
An Update of Kaempferol Protection against Brain Damage Induced by Ischemia-Reperfusion and by 3-Nitropropionic Acid

 ,
,  ,
,  , , and
, , and 
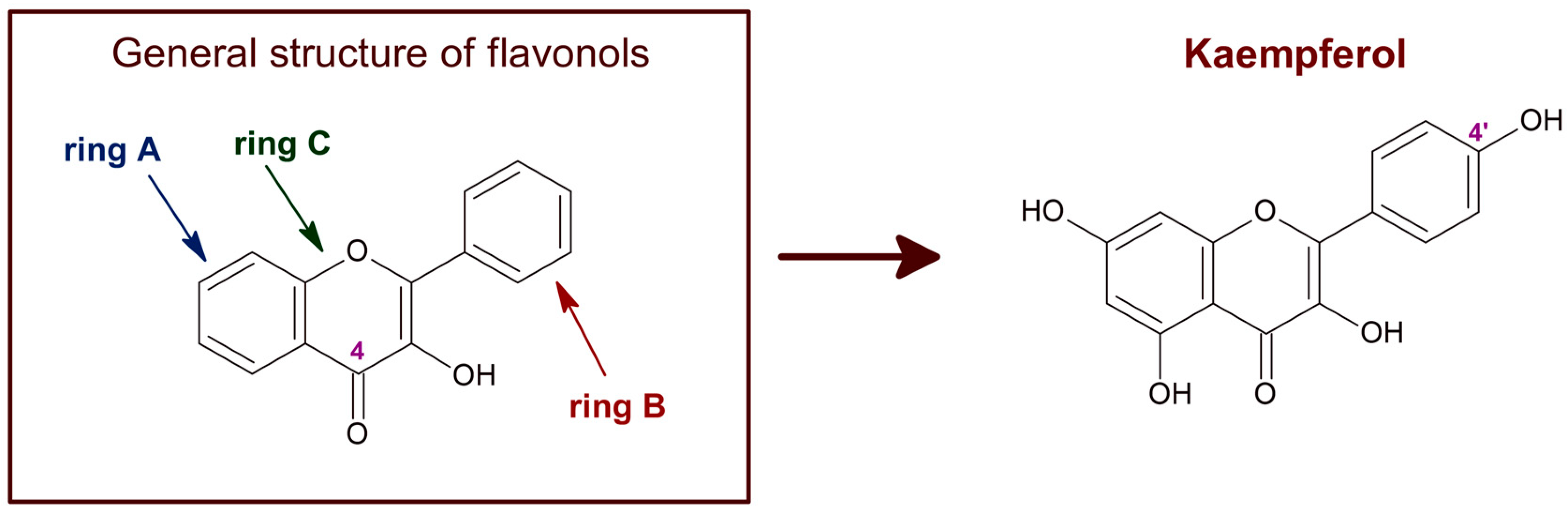{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
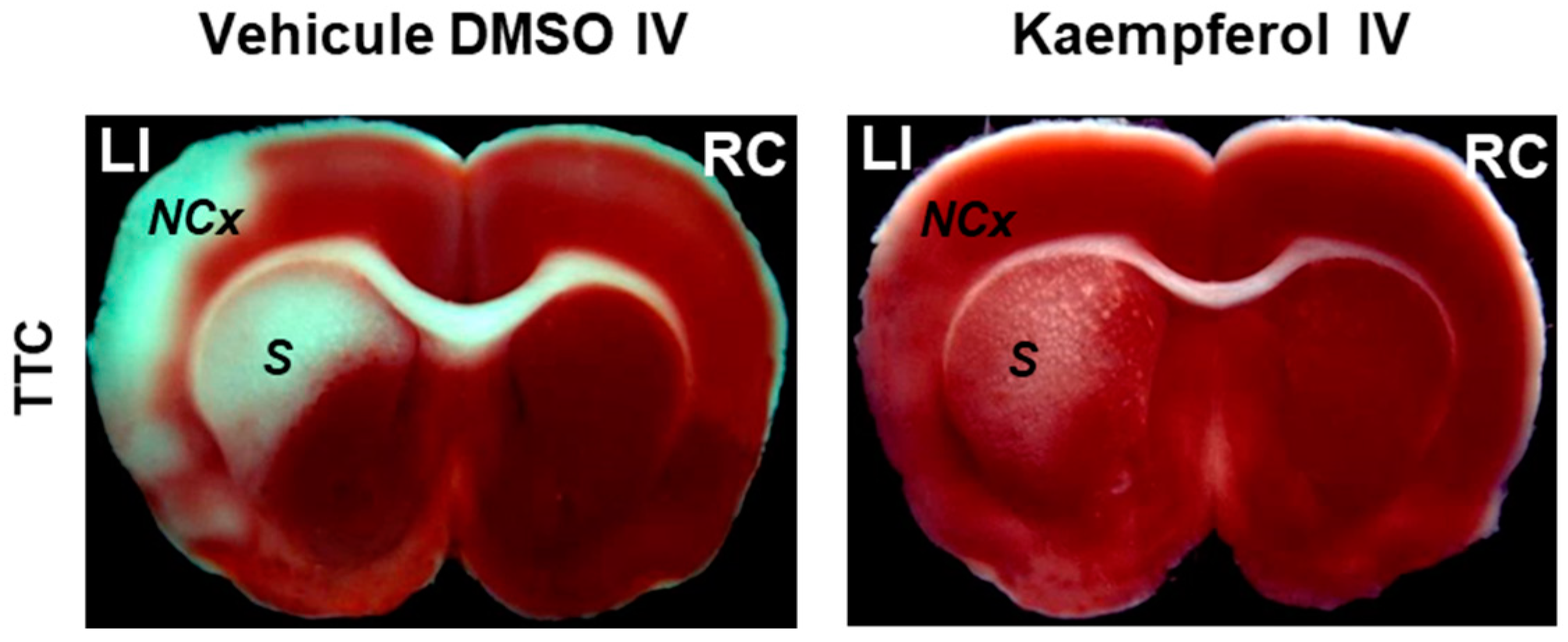
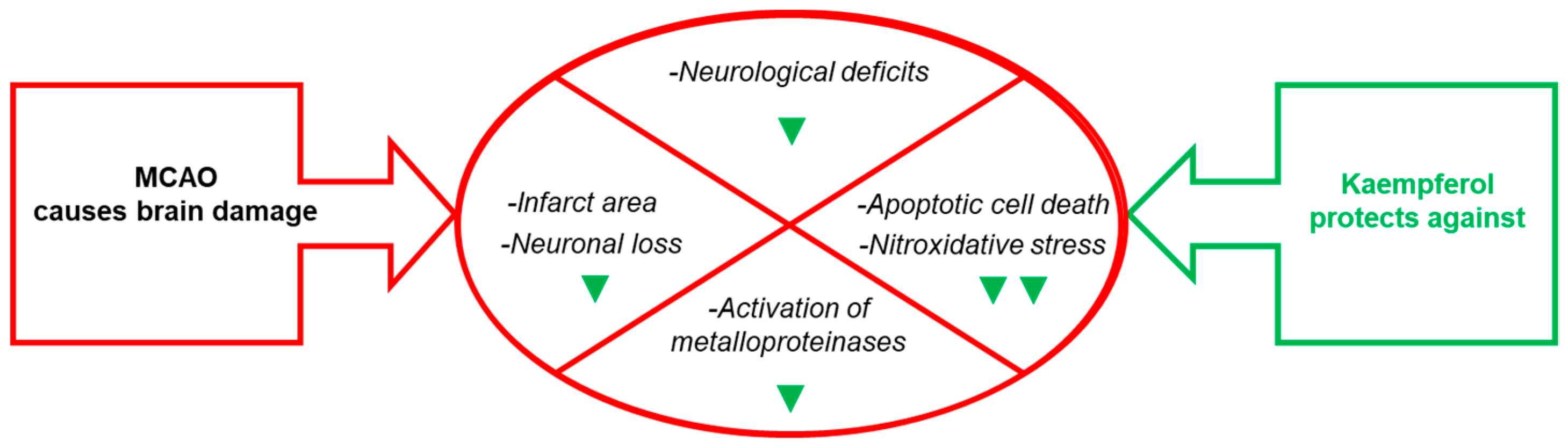
2. Kaempferol Administration Efficiently Prevents Brain Damage Induced by Ischemic Insults
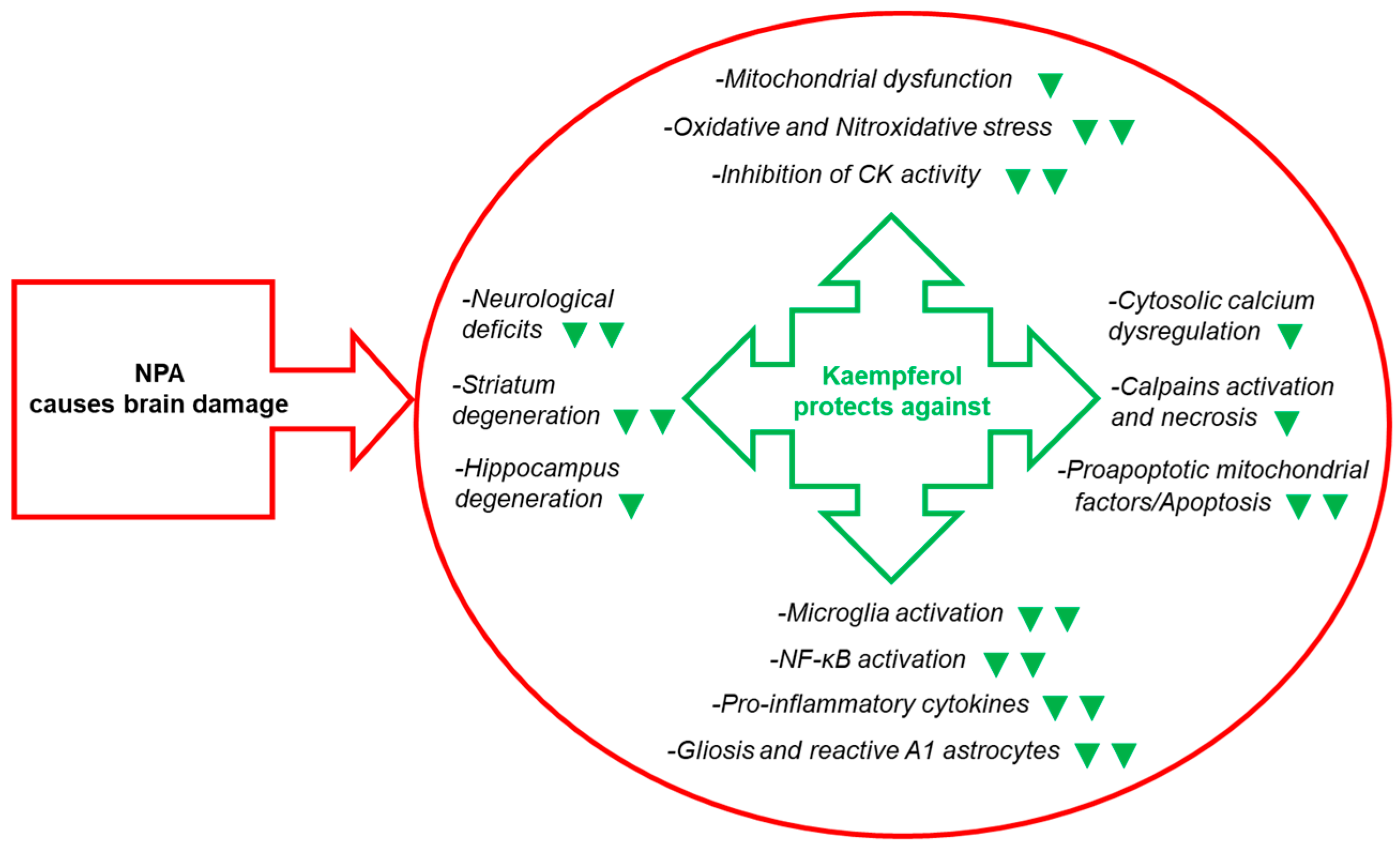
3. Kaempferol Administration Efficiently Prevents Brain Damage Induced by 3-Nitropropionic Acid
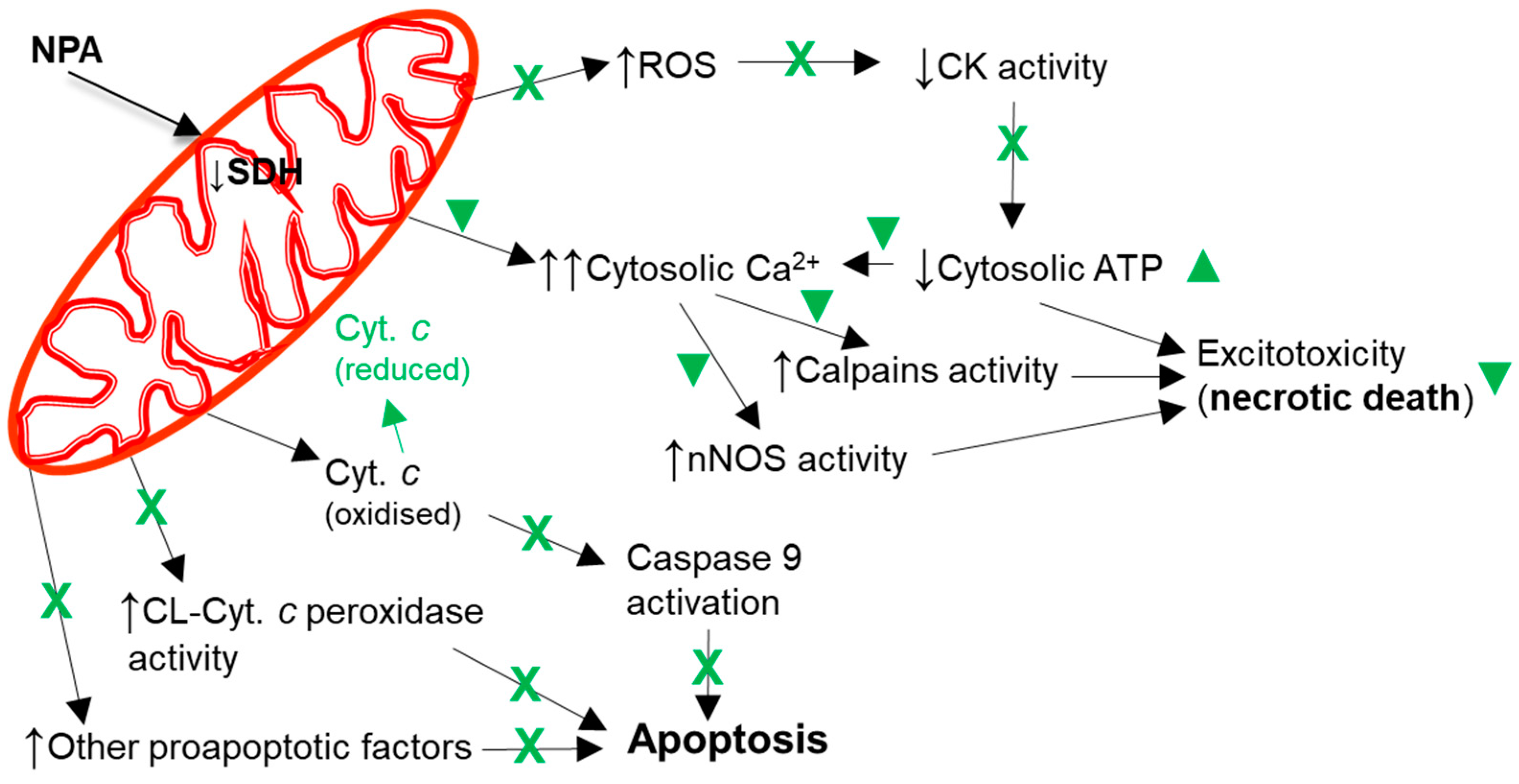
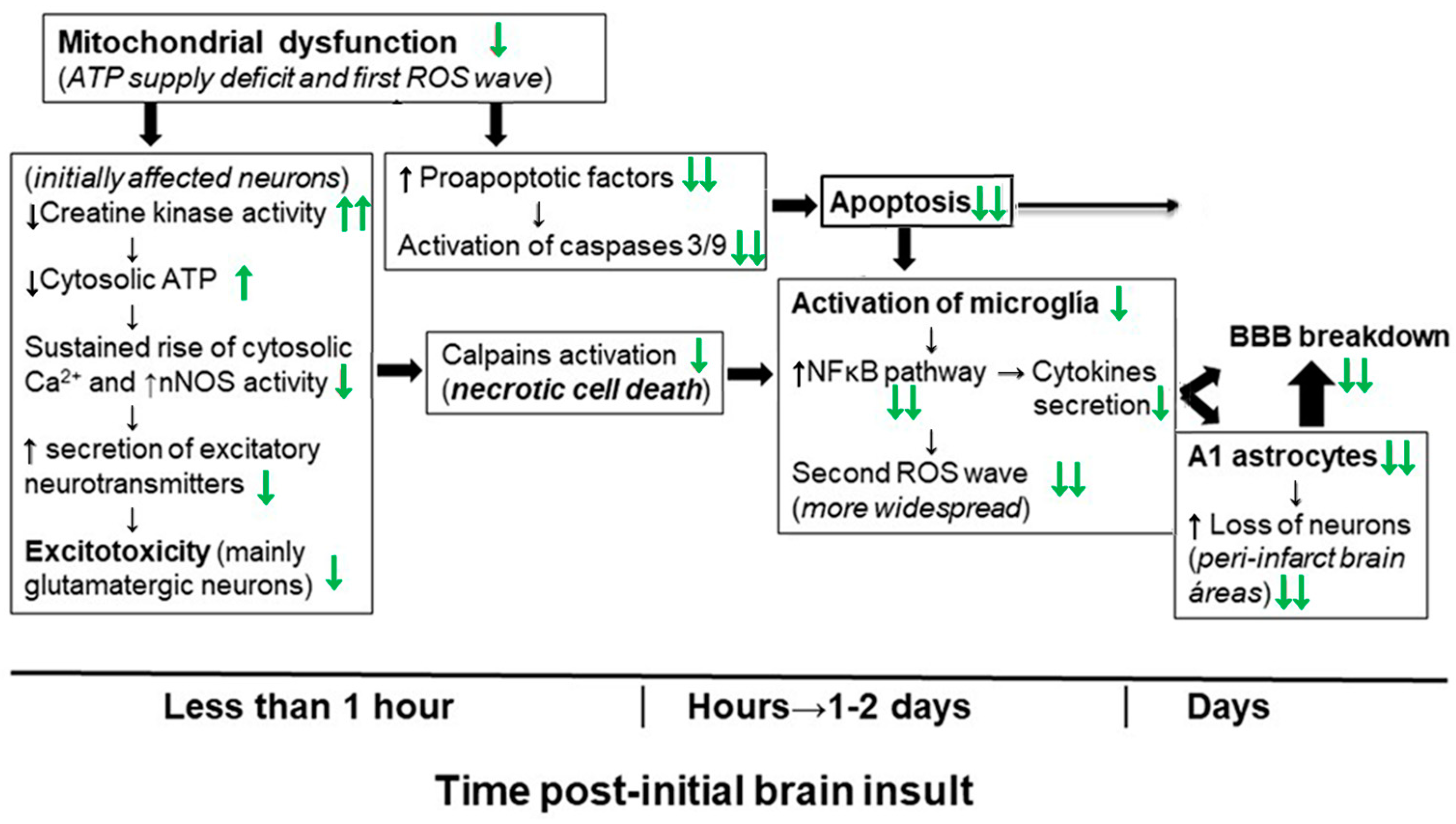
4. Molecular and Cellular Mechanisms That Contribute to Kaempferol Protection against the Brain Damage Produced by Ischemia-Reperfusion and by NPA Administration
5. Conclusions
6. Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| C1q | complement component 1q |
| GSH | reduced glutathione |
| HD | Huntington’s disease |
| IP | intraperitoneal |
| IV | intravenous |
| MCAO | middle cerebral artery occlusion |
| NF-κB | nuclear factor kappa light-chain enhancer of activated B cells |
| NOS (eNOS, iNOS and nNOS) | nitric oxide synthase (endotelial, inducible and neuronal isoforms) |
| NPA | 3-nitropropionic acid |
| ROS | reactive oxygen species |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| TUNEL | terminal deoxyribonucleotidyl transferase-mediated dUTP-fluorescein nick-end labeling |
References
- Gutierrez-Merino, C.; Lopez-Sanchez, C.; Lagoa, R.; Samhan-Arias, A.K.; Bueno, C.; Garcia-Martinez, V. Neuroprotective actions of flavonoids. Curr. Med. Chem. 2011, 18, 1195–1212. [Google Scholar] [CrossRef] [PubMed]
- Lagoa, R.; Samhan-Arias, A.K.; Gutierrez-Merino, C. Correlation between the potency of flavonoids for cytochrome c reduction and inhibition of cardiolipin-induced peroxidase activity. BioFactors 2017, 43, 451–468. [Google Scholar] [CrossRef]
- Harborne, J.B. Nature, distribution and function of plant flavonoids. Prog. Clin. Biol. Res. 1986, 213, 15–24. [Google Scholar] [PubMed]
- Li, Z.; Lee, H.W.; Liang, X.; Liang, D.; Wang, Q.; Huang, D.; Ong, C.N. Profiling of phenolic compounds and antioxidant activity of 12 cruciferous vegetables. Molecules 2018, 23, 1139. [Google Scholar] [CrossRef] [PubMed]
- Kluska, M.; Juszczak, M.; Żuchowski, J.; Stochmal, A.; Woźniak, K. Kaempferol and its glycoside derivatives as modulators of etoposide activity in HL-60 cells. Int. J. Mol. Sci. 2021, 22, 3520. [Google Scholar] [CrossRef] [PubMed]
- Rice-Evans, C. Flavonoid antioxidants. Curr. Med. Chem. 2001, 8, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; Pérez-Guerrero, C.; López-Lázaro, M. A review on the dietary flavonoid kaempferol. Mini Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Chen, Y.C. A review of the dietary flavonoid, kaempferol on human health and cancer chemoprevention. Food Chem. 2013, 138, 2099–2107. [Google Scholar] [CrossRef]
- Imran, M.; Salehi, B.; Sharifi-Rad, J.; Aslam Gondal, T.; Saeed, F.; Imran, A.; Shahbaz, M.; Tsouh Fokou, P.V.; Umair Arsha, M.; Khan, H.; et al. Kaempferol: A key emphasis to its anticancer potential. Molecules 2019, 24, 2277. [Google Scholar] [CrossRef]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef]
- Williamson, G.; Manach, C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am. J. Clin. Nutr. 2005, 81, 243S–255S. [Google Scholar] [CrossRef]
- Walle, T. Absorption and metabolism of flavonoids. Free Radic. Biol. Med. 2004, 36, 829–837. [Google Scholar] [CrossRef]
- Kuhnle, G.; Spencer, J.P.E.; Schroeter, H.; Shenoy, B.; Debnam, E.S.; Srai, S.K.S.; Rice-Evans, C.; Hahn, U. Epicatechin and catechin are O-methylated and glucuronidated in the small intestine. Biochem. Biophys. Res. Commun. 2000, 277, 507–512. [Google Scholar] [CrossRef]
- Moon, J.H.; Nakata, R.; Oshima, S.; Inakuma, T.; Terao, J. Accumulation of quercetin conjugates in blood plasma after the short-term ingestion of onion by women. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R461–R467. [Google Scholar] [CrossRef] [PubMed]
- Vafeiadou, K.; Vauzour, D.; Rodriguez-Mateos, A.; Whiteman, M.; Williams, R.J.; Spencer, J.P.E. Glial metabolism of quercetin reduces its neurotoxic potential. Arch. Biochem. Biophys. 2008, 478, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Morand, C.; Crespy, V.; Demigné, C.; Texier, O.; Régérat, F.; Rémésy, C. Quercetin is recovered in human plasma as conjugated derivatives which retain antioxidant properties. FEBS Lett. 1998, 426, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Moon, J.H.; Tsushida, T.; Nagao, A.; Terao, J. Inhibitory effect of quercetin metabolites and their related derivatives on copper ion induced lipid peroxidation in human low-density lipoprotein. Arch. Biochem. Biophys. 1999, 372, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Tsushida, T.; Nakahara, K.; Terao, J. Identification of quercetin 3-O-D-glucuronide as an antioxidative metabolite in rat plasma after oral administration of quercetin. Free Radic. Biol. Med. 2001, 30, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Shafek, R.E.; Shafik, N.H.; Michael, H.N. Antibacterial and antioxidant activities of two new kaempferol glycosides isolated from Solenostemma argel stem extract. Asian J. Plant Sci. 2012, 11, 143–147. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Fang, X.; Ge, L.; Cao, F.; Zhao, L.; Wang, Z.; Xiao, W. Antitumor, antioxidant and anti-inflammatory activities of kaempferol and its corresponding glycosides and the enzymatic preparation of kaempferol. PLoS ONE 2018, 13, e0197563. [Google Scholar] [CrossRef] [PubMed]
- Bors, W.; Michel, C.; Schikora, S. Interactions of flavonoids with ascorbate and determination of their univalent redox potentials: A pulse radiolysis study. Free Radic. Biol. Med. 1995, 19, 45–52. [Google Scholar] [CrossRef] [PubMed]
- JØrgensen, L.V.; Skibsted, L.H. Flavonoid deactivation of ferrylmyoglobin in relation to ease of oxidation as determined by cyclic voltammetry. Free Rad. Res. 1998, 28, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Husain, S.R.; Cillard, J.; Cillard, P. Hydroxyl radical scavenging activity of flavonoids. Phytochemistry 1987, 26, 2489–2491. [Google Scholar] [CrossRef]
