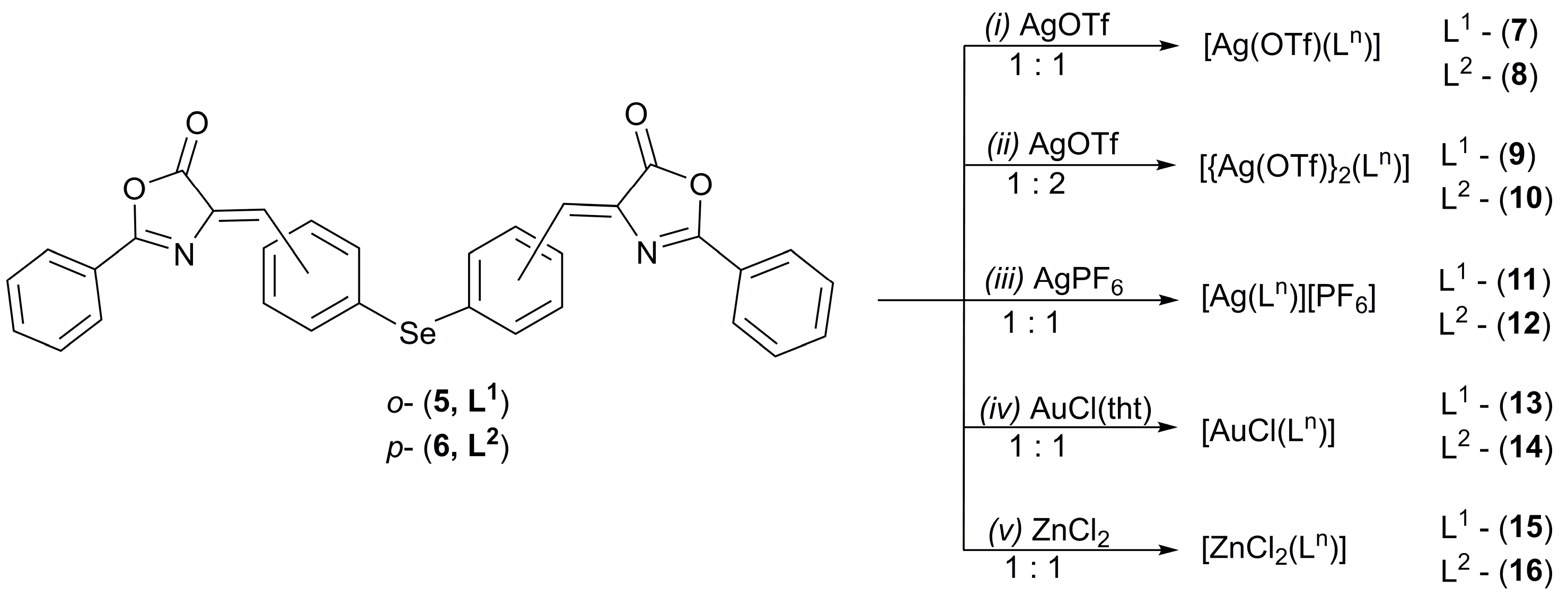
2.1. Synthesis
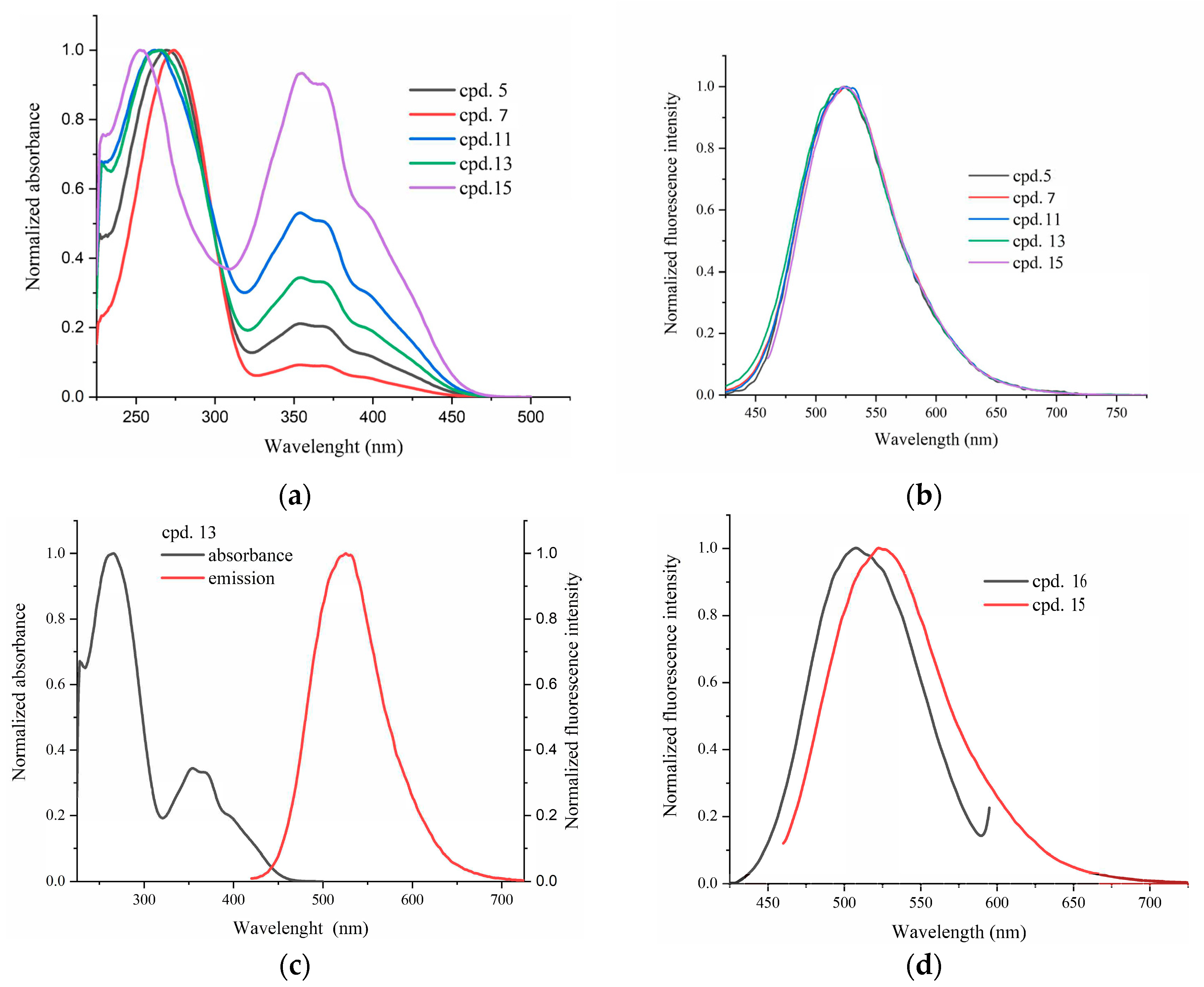
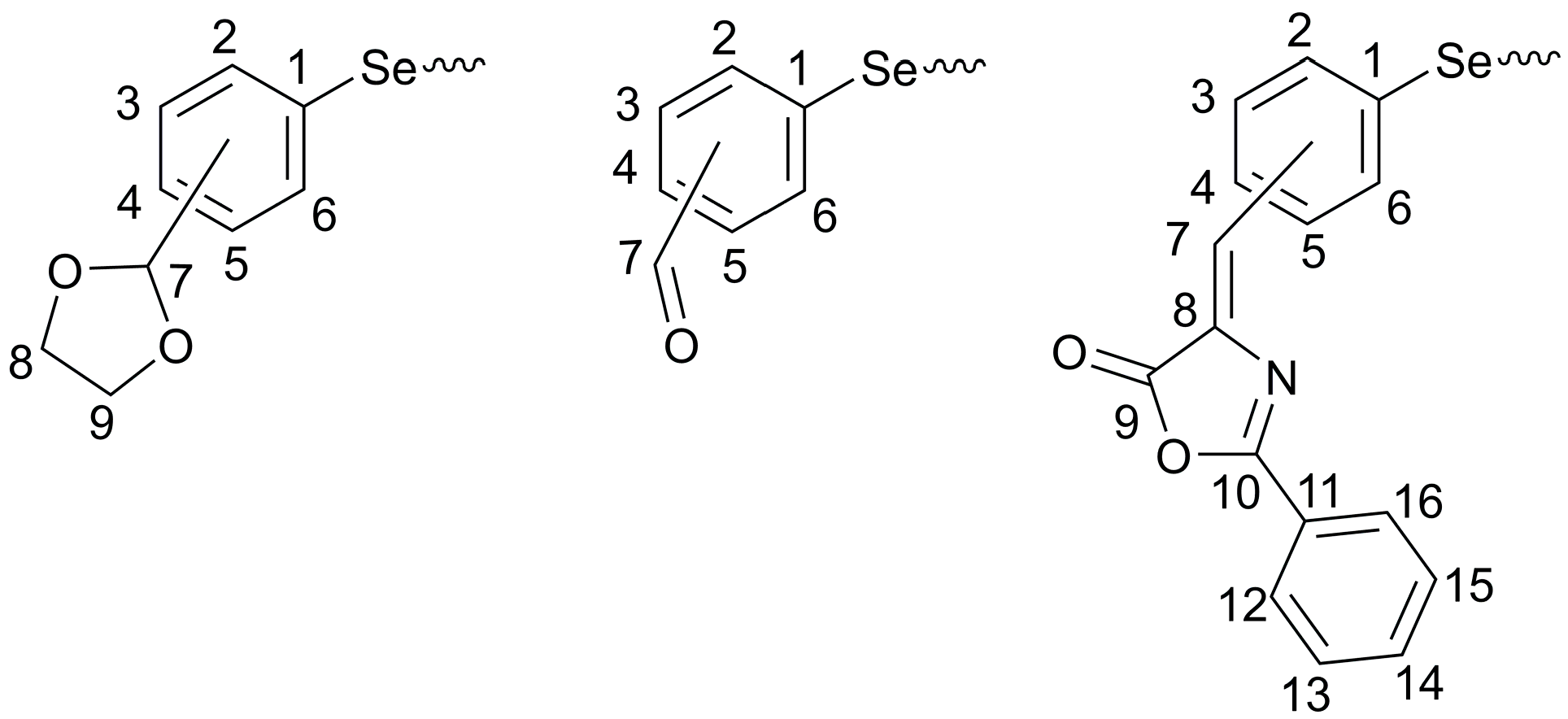
2.2. Spectroscopic Characterization
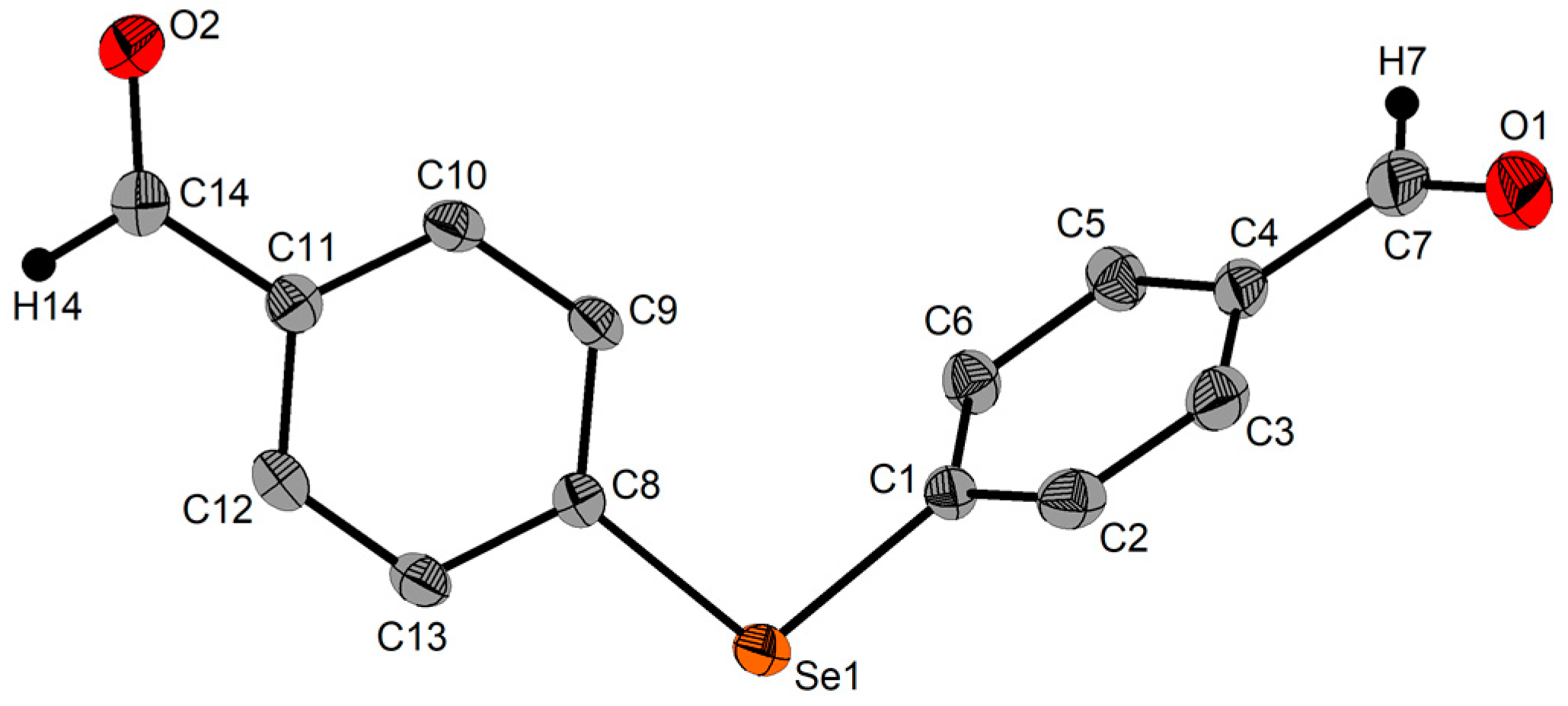
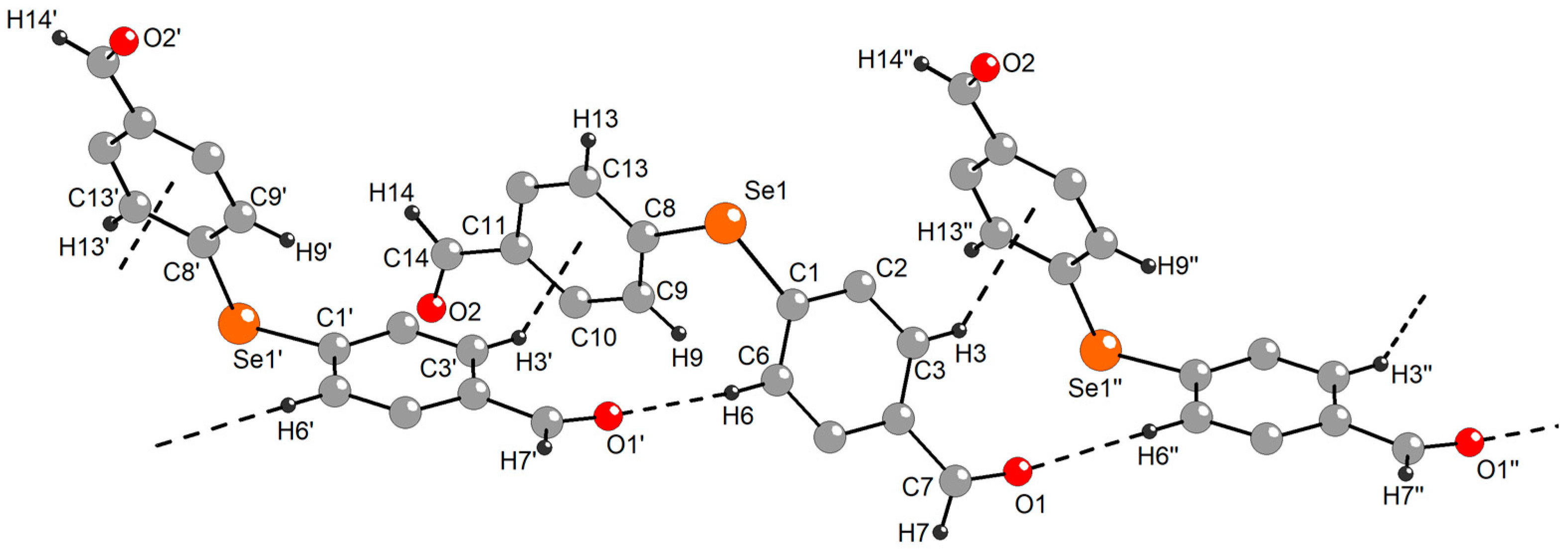
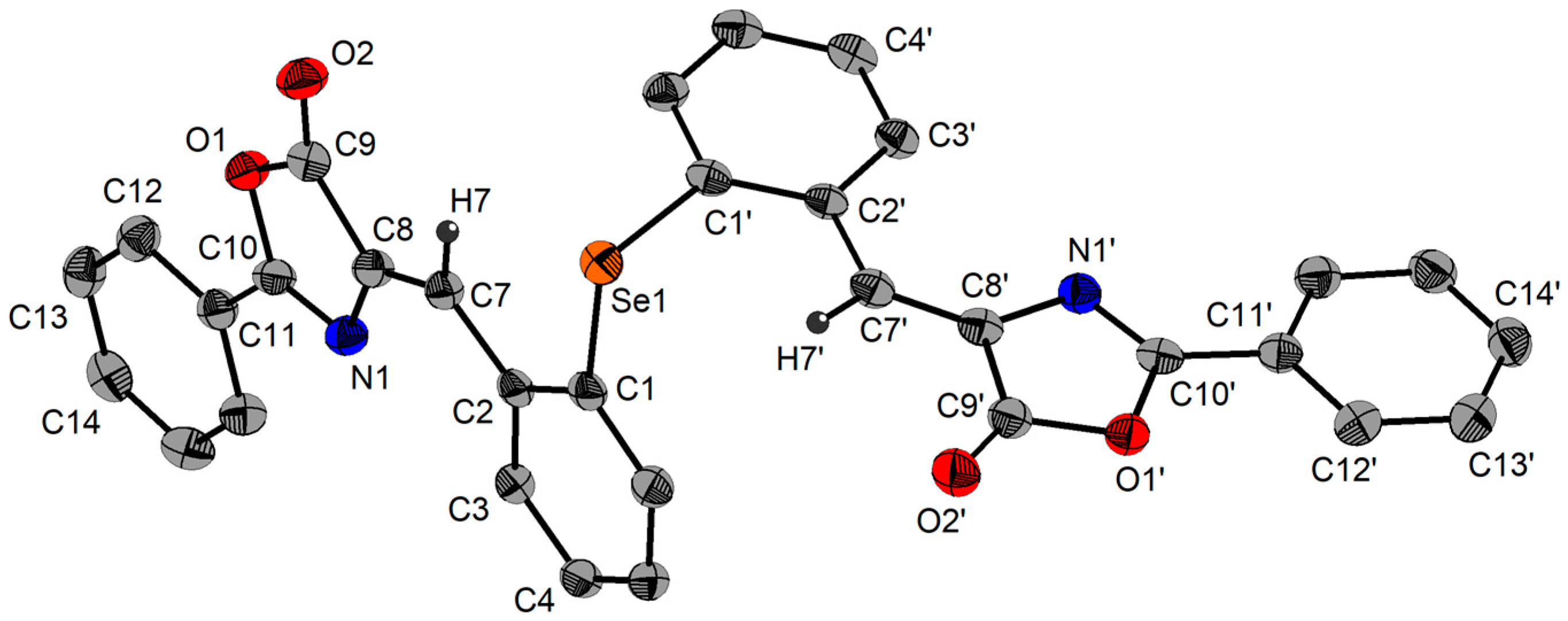
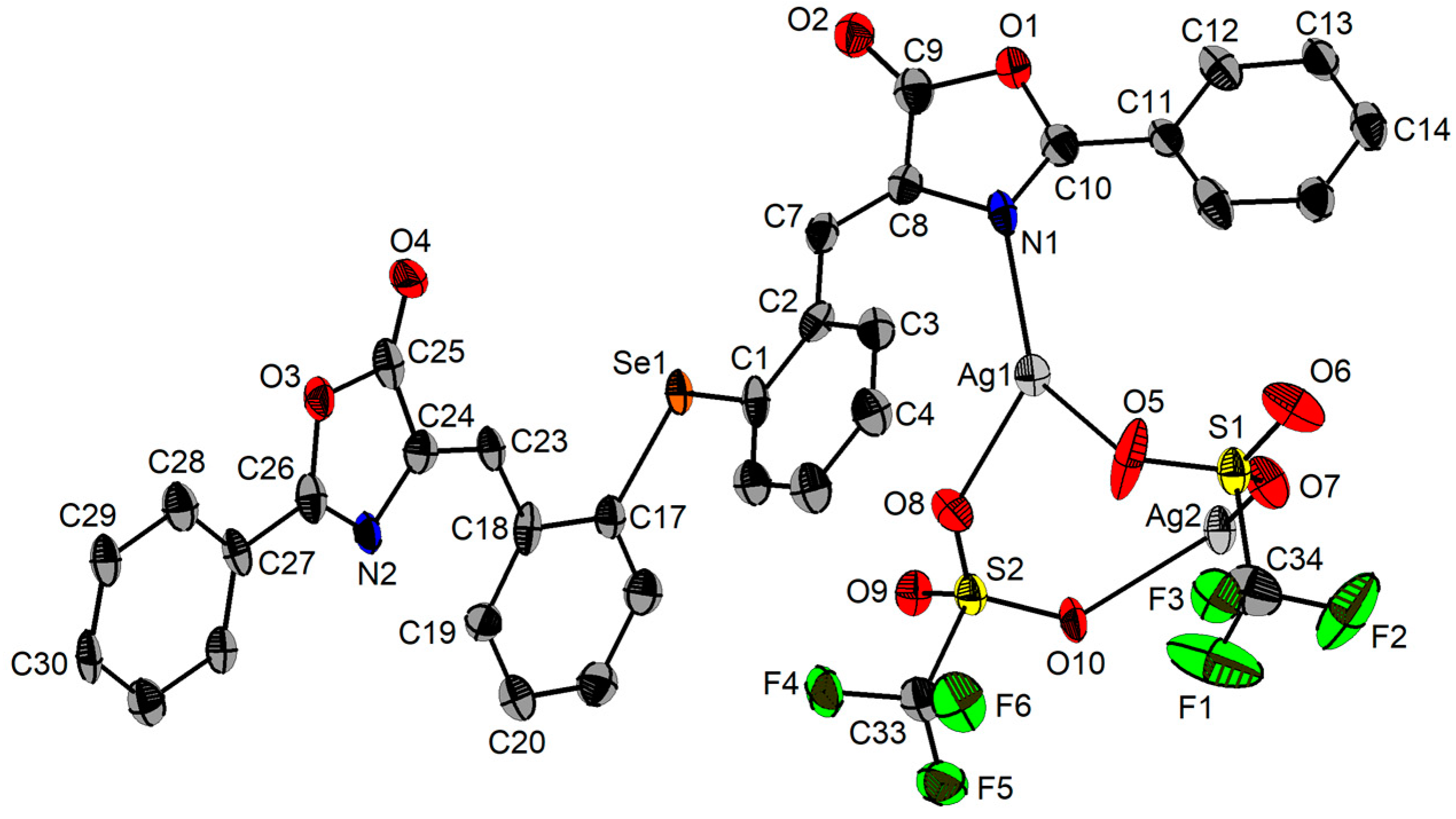
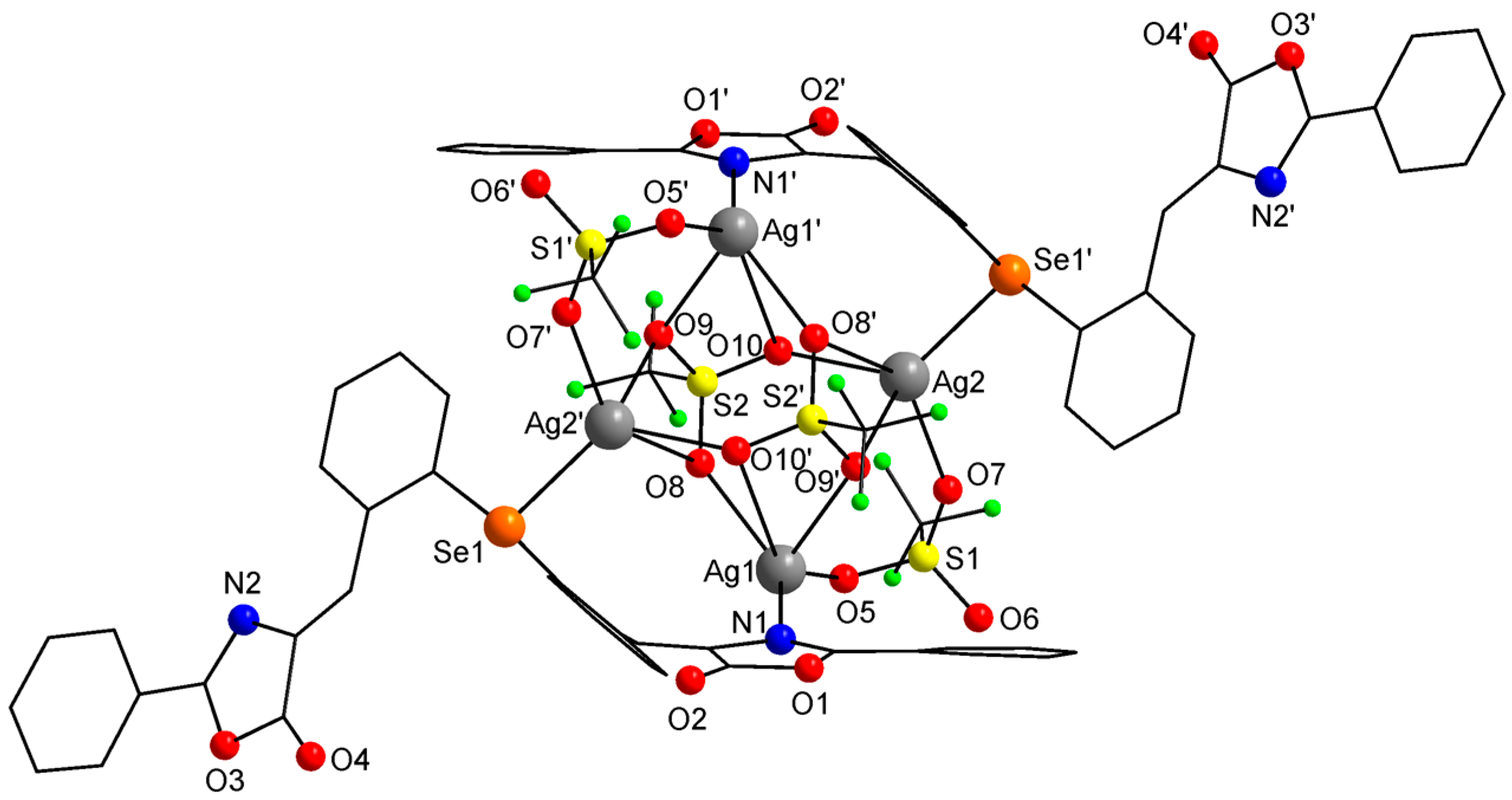
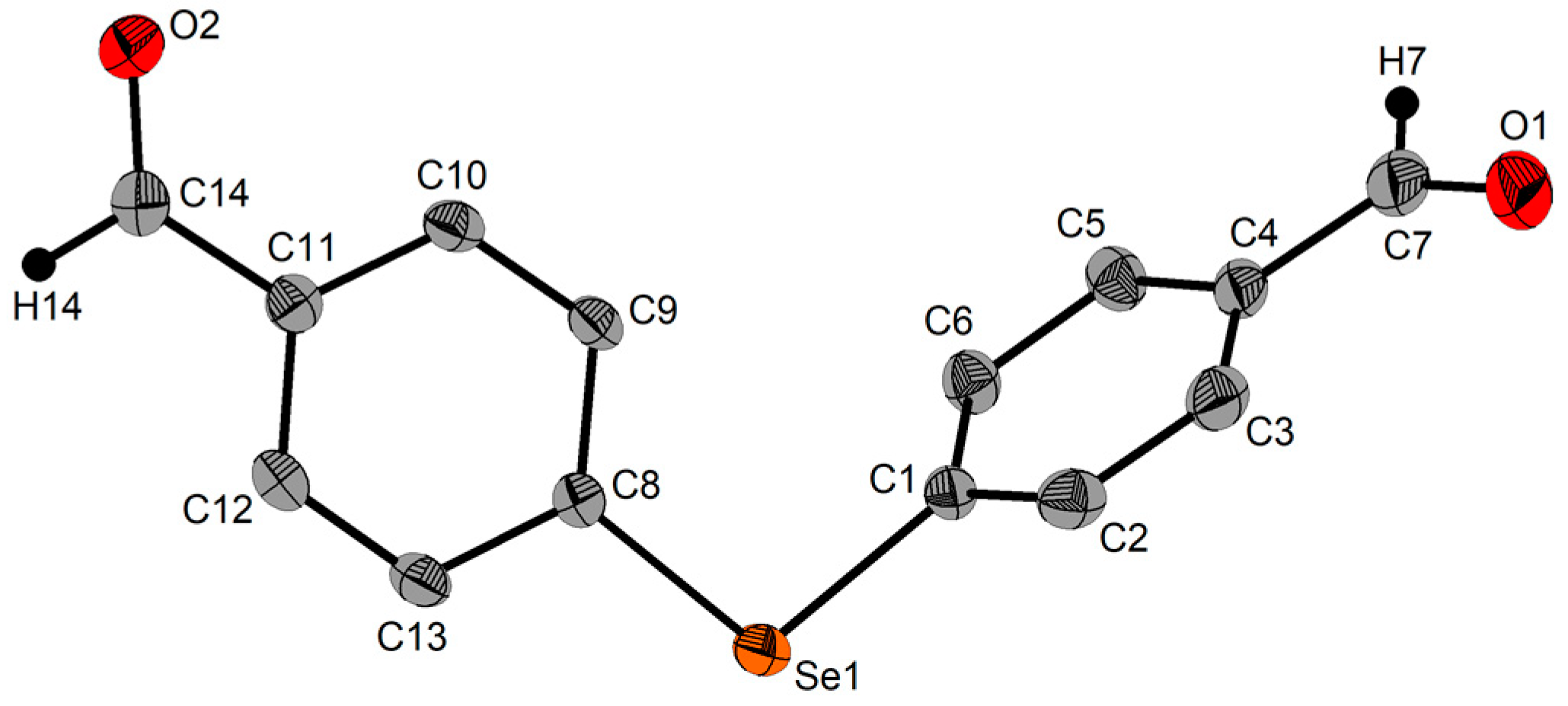
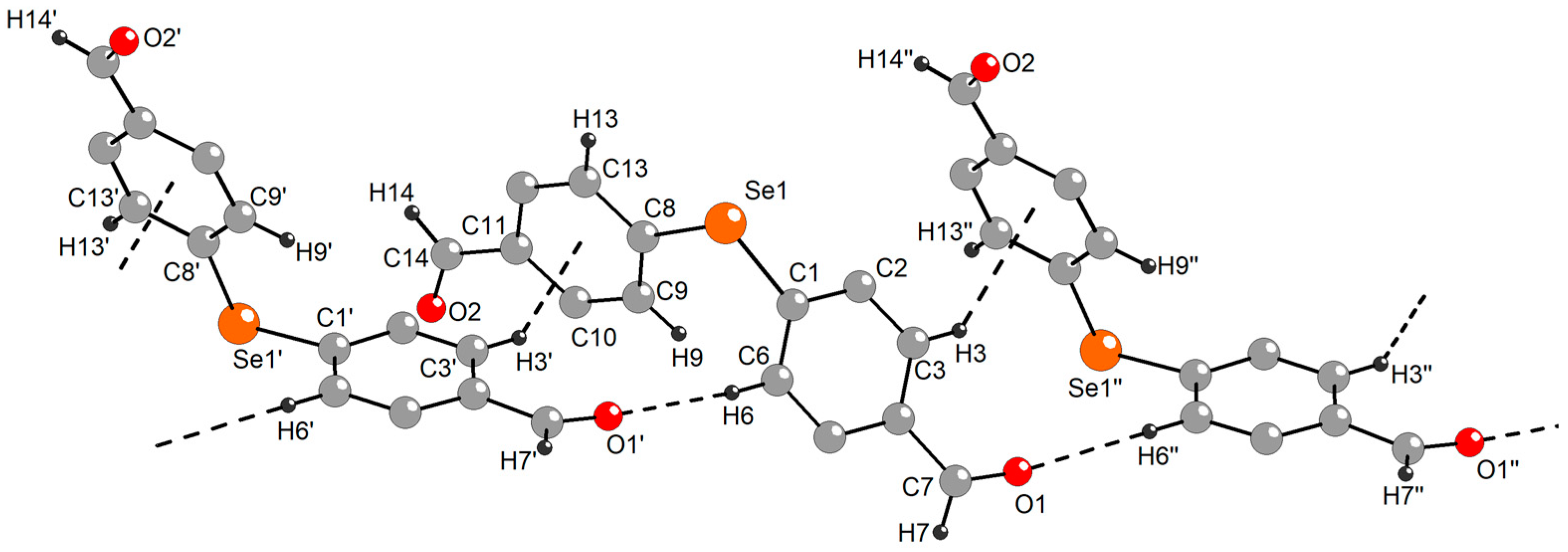
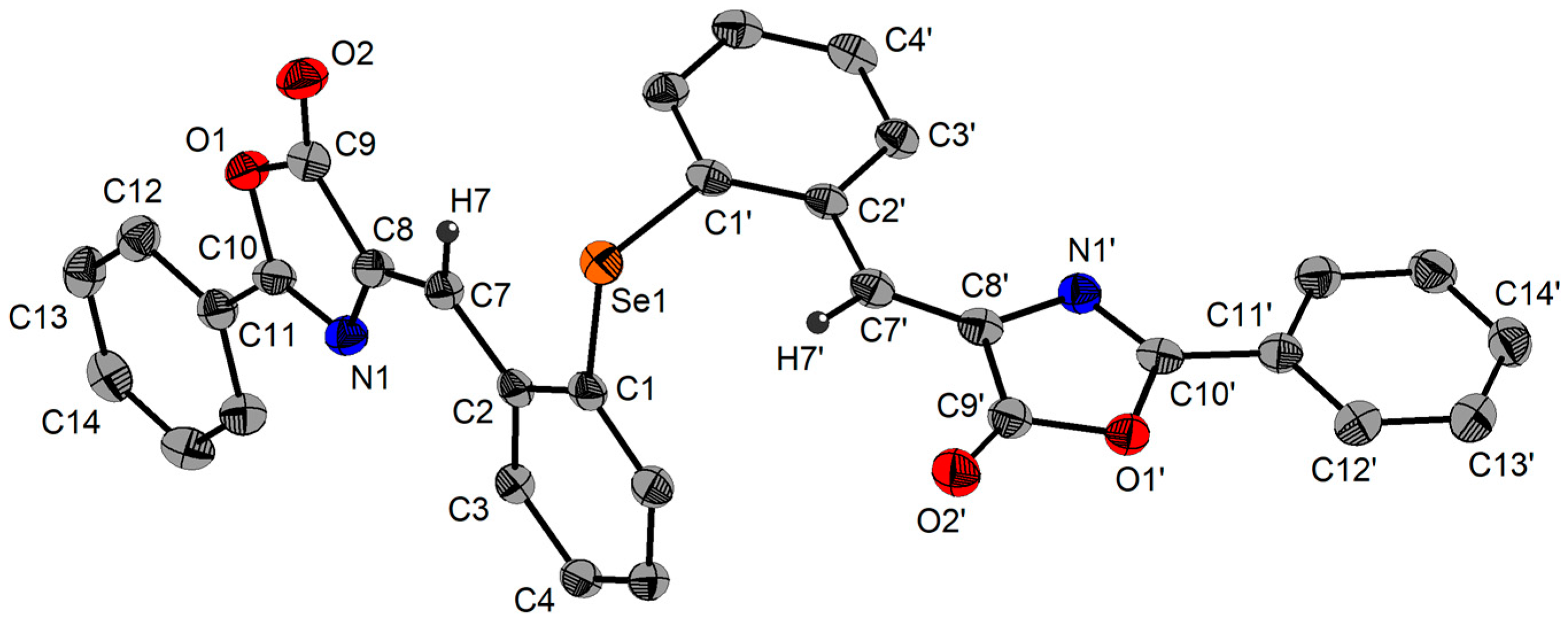
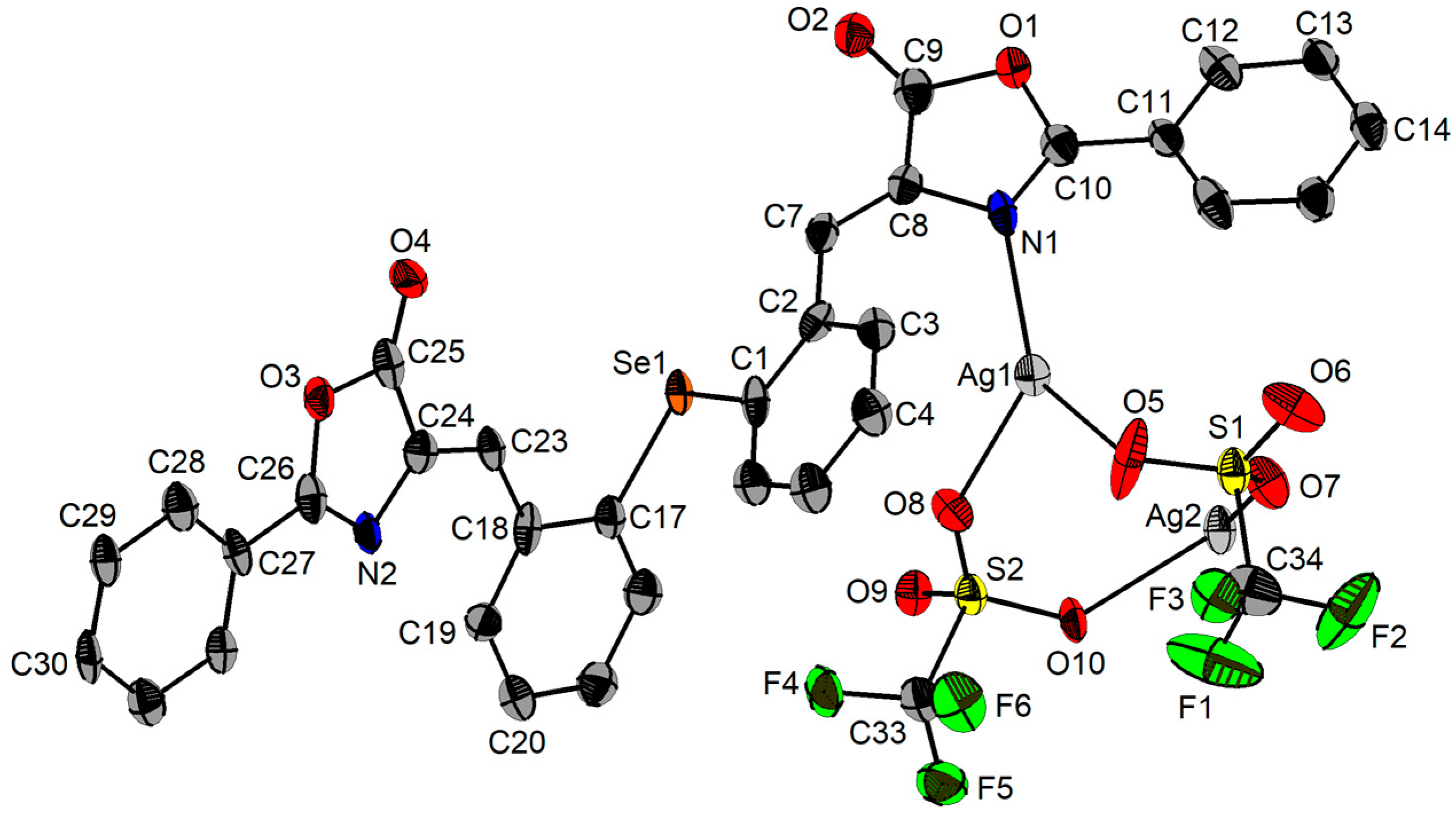
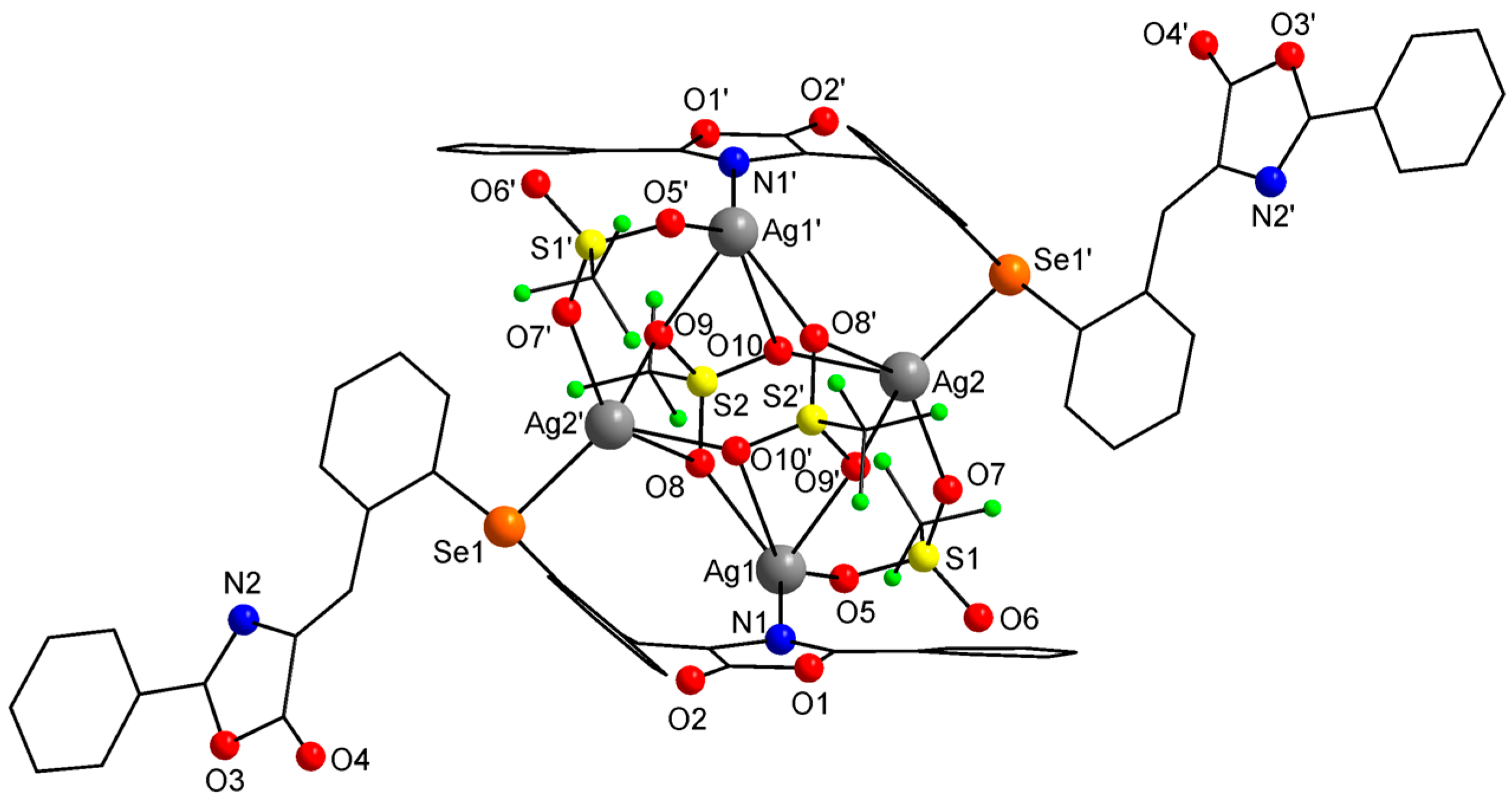
2.3. Single-Crystal X-ray Diffraction Studies
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis and Characterization
- [2-{(CH2O)2CH}C6H4]2Se (1)
- [4-{(CH2O)2CH}C6H4]2Se (2)
- [2-(O=CH)C6H4]2Se (3)
- [4-(O=CH)C6H4]2Se (4)
- [(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se (5)
- [(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se (6)
- [Ag(OTf){[(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (7)
- [Ag(OTf){[(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (8)
- [{Ag(OTf)}2{[(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (9)
- [{Ag(OTf)}2{[(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (10)
- [Ag{[(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}][PF6] (11)
- [Ag{[(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}][PF6] (12)
- [AuCl{[(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (13)
- [AuCl{[(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (14)
- [ZnCl2{[(Z)-2′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (15)
- [ZnCl2{[(Z)-4′-{2-C6H5-(4H)-oxazol-5-one}CHC6H4]2Se}] (16)
3.3. Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jain, V.K.; Priyadarsini, K.I. (Eds.) Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; RSC: Croydon, UK, 2018. [Google Scholar]