- Sichel, G.; Corsaro, C.; Scalia, M.; Di Bilio, A.J.; Bonomo, R.P. In vitro scavenger activity of some flavonoids and melanins against O2−. Free Radic. Biol. Med. 1991, 11, 1–8. [Google Scholar] [CrossRef]
- Bors, W.; Michel, C.; Saran, M. Flavonoid antioxidants: Rate constants for reactions with oxygen radicals. Methods Enzymol. 1994, 234, 420–429. [Google Scholar] [CrossRef]
- Pannala, A.S.; Rice-Evans, C.A.; Halliwell, B.; Singh, S. Inhibition of peroxynitrite-mediated tyrosine nitration by catechin polyphenols. Biochem. Biophys. Res. Commun. 1997, 232, 164–168. [Google Scholar] [CrossRef]
- Arora, A.; Nair, M.G.; Strasburg, G.M. Structure-activity relationships for antioxidant activities of a series of flavonoids in a liposomal system. Free Radic. Biol. Med. 1998, 24, 1355–1363. [Google Scholar] [CrossRef]
- Heijnen, C.G.; Haenen, G.R.; van Acker, F.A.; van der Vijgh, W.J.; Bast, A. Flavonoids as peroxynitrite scavengers: The role of the hydroxyl groups. Toxicol. Vitr. 2001, 15, 3–6. [Google Scholar] [CrossRef]
- Heijnen, C.G.M.; Haenen, G.R.M.M.; Vekemans, J.A.J.M.; Bast, A. Peroxynitrite scavenging of flavonoids: Structure activity relationship. Environ. Toxicol. Pharmacol. 2001, 10, 199–206. [Google Scholar] [CrossRef]
- Santos, M.R.; Mira, L. Protection by flavonoids against the peroxynitrite-mediated oxidation of dihydrorhodamine. Free Radic. Res. 2004, 38, 1011–1018. [Google Scholar] [CrossRef]
- Saija, A.; Scalese, M.; Lanza, M.; Marzullo, D.; Bonina, F.; Castelli, F. Flavonoids as antioxidant agents: Importance of their interaction with biomembranes. Free Radic. Biol. Med. 1995, 19, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, S.V.; Steenken, S.; Simic, M.G.; Hara, H. Flavonoids in Health and Disease; Rice-Evans, C., Packer, L., Eds.; Marcel Dekker: New York, NY, USA, 1998; pp. 137–161. [Google Scholar]
- Ross, J.A.; Kasum, C.M. Dietary flavonoids: Bioavailability, metabolic effects and safety. Ann. Rev. Nutr. 2002, 22, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, L.; Mazza, G. Assessing antioxidant and prooxidant activity of phenolic compounds. J. Agric. Food Chem. 2000, 48, 3597–3604. [Google Scholar] [CrossRef] [PubMed]
- Veitch, N.C.; Grayer, R.J. Flavonoids and their glycosides, including anthocyanins. Nat. Prod. Rep. 2008, 25, 555–611. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, J.A.; Day, A.J.; Morgan, M.R. Experimental determination of octanol-water partition coefficients of quercetin and related flavonoids. J. Agric. Food Chem. 2005, 53, 4355–4360. [Google Scholar] [CrossRef]
- Paulke, A.; Schubert-Zsilavecz, M.; Wurglics, M. Determination of St. John’s wort flavonoid-metabolites in rat brain through high performance liquid chromatography coupled with fluorescence detection. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006, 832, 109–113. [Google Scholar] [CrossRef]
- Rangel-Ordóñez, L.; Nöldner, M.; Schubert-Zsilavecz, M.; Wurglics, M. Plasma levels and distribution of flavonoids in rat brain after single and repeated doses of standardized Ginkgo biloba extract EGb 761®. Planta Med. 2010, 76, 1683–1690. [Google Scholar] [CrossRef]
- Palmer, A.M. The role of the blood-CNS barrier in CNS disorders and their treatment. Neurobiol. Dis. 2010, 37, 3–12. [Google Scholar] [CrossRef]
- Samhan-Arias, A.K.; Martín-Romero, F.J.; Gutiérrez-Merino, C. Kaempferol blocks oxidative stress in cerebellar granule cells and reveals a key role for reactive species production at the plasma membrane in the commitment to apoptosis. Free Radic. Biol. Med. 2004, 37, 48–61. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Martin-Romero, F.J.; Sun, F.; Luis, L.; Samhan-Arias, A.K.; Garcia-Martinez, V.; Gutierrez-Merino, C. Blood micromolar concentrations of kaempferol afford protection against ischemia/reperfusion induced damage in rat brain. Brain Res. 2007, 1182, 123–137. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Martín-Romero, F.J.; Sun, F.; Luis, L.; Samhan-Arias, A.K.; García-Martínez, V.; Gutierrez-Merino, C. Intravenous injections of kaempferol afford protection against rat brain damage induced by transient focal ischemia. In Proceedings of the SFFR-Europe 2008; Grune, T., Ed.; Medimond, S.r.l.: Bologna, Italy, 2008; pp. 59–62. ISBN 978-88-7587-438-4. [Google Scholar]
- Lagoa, R.; Lopez-Sanchez, C.; Samhan-Arias, A.K.; Gañán, C.M.; García-Martínez, V.; Gutierrez-Merino, C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. J. Neurochem. 2009, 111, 473–487. [Google Scholar] [CrossRef]
- Lagoa, R.; Lopez-Sanchez, C.; Samhan-Arias, A.K.; Gañán, C.M.; García-Martínez, V.; Gutierrez-Merino, C. Neuroprotective effects of kaempferol in the 3-nitropropionic acid model of Huntington’s disease. In Free Radicals, Health and Lifestyle (Proceedings of the SFFR-Europe 2009); Caporossi, D., Pigozzi, F., Sabatini, S., Eds.; Medimond, S.r.l.: Bologna, Italy, 2009; pp. 85–88. ISBN 978-88-7587-515-2. [Google Scholar]
- Lagoa, R.; Graziani, I.; Lopez-Sanchez, C.; Garcia-Martinez, V.; Gutierrez-Merino, C. Complex I and cytochrome c are molecular targets of flavonoids that inhibit hydrogen peroxide production by mitochondria. BBA Bioenerg. 2011, 1807, 1562–1572. [Google Scholar] [CrossRef]
- Lagoa, R.; Gutierrez-Merino, C. Cytochrome c reducing agents and antiapoptotic action of antioxidant. In Cytochrome C: Roles and Therapeutic Implications; Arias, N., Ed.; Nova Science Publishers: Hauppage, NY, USA, 2019; Chapter 1; pp. 1–49. ISBN 978-1-53614-907-4. [Google Scholar]
- Lopez-Sanchez, C.; Garcia-Martinez, V.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Gutierrez-Merino, C. Early reactive A1 astrocytes induction by the neurotoxin 3-nitropropionic acid in rat brain. Int. J. Mol. Sci. 2020, 21, 3609. [Google Scholar] [CrossRef]
- Lopez-Sanchez, C.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Garcia-Martinez, V.; Gutierrez-Merino, C. Kaempferol prevents the activation of complement C3 protein and the generation of reactive A1 astrocytes that mediate rat brain degeneration induced by 3-nitropropionic acid. Food Chem. Toxicol. 2022, 164, 113017. [Google Scholar] [CrossRef]