- Laitinen, R.; Oilunkaniemi, R. (Eds.) Selenium and Tellurium Reagents in Chemistry and Materials Science; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2019. [Google Scholar]
- Wirth, T. Organoselenium Chemistry—Modern Developments in Organic Synthesis, Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2000. [Google Scholar]
- Arora, A.; Singh, S.; Oswal, P.; Nautiyal, D.; Rao, G.K.; Kumar, S.; Kumar, A. Preformed molecular complexes of metals with organoselenium ligands: Syntheses and applications in catalysis. Coord. Chem. Rev. 2021, 438, 213885. [Google Scholar] [CrossRef]
- Panda, S.; Panda, A.; Zade, S.S. Organoselenium compounds as fluorescent probes. Coord. Chem. Rev. 2015, 300, 86–100. [Google Scholar] [CrossRef]
- Mamgain, R.; Singh, F.V. Selenium-based fluorescence probes for the detection of bioactive molecules. ACS Org. Inorg. Au 2022, 2, 262–288. [Google Scholar] [CrossRef]
- Shi, W.; Sun, S.; Li, X.; Ma, H. Imaging different interactions of mercury and silver with live cells by a designed fluorescence probe rhodamine B selenolactone. Inorg. Chem. 2010, 49, 1206–1210. [Google Scholar] [CrossRef]
- Ramos-Inza, S.; Plano, D.; Sanmartín, C. Metal-based compounds containing selenium: An appealing approach towards novel therapeutic drugs with anticancer and antimicrobial effects. Eur. J. Med. Chem. 2022, 244, 114834. [Google Scholar] [CrossRef]
- Cargnelutti, R.; Schumacher, R.F.; Belladona, A.L.; Kazmierczak, J.C. Coordination chemistry and synthetic approaches of pyridyl-selenium ligands: A decade update. Coord. Chem Rev. 2021, 426, 213537. [Google Scholar] [CrossRef]
- Malczewska-Jaskóła, K.; Jankowski, W.; Warzajtis, B.; Jasiewicz, B.; Hoffmann, M.; Rychlewska, U. Chalcogenated (S)-(–)-nicotine derivatives as chiral linkers for 1D coordination polymers. Polyhedron 2015, 100, 404–411. [Google Scholar] [CrossRef]