- Broussalis, E.; Killer, M.; McCoy, M.; Harrer, A.; Trinka, E.; Kraus, J. Current therapies in ischemic stroke. Part, A. Recent developments in acute stroke treatment and in stroke prevention. Drug Discov. Today 2012, 17, 296–309. [Google Scholar] [CrossRef]
- Varghese, C.; Oyere, O.; Cowan, M.; Davis, S.; Norrving, B. World Health Organization. Stroke 2016, 47, e210. [Google Scholar] [CrossRef][Green Version]
- Iadecola, C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci. 1997, 20, 132–139. [Google Scholar] [CrossRef]
- Zhang, S.; Qi, Y.; Xu, Y.; Han, X.; Peng, J.; Liu, K.; Sun, C.K. Protective effect of flavonoid-rich extract from Rosa laevigataMichx on cerebral ischemia- reperfusion injury through suppression of apoptosis and inflammation. Neurochem. Int. 2013, 63, 522–532. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, H.Y.; Wu, B.; Cheng, C.Y.; Xiao, W.; Wang, Z.Z.; Yang, Y.Y.; Li, P.; Yang, H. Ginkgolide K attenuates neuronal injury after ischemic stroke by inhibiting mitochondrial fission and GSK-3beta-dependent increases in mitochondrial membrane permeability. Oncotarget 2017, 8, 44682–44693. [Google Scholar] [CrossRef]
- Li, Z.; Yulei, J.; Yaqing, J.; Jinmin, Z.; Xinyong, L.; Jing, G.; Min, L. Protective effects of tetramethylpyrazine analogue Z-11 on cerebral ischemia reperfusion injury. Eur. J. Pharmacol. 2019, 844, 156–164. [Google Scholar] [CrossRef]
- Xie, L.; Wang, Z.; Li, C.; Yang, K.; Liang, Y. Protective effect of nicotinamide adenine dinucleotide (NAD+) against spinal cord ischemia-reperfusion injury via reducing oxidative stress-induced neuronal apoptosis. J. Clin. Neurosci. 2017, 36, 114–119. [Google Scholar] [CrossRef]
- Fu, J.; Sun, H.; Zhang, Y.; Xu, W.; Wang, C.; Fang, Y.; Zhao, J. Neuroprotective effects of luteolin against spinal cord ischemia-reperfusion injury by attenuation of oxidative stress, inflammation, and apoptosis. J. Med. Food 2018, 21, 13–20. [Google Scholar] [CrossRef]
- Hartings, J.A.; Rolli, M.L.; Lu, X.C.M.; Tortella, F.C. Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: Relation to infarct growth and neuroprotection. J. Neurosci. 2003, 23, 11602–11610. [Google Scholar] [CrossRef]
- Hossmann, K.A. Pathophysiology and therapy of experimental stroke. Cell. Mol. Neurobiol. 2006, 26, 1057–1083. [Google Scholar] [CrossRef]
- Chan, P.H. Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 2001, 21, 2–14. [Google Scholar] [CrossRef]
- Saito, A.; Hayashi, T.; Okuno, S.; Nishi, T.; Chan, P.H. Oxidative stress is associated with XIAP and Smac/DIABLO signaling pathways in mouse brains after transient focal cerebral ischemia. Stroke 2004, 35, 1443–1448. [Google Scholar] [CrossRef]
- Romanic, A.M.; White, R.F.; Arleth, A.J.; Ohlstein, E.H.; Barone, F.C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: Inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 1998, 9, 1020–1030. [Google Scholar] [CrossRef]
- Heo, J.H.; Lucero, J.; Abumiya, T.; Koziol, J.A.; Copeland, B.R.; del Zoppo, G.J. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 624–633. [Google Scholar] [CrossRef]
- Asahi, M.; Asahi, K.; Jung, J.C.; del Zoppo, G.J.; Fini, M.E.; Lo, E.H. Role for matrix metalloproteinase 9 after focal cerebral ischemia. Effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 2000, 20, 1681–1689. [Google Scholar] [CrossRef]
- Gu, Z.Z.; Cui, J.; Brown, S.; Fridman, R.; Mobashery, S.; Strongin, A.Y.; Lipton, S.A. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J. Neurosci. 2005, 25, 6401–6408. [Google Scholar] [CrossRef]
- Krajewski, S.; Krajewska, M.; Ellerby, L.M.; Welsh, K.; Xie, Z.H.; Deveraux, Q.L.; Salvesen, G.S.; Bredesen, D.E.; Rosenthal, R.E.; Fiskum, G.; et al. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 5752–5757. [Google Scholar] [CrossRef]
- Cho, S.; Liu, D.; Gonzales, C.; Zaleska, M.M.; Wood, A. Temporal assessment of caspase activation in experimental models of focal and global ischemia. Brain Res. 2003, 982, 146–155. [Google Scholar] [CrossRef]
- Saleem, S.; Zhuang, H.; Biswal, S.; Christen, Y.; Doré, S. Ginkgo biloba extract neuroprotective action is dependent on heme oxygenase 1 in ischemic reperfusion brain injury. Stroke 2008, 39, 3389–3396. [Google Scholar] [CrossRef]
- Hong, J.T.; Yen, J.H.; Wang, L.; Lo, Y.H.; Chen, Z.T.; Wu, M.J. Regulation of heme oxygenase-1 expression and MAPK pathways in response to kaempferol and rhamnocitrin in PC12 cells. Toxicol. Appl. Pharmacol. 2009, 237, 59–68. [Google Scholar] [CrossRef]
- Gao, S.S.; Choi, B.M.; Chen, X.Y.; Zhu, R.Z.; Kim, Y.; So, H.; Park, R.; Sung, M.; Kim, B.R. Kaempferol suppresses cisplatin-induced apoptosis via inductions of heme oxygenase-1 and glutamate-cysteine ligase catalytic subunit in HEI-OC1 cells. Pharm. Res. 2010, 27, 235–245. [Google Scholar] [CrossRef]
- Li, R.P.; Guo, M.L.; Zhang, G.; Xu, X.F.; Li, Q. Neuroprotection of nicotiflorin in permanent focal cerebral ischemia and in neuronal cultures. Biol. Pharm. Bull. 2006, 29, 1868–1872. [Google Scholar] [CrossRef]
- Li, R.P.; Guo, M.L.; Zhang, G.; Xu, X.F.; Li, Q. Nicotiflorin reduces cerebral ischemic damage and upregulates endothelial nitric oxide synthase in primarily cultured rat cerebral blood vessel endothelial cells. J. Ethnopharmacol. 2006, 107, 143–150. [Google Scholar] [CrossRef]
- Sun, F.; Lopez-Sanchez, C.; Martin-Romero, F.J.; Luis, L.; Gutierrez-Merino, C.; Garcia-Martinez, V. Transfemoral selective “intraluminal wiring” technique for transient middle cerebral artery occlusion in rats. J. Neurosci. Methods. 2005, 149, 82–89. [Google Scholar] [CrossRef]
- Lehrmann, E.; Christensen, T.; Zimmer, J.; Diemer, N.H.; Finsen, B. Microglial and macrophage reactions mark progressive changes and define the penumbra in the rat neocortex and striatum after transient middle cerebral artery occlusion. J. Comp. Neurol. 1997, 386, 461–476. [Google Scholar] [CrossRef]
- Yu, L.; Chen, C.; Wang, L.F.; Kuang, X.; Liu, K.; Zhang, H.; Du, J.R. Neuroprotective effect of kaempferol glycosides against brain injury and neuroinflammation, inhibiting the activation of NF-κB and STAT3 in transient focal stroke. PLoS ONE 2013, 8, e55839. [Google Scholar] [CrossRef]
- Abd El Mohsen, M.M.; Kuhnle, G.; Rechner, A.R.; Schroeter, H.; Rose, S.; Jenner, P.; Rice-Evans, C.A. Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Radic. Biol. Med. 2002, 33, 1693–1702. [Google Scholar] [CrossRef]
- Zhang, S.S.; Liu, M.; Liu, D.N.; Shang, Y.F.; Du, G.H.; Wang, Y.H. Network pharmacology analysis and experimental validation of kaempferol in the treatment of ischemic stroke by inhibiting apoptosis and regulating neuroinflammation involving neutrophils. Int. J. Mol. Sci. 2022, 23, 12694. [Google Scholar] [CrossRef]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.Y.; Lin, L.; Liu, B.L.; Liu, K.; Li, P.; Yang, H. Succinate induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef]
- Li, W.H.; Cheng, X.; Yang, Y.L.; Liu, M.; Zhang, S.S.; Wang, Y.H.; Du, G.H. Kaempferol attenuates neuroinflammation and blood brain barrier dysfunction to improve neurological deficits in cerebral ischemia/reperfusion rats. Brain Res. 2019, 1722, 146361. [Google Scholar] [CrossRef]
- Wang, S.; Xu, H.; Xin, Y.; Li, M.; Fu, W.; Wang, Y.; Lu, Z.; Yu, X.; Sui, D. Neuroprotective effects of Kaempferide-7-O-(4″-O-acetylrhamnosyl)-3-O-rutinoside on cerebral ischemia-reperfusion injury in rats. Eur. J. Pharmacol. 2016, 788, 335–342. [Google Scholar] [CrossRef]
- Manach, C.; Régérat, F.; Texier, G.; Agullo, G.; Demigné, C.; Rémésy, C. Bioavailability, metabolism and physiological impact of 4-oxo-flavonoids. Nutr. Res. 1996, 16, 517–544. [Google Scholar] [CrossRef]
- Hollman, P.C.; Katan, M.B. Absorption, metabolism and health effects of dietary flavonoids in man. Biomed. Pharmacother. 1997, 51, 305–310. [Google Scholar] [CrossRef]
- Hackett, A.M. The metabolism of flavonoid compounds in mammals. Prog. Clin. Biol. Res. 1986, 213, 177–194. [Google Scholar]
- Ishige, K.; Schubert, D.; Sagara, Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001, 30, 433–446. [Google Scholar] [CrossRef]
- Benveniste, H.; Drejer, J.; Schousboe, A.; Diemer, N.H. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984, 43, 1369–1374. [Google Scholar] [CrossRef]
- Szatkowski, M.; Atwell, D. Triggering and execution of neuronal death in brain ischemia: Two phases of glutamate release by different mechanisms. Trends Neurosci. 1994, 17, 359–365. [Google Scholar] [CrossRef]
- Uzdensky, A.B. Apoptosis regulation in the penumbra after ischemic stroke: Expression of pro- and antiapoptotic proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef]
- Contestabile, A. Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 2002, 1, 41–55. [Google Scholar] [CrossRef]
- Gutierrez-Merino, C.; Marques-da-Silva, D.; Fortalezas, S.; Samhan-Arias, A.K. The critical role of lipid rafts nanodomains in the cross-talk between calcium and reactive oxygen and nitrogen species in cerebellar granule neurons apoptosis by extracellular potassium deprivation. AIMS Mol. Sci. 2016, 3, 12–29. [Google Scholar] [CrossRef]
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190. [Google Scholar] [CrossRef]
- Ludolph, A.C.; He, F.; Spencer, P.S.; Hammerstad, J.; Sabri, M. 3-Nitropropionic acid—Exogenous animal neurotoxin and possible human striatal toxin. Can. J. Neurol. Sci. 1991, 18, 492–498. [Google Scholar] [CrossRef]
- He, F.; Zhang, S.; Qian, F.; Zhang, C. Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid). Neurology 1995, 45, 2178–2183. [Google Scholar] [CrossRef]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef]
- Brouillet, E.; Jenkins, B.G.; Hyman, B.T.; Ferrante, R.J.; Kowall, N.W.; Srivastava, R.; Roy, D.S.; Rosen, B.R.; Beal, M.F. Age dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J. Neurochem. 1993, 60, 356–359. [Google Scholar] [CrossRef]
- Brouillet, E.; Hantraye, P.; Ferrante, R.J.; Dolan, R.; Leroy-Willig, A.; Kowall, N.W.; Beal, M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl. Acad. Sci. USA 1995, 92, 7105–7109. [Google Scholar] [CrossRef]
- Brouillet, E.; Conde, F.; Beal, M.F.; Hantraye, P. Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Tsang, T.M.; Haselden, J.N.; Holmes, E. Metabolomic characterization of the 3-nitropropionic acid rat model of Huntington’s disease. Neurochem. Res. 2009, 34, 1261–1271. [Google Scholar] [CrossRef]
- Menze, E.; Esmat, A.; Tadros, M.G.; Abdel-Naim, A.B.; Khalifa, A.E. Genistein improves 3-NPA-induced memory impairment in ovariectomized rats: Impact of its antioxidant, anti-inflammatory and acetylcholinesterase modulatory properties. PLoS ONE 2015, 10, e0117223. [Google Scholar] [CrossRef]
- Ho, A.K.; Sahakian, B.J.; Brown, R.G.; Barker, R.A.; Hodges, J.R.; Ané, M.N.; Snowden, J.; Thompson, J.; Esmonde, T.; Gentry, R.; et al. Profile of cognitive progression in early Huntington’s disease. Neurology 2003, 61, 1702–1706. [Google Scholar] [CrossRef]
- Phillips, W.; Shannon, K.M.; Barker, R.A. The current clinical management of Huntington’s disease. Mov. Disord. 2008, 23, 1491–1504. [Google Scholar] [CrossRef]
- Hylin, J.W.; Matsumoto, H. Inhibition of succinic dehydrogenase by 3-nitropropanoate. Toxicol. Appl. Pharmacol. 1964, 6, 168–171. [Google Scholar] [CrossRef]
- Alston, T.A.; Mela, L.; Bright, H.J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef]
- Huang, L.S.; Sun, G.; Cobessi, D.; Wang, A.C.; Shen, J.T.; Tung, E.Y.; Anderson, V.E.; Berry, E.A. 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J. Biol. Chem. 2006, 281, 5965–5972. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Derr-Yellin, E.; Nicklas, W.J. NMDA receptor involvement in toxicity to dopamine neurons in vitro caused by the succinate dehydrogenase inhibitor 3-nitropropionic acid. J. Neurochem. 1995, 64, 455–458. [Google Scholar] [CrossRef]
- Nasr, P.; Gursahani, H.I.; Pang, Z.; Bondada, V.; Lee, J.; Hadley, R.W.; Geddes, J.W. Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem. Int. 2003, 43, 89–99. [Google Scholar] [CrossRef]
- Beal, M.F.; Ferrante, R.J.; Henshaw, R.; Matthews, R.T.; Chan, P.H.; Kowall, N.W.; Epstein, C.J.; Schulz, J.B. 3-Nitropropionic acid neurotoxicity is attenuated in copper/zinc superoxide dismutase transgenic mice. J. Neurochem. 1995, 65, 919–922. [Google Scholar] [CrossRef]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Schultz, J.B.; Henshaw, D.R.; MacGarvey, U.A.; Beal, M.F. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem. Int. 1996, 29, 167–171. [Google Scholar] [CrossRef]