- Williams, D.J.; White, K.M.; VanDerveer, D.; Wilkinson, A.P. Dichlorobis[1,3-dimethyl-2(3H)-imidazoleselone]zinc(II): A potential zinc selenide synthon. Inorg. Chem. Commun. 2002, 5, 124–126. [Google Scholar] [CrossRef]
- Pop, A.; Mitea, R.; Silvestru, A. Diorganochalcogen(II) ligands of type [R2C(OH)CH2](2-Me2NCH2C6H4) E (E = S, Se, Te; R = Me, Ph), and their silver(I) complexes. J. Organomet. Chem. 2014, 768, 121–127. [Google Scholar] [CrossRef]
- Popa, R.A.; Silvestru, A.; Pop, A. Silver(I) complexes of a new multidentate macrocyclic ligand with N/S/Se donor atoms. Polyhedron 2016, 110, 197–202. [Google Scholar] [CrossRef]
- Dumitraș, D.; Pop, A.; Silvestru, A. Silver(I) and gold(I) complexes of multidentate ligands based on functionalized pyridine. Polyhedron 2022, 220, 115801. [Google Scholar] [CrossRef]
- Pop, A.; Bellini, C.; Şuteu, R.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Silvestru, A.; Sarazin, Y. Cadmium complexes bearing Me2N^E^O− (E = S, Se) organochalcogenoalkoxides and their zinc and mercury analogues. Dalton Trans. 2017, 46, 3179–3191. [Google Scholar] [CrossRef]
- Hodorogea, A.M.; Silvestru, A.; Lippolis, V.; Pop, A. Group 12 metal complexes of mixed thia/aza and thia/oxa/aza macrocyclic ligands. Polyhedron 2022, 216, 115650. [Google Scholar] [CrossRef]
- Pop, A.; Wang, L.; Dorcet, V.; Roisnel, T.; Carpentier, J.-F.; Silvestru, A.; Sarazin, Y. On the coordination chemistry of organochalcogenolates RNMe2∧E− and RNMe2∧E∧O− (E = S, Se) onto lead(II) and lighter divalent tetrel elements. Dalton Trans. 2014, 43, 16459–16474. [Google Scholar] [CrossRef]
- Icli, S.; Doroshenko, A.O.; Alp, S.; Abmanova, N.A.; Egorova, S.I.; Astley, S.T. Structure and luminescent properties of the 4-arylidene-2-aryl-5-oxazolones (azlactones) in solution and crystalline state. Spectrosc. Lett. 1999, 32, 553–569. [Google Scholar] [CrossRef]