- Kim, G.W.; Copin, J.C.; Kawase, M.; Chen, S.F.; Sato, S.; Gobbel, G.T.; Chan, P.H. Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J. Cereb. Blood Flow Metab. 2000, 20, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, T.R.; Carvalho, A.C.P.; Jurkiewicz, A.; Frussa-Filho, R.; Smaili, S.S. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J. Neurochem. 2004, 88, 1220–1228. [Google Scholar] [CrossRef]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.; Jenkins, B.G.; Ferrante, R.J.; Rosen, B.R.; Kaddurah-Daouk, R.; Beal, M.F. Neuroprotective effects of creatine and cyclocreatine in animal models of Huntington’s disease. J. Neurosci. 1998, 18, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Blamire, A.M.; Manners, D.N.; Rajagopalan, B.; Styles, P.; Schapira, A.H.V.; Warner, T.T. High-dose creatine therapy for Huntington disease: A 2-year clinical and MRS study. Neurology 2005, 64, 1655–1656. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef]
- Aksenova, M.V.; Aksenov, M.Y.; Payne, R.M.; Trojanowski, J.Q.; Schmidt, M.L.; Carney, J.M.; Butterfield, D.A.; Markesbery, W.R. Oxidation of cytosolic proteins and expression of creatine kinase BB in frontal lobe in different neurodegenerative disorders. Dement. Geriatr. Cogn. Disord. 1999, 10, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, M.; Aksenova, M.; Butterfield, D.A.; Markesbery, W.R. Oxidative modification of creatine kinase BB in Alzheimer’s disease brain. J. Neurochem. 2000, 74, 2520–2527. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000, 23, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Merino, C.; Marques-da-Silva, D.; Fortalezas, S.K.A. Cytosolic calcium homeostasis in neurons—Control systems, modulation by reactive oxygen and nitrogen species, and space and time fluctuations. In Neurochemistry; Heinbockel, T., Ed.; InTech.: Rijeka, Croatia, 2014; Chapter 3; pp. 59–110. [Google Scholar] [CrossRef]
- Darley-Usmar, V.; Wiseman, H.; Halliwell, B. Nitric oxide and oxygen radicals: A question of balance. FEBS Lett. 1995, 369, 131–135. [Google Scholar] [CrossRef]
- Sies, H.; Arteel, G.E. Interaction of peroxynitrite with selenoproteins and glutathione peroxidase mimics. Free Radic. Biol. Med. 2000, 28, 1451–1455. [Google Scholar] [CrossRef]
- Chen, J.; Berry, M.J. Selenium and selenoproteins in the brain and brain diseases. J. Neurochem. 2003, 86, 1–12. [Google Scholar] [CrossRef]
- Gutierrez-Merino, C. Redox modulation of neuronal calcium homeostasis and its deregulation by reactive oxygen species. In Free Radicals in Biology and Medicine; Gutiérrez-Merino, C., Leeuwenburgh, C., Eds.; Research Signpost: Kerala, India, 2008; pp. 67–101. [Google Scholar]
- Hidalgo, C.; Donoso, P. Crosstalk between calcium and redox signaling: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1275–1312. [Google Scholar] [CrossRef]
- Nishino, H.; Fujimoto, I.; Shimano, Y.; Hida, H.; Kumazaki, M.; Fukuda, A. 3-Nitropropionic acid produces striatum selective lesions accompanied by iNOS expression. J. Chem. Neuroanat. 1996, 10, 209–212. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Park, J.E.; Hong, N.H.; Im, W.S.; Kang, L.; Han, Z.; Jung, K.H.; Kim, M.W.; Kim, M. Atorvastatin attenuates mitochondrial toxin-induced striatal degeneration, with decreasing iNOS/c-Jun levels and activating ERK/Akt pathways. J. Neurochem. 2008, 104, 1190–1200. [Google Scholar] [CrossRef]
- Deshpande, S.B.; Hida, H.; Takei-Io, N.; Masuda, T.; Baba, H.; Nishino, H. Involvement of nitric oxide in 3-nitropropionic acid induced striatal toxicity in rats. Brain Res. 2006, 1108, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Stewart, V.C.; Sharpe, M.A.; Clark, J.B.; Heales, S.J.R. Astrocyte-derived nitric oxide causes both reversible and irreversible damage to the neuronal mitochondrial respiratory chain. J. Neurochem. 2002, 75, 694–700. [Google Scholar] [CrossRef] [PubMed]
- La Fontaine, M.A.; Geddes, J.W.; Banks, A.; Butterfield, D.A. Effect of exogenous and endogenous antioxidants on 3-nitropropionic acid-induced in vivo oxidative stress and striatal lesions: Insights into Huntington’s disease. J. Neurochem. 2000, 75, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Mundo, M.N.; Silva-Adaya, D.; Maldonado, P.D.; Galvan-Arzate, S.; Andres-Martinez, L.; Perez-De La Cruz, V.; Pedraza-Chaverri, J.; Santamaria, A. S-Allylcysteine prevents the rat from 3-nitropropionic acid-induced hyperactivity, early markers of oxidative stress and mitochondrial dysfunction. Neurosci. Res. 2006, 56, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Bizat, N.; Hermel, J.M.; Boyer, F.; Jacquard, C.; Creminon, C.; Ouary, S.; Escartin, C.; Hantraye, P.; Kajewski, S.; Brouillet, E. Calpain is a major cell death effector in selective striatal degeneration induced in vivo by 3-nitropropionate: Implications for Huntington’s disease. J. Neurosci. 2003, 23, 5020–5030. [Google Scholar] [CrossRef]
- Artal-Sanz, M.; Tavernarakis, N. Proteolytic mechanisms in necrotic cell death and neurodegeneration. FEBS Lett. 2005, 579, 3287–3296. [Google Scholar] [CrossRef]
- Volbracht, C.; Chua, B.T.; Ng, C.P.; Bahr, B.A.; Hong, W.; Li, P. The critical role of calpain versus caspase activation in excitotoxic injury induced by nitric oxide. J. Neurochem. 2005, 93, 1280–1292. [Google Scholar] [CrossRef]
- Greene, J.G.; Sheu, S.S.; Gross, R.A.; Greenamyre, J.T. 3-Nitropropionic acid exacerbates N-methyl-D-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience 1998, 84, 503–510. [Google Scholar] [CrossRef]
- Calabresi, P.; Gubellini, P.; Picconi, B.; Centonze, D.; Pisani, A.; Bonsi, P.; Greengard, P.; Hipskind, R.A.; Borrelli, E.; Bernardi, G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J. Neurosci. 2001, 21, 5110–5120. [Google Scholar] [CrossRef]
- Crespo-Biel, N.; Camins, A.; Pelegrı, C.; Vilaplana, J.; Palla´s, M.; Canudas, A.M. 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci. Lett. 2007, 421, 77–81. [Google Scholar] [CrossRef]
- Akashiba, H.; Ikegaya, Y.; Nishiyama, N.; Matsuki, N. Differential involvement of cell cycle reactivation between striatal and cortical neurons in cell death induced by 3-nitropropionic acid. J. Biol. Chem. 2008, 283, 6594–6606. [Google Scholar] [CrossRef]
- Ryu, J.K.; Nagai, A.; Kim, J.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: In vitro and in vivo studies. Neurobiol. Dis. 2003, 12, 121–132. [Google Scholar] [CrossRef]
- Chakraborty, J.; Singh, R.; Dutta, D.; Naskar, A.; Rajamma, U.; Mohanakumar, K.P. Quercetin improves behavioral deficiencies, restores astrocytes and microglia, and reduces serotonin metabolism in 3-nitropropionic acid-induced rat model of Huntington’s disease. CNS Neurosci. Ther. 2014, 20, 10–19. [Google Scholar] [CrossRef]
- Jin, X.; Riew, T.R.; Kim, H.L.; Choi, J.H.; Lee, M.Y. Morphological characterization of NG2 glia and their association with neuroglial cells in the 3-nitropropionic acid–lesioned striatum of rat. Sci. Rep. 2018, 8, 5942. [Google Scholar] [CrossRef]
- Niccolini, F.; Politis, M. Neuroimaging in Huntington’s disease. World J. Radiol. 2014, 6, 301–312. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.I.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen. Res. 2020, 15, 25–29. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Ekdahl, K.N.; Wiegner, R.; Fromell, K.; Nilsson, B. Auxiliary activation of the complement system and its importance for the pathophysiology of clinical conditions. Semin Immunopathol. 2018, 40, 87–102. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Fu, Y. 3-Nitropropionic acid produces indirect excitotoxic damage to rat striatum. Neurotoxicol. Teratol. 1995, 17, 333–339. [Google Scholar] [CrossRef]
- Nishino, H.; Kumazaki, M.; Fukuda, A.; Fujimoto, I.; Shimano, Y.; Hida, H.; Sakurai, T.; Deshpande, S.B.; Shimizu, H.; Morikawa, S.; et al. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: Involvement of dopamine toxicity. Neurosci. Res. 1997, 27, 343–355. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef]
- Park, S.H.; Oh, S.R.; Jung, K.Y.; Lee, I.S.; Ahn, K.S.; Kim, J.H.; Kim, Y.S.; Lee, J.J.; Lee, H.K. Acylated flavonol glycosides with anti-complement activity from Persicaria lapathifolia. Chem. Pharm. Bull. 1999, 47, 1484–1486. [Google Scholar] [CrossRef]
- Schroeter, H.; Spencer, J.P.E.; Rice-Evans, C.; Williams, R.J. Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem. J. 2001, 358, 547–557. [Google Scholar] [CrossRef]
- Silva, B.; Oliveira, P.J.; Dias, A.; Malva, J.O. Quercetin, kaempferol and biapigenin from hypericum perforatum are neuroprotective against excitotoxic insults. Neurotox. Res. 2008, 13, 265–279. [Google Scholar] [CrossRef]
- Schroeder, E.K.; Kelsey, N.A.; Doyle, J.; Breed, E.; Bouchard, R.J.; Loucks, F.A.; Harbison, R.A.; Linseman, D.A. Green tea epigallocatechin 3-gallate accumulates in mitochondria and displays a selective antiapoptotic effect against inducers of mitochondrial oxidative stress in neurons. Antoxid. Redox Signal. 2009, 11, 469–480. [Google Scholar] [CrossRef]
- Nichols, M.; Zhang, J.; Polster, B.; Elustondo, P.; Thirumaran, A.; Pavlov, E.V.; Robertson, G.S. Synergistic neuroprotection by epicatechin and quercetin: Activation of convergent mitochondrial signaling pathways. Neuroscience 2015, 308, 75–94. [Google Scholar] [CrossRef]
- Chitturi, J.; Santhakumar, V.; Kannurpatti, S.S. Beneficial effects of kaempferol after developmental traumatic brain injury is through protection of mitochondrial function, oxidative metabolism, and neural viability. J. Neurotrauma 2019, 36, 1264–1278. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Lesnefsky, E.J.; Moghaddas, S.; Tandler, B.; Kerner, J.; Hoppel, C.L. Mitochondrial dysfunction in cardiac disease: Ischemia–reperfusion, aging, and heart failure, J. Mol. Cell. Cardiol. 2001, 33, 1065–1089. [Google Scholar] [CrossRef]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: Implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Clayton, R.; Clark, J.B.; Sharpe, M. Cytochrome c release from rat brain mitochondria is proportional to the mitochondrial functional deficit: Implications for apoptosis and neurodegenerative disease. J. Neurochem. 2005, 92, 840–849. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 877–881. [Google Scholar] [CrossRef]
- Wu, C.C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antoxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef]
- Borutaite, V.; Brown, G.C. Mitochondrial regulation of caspase activation by cytochrome oxidase and tetramethylphenylenediamine via cytosolic cytochrome c redox state. J. Biol. Chem. 2007, 282, 31124–31130. [Google Scholar] [CrossRef]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef]
- Barauskaite, J.; Grybauskiene, R.; Morkuniene, R.; Borutaite, V.; Brown, G.C. Tetramethylphenylenediamine protects the isolated heart against ischaemia-induced apoptosis and reperfusion-induced necrosis. Br. J. Pharmacol. 2011, 162, 1136–1142. [Google Scholar] [CrossRef]
- Skemiene, K.; Rakauskaite, G.; Trumbeckaite, S.; Liobikas, J.; Brown, G.C.; Borutaite, V. Anthocyanins block ischemia-induced apoptosis in the perfused heart and support mitochondrial respiration potentially by reducing cytosolic cytochrome c. Int. J. Biochem. Cell Biol. 2013, 45, 23–29. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, A.; Bayir, H.; Stoyanovsky, D.; Borisenko, G.G.; Tyurina, Y.Y.; Wipf, P.; Atkinson, J.; Greenberger, J.S.; Chapkin, R.S.; et al. Mitochondria-targeted disruptors and inhibitors of cytochrome c/cardiolipin peroxidase complexes: A new strategy in anti-apoptotic drug discovery. Mol. Nutr. Food Res. 2009, 53, 104–114. [Google Scholar] [CrossRef]
- Abe, M.; Niibayashi, R.; Koubori, S.; Moriyama, I.; Miyoshi, H. Molecular mechanisms for the induction of peroxidase activity of the cytochrome c-cardiolipin complex. Biochemistry 2011, 50, 8383–8391. [Google Scholar] [CrossRef]
- Patriarca, A.; Polticelli, F.; Piro, M.C.; Sinibaldi, F.; Mei, G.; Bari, M.; Santucci, R.; Fiorucci, L. Conversion of cytochrome c into a peroxidase: Inhibitory mechanisms and implication for neurodegenerative diseases. Arch. Biochem. Biophys. 2012, 522, 62–69. [Google Scholar] [CrossRef]
- Muenzner, J.; Toffey, J.R.; Hong, Y.N.; Pletneva, E.V. Becoming a peroxidase: Cardiolipin-induced unfolding of cytochrome c. J. Phys. Chem. B. 2013, 117, 12878–12886. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Basova, L.V.; Kurnikov, I.V.; Wang, L.; Ritov, V.B.; Belikova, N.A.; Vlasova, I.I.; Pacheco, A.A.; Winnica, D.E.; Peterson, J.; Bayir, H.; et al. Cardiolipin switch in mitochondria: Shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 2007, 46, 3423–3434. [Google Scholar] [CrossRef]