- Rodrigues, C.A.B.; Mariz, I.F.A.; Maçôas, E.M.S.; Afonso, C.A.M. Two-photon absorption properties of push-pull oxazolones derivatives. Dye. Pigm. 2012, 95, 713–722. [Google Scholar] [CrossRef]
- Rodrigues, C.A.B.; Mariz, I.F.A.; Maçôas, E.M.S.; Afonso, C.A.M.; Martinho, J.M.G. Unsaturated oxazolones as nonlinear fluorophores. Dye. Pigm. 2013, 99, 642–652. [Google Scholar] [CrossRef]
- Blanco-Lomas, M.; Funes-Ardoiz, I.; Campos, P.J.; Sampedro, D. Oxazolone-based photoswitches: Synthesis and properties. Eur. J. Org. Chem. 2013, 2013, 6611–6618. [Google Scholar] [CrossRef]
- Szukalski, A.; Krawczyk, P.; Sahraoui, B.; Jędrzejewska, B. Multifunctional Oxazolone Derivative as an Optical Amplifier, Generator, and Modulator. J. Phys. Chem. B 2022, 126, 1742–1757. [Google Scholar] [CrossRef] [PubMed]
- Butuza, R.A.; Dumitraș, D.; Bohan, C.; Pop, A. Silver(I) complexes containing heteroleptic diorganochalcogen(II) ligands. New J. Chem. 2023, 47, 2202–2210. [Google Scholar] [CrossRef]
- Laguna, M.; Villacampa, M.D. Hexanuclear Mercury-Silver Complexes. Novel Coordination for Bridging Mesityl and Triflate Groups. Inorg. Chem. 1998, 37, 133–135. [Google Scholar]
- Panda, A.; Menon, S.C.; Singh, H.B.; Butcher, R.J. Synthesis of some macrocycles/bicycles from bis(o-formylphenyl) selenide: X-ray crystal structure of bis(o-formylphenyl) selenide and the first 28-membered selenium containing macrocyclic ligand. J. Organomet. Chem. 2001, 623, 87–94. [Google Scholar] [CrossRef]
- Kiss, L.; Pop, A.; Sergiu, S.; Raț, C.I.; Silvestru, C. Synthesis and characterization of [4-{(CH2O)2CH}C6H4]2Hg, [4-(O=CH)C6H4]2Hg and [(E)-4-(RN=CH)C6H4]2Hg (R = 2′-py, 4′-py, 2′-pyCH2, 4′-pyCH2). Appl. Organomet. Chem. 2021, 35, e6339. [Google Scholar] [CrossRef]
- Yadav, D.; Dixit, A.K.; Raghothama, S.; Awasthi, S.K. Ni nanoparticle-confined covalent organic polymer directed diaryl-selenides synthesis. Dalton Trans. 2020, 49, 12266–12272. [Google Scholar] [CrossRef]
- Plöchl, J. Ueber phenylglycidasäure (phenyloxacrylsäure). Ber. Dtsch. Chem. Ges. 1883, 16, 2815. [Google Scholar] [CrossRef]
- Plöchl, J. Ueber einige derivate der benzoylimidozimmtsäure. Ber. Dtsch. Chem. Ges. 1884, 17, 1623. [Google Scholar] [CrossRef]
- Erlenmeyer, E. Ueber die condensation der hippursäure mit phtalsäureanhydrid und mit benzaldehyde. Justus Liebigs Ann. Chem. 1893, 275, 1. [Google Scholar]
- Buck, J.S.; Ide, W.S. Azlactone of α-benzoylamino-β-(3,4-dimethoxyphenyl)-acrylic acid. Org. Synth. 1933, 13, 8–9. [Google Scholar]
- Blanco-Lomas, M.; Campos, P.J.; Sampedro, D. Benzylidene-oxazolones as molecular photoswitches. Org. Lett. 2012, 14, 4334–4337. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-Z.; Li, X.-H.; Zhang, X.-Z. Molecular structure, vibrational spectra and theoretical NBO, HOMO-LUMO analysis of N-benzoyl glycine by DFT and ab-initio HF methods. Indian J. Pure Appl. Phys. 2012, 50, 719–726. [Google Scholar]
- Saour, K.Y.; Al-Bayati, R.I.H.; Shia, J.S. Synthesis of 4-benzylidene-2-(4-nitro-phenyl)-4H-oxazol-5-one derivatives with suspected biological activity. Chem. Mat. Res. 2015, 7, 105–109. [Google Scholar]
- Mavridis, E.; Bermperoglou, E.; Pontiki, E.; Hadjipavlou-Litina, D. 5-(4H)-Oxazolones and Their Benzamides as Potential Bioactive Small Molecules. Molecules 2020, 25, 3173. [Google Scholar] [CrossRef]
- Barbuceanu, S.-F.; Rosca, E.-V.; Apostol, T.-V.; Socea, L.-I.; Draghici, C.; Farcasanu, I.C.; Ruta, L.L.; Nitulescu, G.M.; Iscrulescu, L.; Pahontu, E.-M.; et al. New Heterocyclic Compounds from Oxazol-5(4H)-one and 1,2,4-Triazin-6(5H)-one Classes: Synthesis, Characterization and Toxicity Evaluation. Molecules 2023, 28, 4834. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlman, P.; Affolter, C. Structure Determination of Organic Compounds. Tables of Spectral Data, 4th ed.; Springer: New York, NY, USA, 2000. [Google Scholar]