- Hanske, J.; Toffey, J.R.; Morenz, A.M.; Bonilla, A.J.; Schiavoni, K.H.; Pletneva, E.V. Conformational properties of cardiolipin-bound cytochrome c. Proc. Natl. Acad. Sci. USA 2012, 109, 125–130. [Google Scholar] [CrossRef]
- Sinibaldi, F.; Howes, B.D.; Droghetti, E.; Polticelli, F.; Piro, M.C.; Di Pierro, D.; Fiorucci, L.; Coletta, M.; Smulevich, G.; Santucci, R. Role of lysines in cytochrome c-cardiolipin interaction. Biochemistry 2013, 52, 4578–4588. [Google Scholar] [CrossRef]
- Petrosillo, G.; Ruggiero, F.M.; Pistolese, M.; Paradies, G. Reactive oxygen species generated from the mitochondrial electron transport chain induce cytochrome c dissociation from beef-heart submitochondrial particles via cardiolipin peroxidation. Possible role in the apoptosis. FEBS Lett. 2001, 509, 435–438. [Google Scholar] [CrossRef]
- Bergstrom, C.L.; Beales, P.A.; Lv, Y.; Vanderlick, T.K.; Groves, J.T. Cytochrome c causes pore formation in cardiolipin-containing membranes. Proc. Natl. Acad. Sci. USA 2013, 110, 6269–6274. [Google Scholar] [CrossRef]
- Ramassamy, C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets, Eur. J. Pharmacol. 2006, 545, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, K.; Kihara, T.; Shen, H.; Shimmyo, Y.; Niidome, T.; Sugimoto, H. Distinct mechanisms underlie distinct polyphenol-induced neuroprotection. FEBS Lett. 2006, 580, 6623–6628. [Google Scholar] [CrossRef]
- Jovanovic, S.V.; Steenken, S.; Tosic, M.; Marjanovic, B.; Simic, M.G. Flavonoids as antioxidants. J. Am. Chem. Soc. 1994, 116, 4846–4851. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Kaiserová, H.; Šimûnek, T.; van der Vijgh, W.; Bast, A.; Kvasničková, E. Flavonoids as protectors against doxorubicin cardiotoxicity: Role of iron chelation, antioxidant activity and inhibition of carbonyl reductase. Biochim. Biophys. Acta-Mol. Basis Dis. 2007, 1772, 1065–1074. [Google Scholar] [CrossRef]
- Morre, D.J.; Bridge, A.; Wu, L.Y.; Morre, D.M. Preferential inhibition by (-)-epigallocatechin-3-gallate of the cell surface NADH oxidase and growth of transformed cells in culture. Biochem. Pharmacol. 2000, 60, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Van Hoorn, D.E.; Nijveldt, R.J.; Van Leeuwen, P.A.; Hofman, Z.; M’Rabet, L.; De Bont, D.B.; Van Norren, K. Accurate prediction of xanthine oxidase inhibition based on the structure of flavonoids. Eur. J. Pharmacol. 2002, 451, 111–118. [Google Scholar] [CrossRef]
- Potapovich, A.I.; Kostyuk, V.A. Comparative study of antioxidant properties and cytoprotective activity of flavonoids. Biochemistry 2003, 68, 514–519. [Google Scholar] [CrossRef]
- Schneider, I.; Bucar, F. Lipoxygenase inhibitors from natural plant sources. Part 1: Medicinal plants with inhibitory activity on arachidonate 5-lipoxygenase and 5-lipoxygenase[sol]cyclooxygenase. Phytother. Res. 2005, 19, 81–102. [Google Scholar] [CrossRef]
- Huber, A.; Bürkle, A.; Münch, G. Degenerative Diseases of the Nervous System. In Handbook of Neurochemistry and Molecular Neurobiology; Lajtha, A.Y., Riederer, P., Mandel, S.A., Battistin, L., Eds.; Springer: Berlin, Germany, 2007; pp. 77–102. [Google Scholar]
- Dreiseitel, A.; Korte, G.; Schreier, P.; Oehme, A.; Locher, S.; Domani, M.; Hajak, G.; Sand, P.G. Berry anthocyanins and their aglycones inhibit monoamine oxidases A and B. Pharmacol. Res. 2009, 59, 306–311. [Google Scholar] [CrossRef]
- Mulabagal, V.; Lang, G.A.; DeWitt, D.L.; Dalavoy, S.S.; Nair, M.G. Anthocyanin content, lipid peroxidation and cyclooxygenase enzyme inhibitory activities of sweet and sour cherries. J. Agric. Food Chem. 2009, 57, 1239–1246. [Google Scholar] [CrossRef]
- Chen, I.F.; Breen, K. On the ability of four flavonoids, baicilein, luteolin, naringenin, and quercetin, to suppress the Fenton reaction of the iron-ATP complex. Biometals 2000, 13, 77–83. [Google Scholar] [CrossRef]
- Mira, L.; Fernandez, M.T.; Santos, M.; Rocha, R.; Florêncio, M.H.; Jennings, K.R. Interactions of flavonoids with iron and copper ions: A mechanism for their antioxidant activity. Free Radic. Res. 2002, 36, 1199–1208. [Google Scholar] [CrossRef]
- Silva, M.M.; Santos, M.R.; Caroço, G.; Rocha, R.; Justino, G.; Mira, L. Structure-antioxidant activity relationships of flavonoids: A re-examination. Free Radic. Res. 2002, 36, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Campos-Esparza, M.R.; Sanchez-Gomez, M.V.; Matute, C. Molecular mechanism of neuroprotection by two natural antioxidant polyphenols. Cell Calcium 2009, 45, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Young, A.B.; Penney, J.B.; Beal, M.F. 3-Nitropropionic acid toxicity in the striatum. J. Neurochem. 1994, 63, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Prosperetti, C.; Barone, I.; Rossi, S.; Picconi, B.; Tscherter, A.; De Chiara, V.; Bernardi, G.; Calabresi, P. NR2B-containing NMDA receptors promote the neurotoxic effects of 3-nitropropionic acid but not of rotenone in the striatum. Exp. Neurol. 2006, 202, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Fatokun, A.A.; Smith, R.A.; Stone, T.W. Resistance to kynurenic acid of the NMDA receptor-dependent toxicity of 3-nitropropionic acid and cyanide in cerebellar granule neurons. Brain Res. 2008, 1215, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.K.; Hung, C.F.; Weng, J.R.; Hsieh, T.Y.; Wang, S.J. Kaempferol 3-rhamnoside on glutamate release from rat cerebrocortical nerve terminals involves P/Q-type Ca2+ channel and Ca2+/calmodulin-dependent protein kinase II-dependent pathway suppression. Molecules 2022, 27, 1342. [Google Scholar] [CrossRef] [PubMed]
- Montero, M.; Lobatón, C.D.; Hernández-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef]
- Al-Khayri, J.M.; Sahana, G.R.; Nagella, P.; Joseph, B.V.; Alessa, F.M.; Al-Mssallem, M.Q. Flavonoids as potential anti-inflammatory molecules: A review. Molecules 2022, 27, 2901. [Google Scholar] [CrossRef]
- Olędzka, A.J.; Czerwińska, M.E. Role of plant-derived compounds in the molecular pathways related to inflammation. Int. J. Mol. Sci. 2023, 24, 4666. [Google Scholar] [CrossRef]