- Angus-Dunne, S.J.; Lee Chin, L.E.P.; Burns, R.C.; Lawrance, G.A. Metallocene and organo-main group trifluoromethanesulfonates. Transit. Met. Chem. 2006, 31, 268–275. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, H.B.; Wolmershauser, G. Protection against peroxynitrite-mediated nitration reaction by intramolecularly coordinated diorganoselenides. Organometallics 2006, 25, 382–393. [Google Scholar] [CrossRef]
- Apte, S.D.; Zade, S.S.; Singh, H.B.; Butcher, R.J. Contrasting behavior of bis[2-(4,4-dimethyl-2-oxazolinyl)phenyl] chalcogenides (Se/Te) toward mercuric chloride: Facile cleavage of the Te–C Bond. Organometallics 2003, 22, 5473–5477. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Saleem, K. Empirical formulae to molecular structures of metal complexes by molar conductance. Synth. React. Inorg. Met. Org. Chem. 2013, 43, 1162–1170. [Google Scholar] [CrossRef]
- Wang, D.-H.; Zhang, Y.; Wang, Y.-T.; Feng, H.-Y.; Chen, Y.; Zhao, D.-Z. Silver(I) complexes of diphenylpyridines: Crystal structures, luminescence studies, theoretical insights, and biological activities. ChemPlusChem 2017, 82, 323–332. [Google Scholar] [CrossRef]
- Du, L.-Y.; Shi, W.-J.; Hou, L.; Wang, Y.-Y.; Shi, Q.-Z.; Zhu, Z. Solvent or temperature induced diverse coordination polymers of silver(I) sulfate and bipyrazole systems: Syntheses, crystal structures, luminescence, and sorption properties. Inorg. Chem. 2013, 52, 14018–14027. [Google Scholar] [CrossRef]
- Baranov, A.Y.; Rakhmanova, M.I.; Samsonenko, D.G.; Malysheva, S.F.; Belogorlova, N.A.; Bagryanskaya, I.Y.u.; Fedin, V.P.; Artem’eva, A.V. Silver(I) and gold(I) complexes with tris[2-(2-pyridyl)ethyl]phosphine. Inorg. Chim. Acta 2019, 494, 78–83. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, K.A.; Wilson, S.R.; Moore, J.S. Coordination networks of 3,3′-dicyanodiphenylacetylene and silver(I) salts: structural diversity through changes in ligand conformation and counterion. Inorg. Chem. 1997, 36, 2960–2968. [Google Scholar] [CrossRef] [PubMed]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Foss, O.; Pitha, J.J. Xanthates and dithiocarbamates of selenium(II) and tellurium(II). Inorg. Synt. 1953, 4, 91–93. [Google Scholar]
- Uson, R.; Laguna, A.; Laguna, M. (Tetrahydrothiophene)gold(I) or gold(III) complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- MestReC; MestReNova; Mestrelab Research S.L. A Coruna 15706, version 14; Mestrelab Research: Santiago de Compostela, Spain, 2020.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Pennington, W.T. DIAMOND—Visual Crystal Structure Information System; Crystal Impact: Bonn, Germany, 2001. [Google Scholar]
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | λabs (nm) [ε (M−1·cm−1)] | λex (nm) | λem (nm) | Stokes Shift (cm−1) |
|---|---|---|---|---|
| 3 | 242 [30,806]; 271 [6805]; 344 [4659] | - | - | - |
| 4 | 242 [18,414]; 313 [13,818]; 337 (sh) | - | - | - |
| 5 | 255 [21,719]; 354 [28,900]; 367 [27,809]; 400 (sh) | 400 | 523 | 5880 |
| 6 | 232 [12,818]; 262 [17,550]; 372 [25,677]; 404 [31,274]; 430 [sh] | 430 | 510 | 3648 |
| 7 | 255 [13,485]; 354 [23,677]; 367 [22,836]; 400 (sh) | 400 | 524 | 5916 |
| 8 | 265 [5357]; 372 [8840]; 399 [10,806]; 418 [11,154] | - | - | - |
| 11 | 258 [23,581]; 365 [21,968]; 354 [22,844]; 400 (sh) | 400 | 523 | 5880 |
| 12 | 264 [21,723]; 367 [29,273]; 401 [34,650]; 430 (sh) | 430 | 511 | 3686 |
| 13 | 266 [16,567]; 354 [13,599]; 366 [13,134]; 400 (sh) | 400 | 526 | 5989 |
| 14 | 265 [17,74]; 374 [21,201]; 399 [24,835]; 430 (sh) | - | - | - |
| 15 | 266 [15,427]; 372 [31,308]; 404 [31,236]; 427 (sh) | 427 | 523 | 4299 |
| 16 | 252 [18,387]; 355 [19,551]; 368 [18,930]; 395 (sh) | 395 | 508 | 5631 |
| Se1‒C1 | 1.943(8) | C1‒Se1‒C17 | 99.3(4) | ||