- Ren, J.; Lu, Y.; Qian, Y.; Chen, B.; Wu, T.; Ji, G. Recent progress regarding kaempferol for the treatment of various diseases. Exp. Ther. Med. 2019, 18, 2759–2776. [Google Scholar] [CrossRef]
- Silva Dos Santos, J.; Gonçalves Cirino, J.P.; de Oliveira Carvalho, P.; Ortega, M.M. The pharmacological action of kaempferol in central nervous system diseases: A Review. Front. Pharmacol. 2021, 11, 565700. [Google Scholar] [CrossRef]
- Lim, H.J.; Prajapati, R.; Seong, S.H.; Jung, H.A.; Choi, J.S. Antioxidant and antineuroinflammatory mechanisms of kaempferol-3-O-β-D-glucuronate on lipopolysaccharide-stimulated BV2 microglial cells through the Nrf2/HO-1 signaling cascade and MAPK/NF-κB pathway. ACS Omega 2023, 8, 6538–6549. [Google Scholar] [CrossRef]
- Muraoka, K.; Shimizu, K.; Sun, X.; Tani, Y.; Izumumi, R.; Miwa, K.; Yamamoto, K. Flavonoids exert diverse inhibitory effects on the activation of NF-kappaB. Transplant. Proc. 2002, 34, 1335–1340. [Google Scholar] [CrossRef]
- Zhang, D.L.; Zhang, Y.T.; Yin, J.J.; Zhao, B.L. Oral administration of Crataegus flavonoids protects against ischemia/reperfusion brain damage in gerbils. J. Neurochem. 2004, 90, 211–219. [Google Scholar] [CrossRef]
- Zhang, J.; Johnston, G.; Stebler, B.; Keller, E.T. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFkappaB-inducing kinase. Antioxid. Redox Signal. 2001, 3, 493–504. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Gloire, G.; Piette, J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009, 11, 2209–2222. [Google Scholar] [CrossRef]
- Stanek, L.M.; Bu, J.; Shihabuddin, L.S. Astrocyte transduction is required for rescue of behavioral phenotypes in the YAC128 mouse model with AAV-RNAi mediated HTT lowering therapeutics. Neurobiol. Dis. 2019, 129, 29–37. [Google Scholar] [CrossRef]
- Wang, T.; Sun, Q.; Yang, J.; Wang, G.; Zhao, F.; Chen, Y.; Jin, Y. Reactive astrocytes induced by 2-chloroethanol modulate microglia polarization through IL-1β, TNF-α, and iNOS upregulation. Food Chem. Toxicol. 2021, 157, 112550. [Google Scholar] [CrossRef]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Lee, K.M.; MacLean, A.G. New advances on glial activation in health and disease. World J. Virol. 2015, 4, 42–55. [Google Scholar] [CrossRef]
- Kim, S.W.; Lee, H.; Lee, H.K.; Kim, I.D.; Lee, J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 2019, 7, 94. [Google Scholar] [CrossRef]
- Li, F.; Zhao, H.; Li, G.; Zhang, S.; Wang, R.; Tao, Z.; Zheng, Y.; Han, Z.; Liu, P.; Ma, Q.; et al. Intravenous antagomiR-494 lessens brain-infiltrating neutrophils by increasing HDAC2-mediated repression of multiple MMPs in experimental stroke. FASEB J. 2020, 34, 6934–6949. [Google Scholar] [CrossRef]
- Gledhill, J.R.; Montgomery, M.G.; Leslie, A.G.W.; Walker, J.E. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc. Natl. Acad. Sci. USA 2007, 104, 13632–13637. [Google Scholar] [CrossRef]
- Qian, Y.S.; Ramamurthy, S.; Candasamy, M.; Shadab, M.; Kumar, R.H.; Meka, V.S. Production, characterization and evaluation of kaempferol nanosuspension for improving oral bioavailability. Curr. Pharm. Biotechnol. 2016, 17, 549–555. [Google Scholar] [CrossRef]
- Li, S.; Pu, X.-P. Neuroprotective effect of kaempferol against a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Biol. Pharm. Bull. 2011, 34, 1291–1296. [Google Scholar] [CrossRef]
- Hussein, R.M.; Mohamed, W.R.; Omar, H.A. A neuroprotective role of kaempferol against chlorpyrifos-induced oxidative stress and memory deficits in rats via GSK3β-Nrf2 signaling pathway. Pestic. Biochem. Physiol. 2018, 152, 29–37. [Google Scholar] [CrossRef]
- Lin, H.; Wang, X.; Zhao, J.; Lin, Z. Protective effect of kaempferol against cognitive and neurological disturbances induced by D-galactose and aluminum chloride in mice. J. Funct. Foods. 2023, 100, 105385. [Google Scholar] [CrossRef]
- Liu, Z.; Yao, X.; Sun, B.; Jiang, W.; Liao, C.; Dai, X.; Chen, Y.; Chen, J.; Ding, R. Pretreatment with kaempferol attenuates microglia-mediated neuroinflammation by inhibiting MAPKs–NF–κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radic. Biol. Med. 2021, 168, 142–154. [Google Scholar] [CrossRef]
- Chang, S.; Li, X.; Zheng, Y.; Shi, H.; Zhang, D.; Jing, B.; Chen, Z.; Qian, G.; Zhao, G. Kaempferol exerts a neuroprotective effect to reduce neuropathic pain through TLR4/NF-ĸB signaling pathway. Phytother. Res. 2022, 36, 1678–1691. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019, 28, 2111–2123. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Lahiani-Cohen, I.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Rosenmann, H. Exposure to 3-nitropropionic acid mitochondrial toxin induces tau pathology in tangle-mouse model and in wild type-mice. Front. Cell Dev. Biol. 2020, 7, 321. [Google Scholar] [CrossRef]
- Babaei, P.; Eyvani, K.; Kouhestani, S. Sex-independent cognition improvement in response to kaempferol in the model of sporadic Alzheimer’s disease. Neurochem. Res. 2021, 46, 1480–1486. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Sánchez, C.; Lagoa, R.; Poejo, J.; García-López, V.; García-Martínez, V.; Gutierrez-Merino, C. An Update of Kaempferol Protection against Brain Damage Induced by Ischemia-Reperfusion and by 3-Nitropropionic Acid. Molecules 2024, 29, 776. https://doi.org/10.3390/molecules29040776
López-Sánchez C, Lagoa R, Poejo J, García-López V, García-Martínez V, Gutierrez-Merino C. An Update of Kaempferol Protection against Brain Damage Induced by Ischemia-Reperfusion and by 3-Nitropropionic Acid. Molecules. 2024; 29(4):776. https://doi.org/10.3390/molecules29040776
Chicago/Turabian StyleLópez-Sánchez, Carmen, Ricardo Lagoa, Joana Poejo, Virginio García-López, Virginio García-Martínez, and Carlos Gutierrez-Merino. 2024. "An Update of Kaempferol Protection against Brain Damage Induced by Ischemia-Reperfusion and by 3-Nitropropionic Acid" Molecules 29, no. 4: 776. https://doi.org/10.3390/molecules29040776
APA StyleLópez-Sánchez, C., Lagoa, R., Poejo, J., García-López, V., García-Martínez, V., & Gutierrez-Merino, C. (2024). An Update of Kaempferol Protection against Brain Damage Induced by Ischemia-Reperfusion and by 3-Nitropropionic Acid. Molecules, 29(4), 776. https://doi.org/10.3390/molecules29040776