| Se1‒C17 | 1.941(9) | ||||
| Ag1−N1 | 2.266(8) | N1−Ag1−O5 | 134.9(3) | Se1’−Ag2−O7 | 115.59(16) |
| Ag1−O5 | 2.254(7) | N1−Ag1−O8 | 126.9(2) | Se1’−Ag2−O10 | 125.86(16) |
| Ag1−O8 | 2.359(6) | N1−Ag1−O9’ | 83.03(22) | Se1’−Ag2−O8’ | 92.83(13) |
| Ag1−O9’ | 2.6514(56) | N1−Ag1−O10’ | 69.19(20) | Se1’−Ag2−O9’ | 119.69(13) |
| Ag1−O10’ | 3.5053(7) | O5−Ag1−O8 | 98.1(3) | O7−Ag2−O10 | 108.1(2) |
| Ag2−Se1’ | 2.5691(1) | O5−Ag1−O9’ | 107.77(24) | O7−Ag2−O8’ | 136.21(21) |
| Ag2−O7 | 2.336(6) | O5−Ag1−O10’ | 146.10(22) | O7−Ag2−O9’ | 86.5(2) |
| Ag2−O10 | 2.341(6) | O8−Ag1−O9’ | 84.02(22) | O10−Ag2−O8’ | 75.43(19) |
| Ag2−O8’ | 3.044(76) | O8−Ag1−O10’ | 66.14(2) | O10−Ag2−O9’ | 92.5(2) |
| Ag2−O9’ | 2.582(6) | O9’−Ag1−O10’ | 43.63(17) | O8’−Ag2−O9’ | 49.82(18) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumitraș, D.; Gal, E.; Silvestru, C.; Pop, A. Metal Complexes Containing Homoleptic Diorganoselenium(II) Ligands: Synthesis, Characterization and Investigation of Optical Properties. Molecules 2024, 29, 792. https://doi.org/10.3390/molecules29040792
Dumitraș D, Gal E, Silvestru C, Pop A. Metal Complexes Containing Homoleptic Diorganoselenium(II) Ligands: Synthesis, Characterization and Investigation of Optical Properties. Molecules. 2024; 29(4):792. https://doi.org/10.3390/molecules29040792
Chicago/Turabian StyleDumitraș, Darius, Emese Gal, Cristian Silvestru, and Alexandra Pop. 2024. "Metal Complexes Containing Homoleptic Diorganoselenium(II) Ligands: Synthesis, Characterization and Investigation of Optical Properties" Molecules 29, no. 4: 792. https://doi.org/10.3390/molecules29040792
APA StyleDumitraș, D., Gal, E., Silvestru, C., & Pop, A. (2024). Metal Complexes Containing Homoleptic Diorganoselenium(II) Ligands: Synthesis, Characterization and Investigation of Optical Properties. Molecules, 29(4), 792. https://doi.org/10.3390/molecules29040792