
Advances for Triangular and Sandwich-Shaped All-Metal Aromatics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Findings of Triangular All-Metal Aromatic Clusters
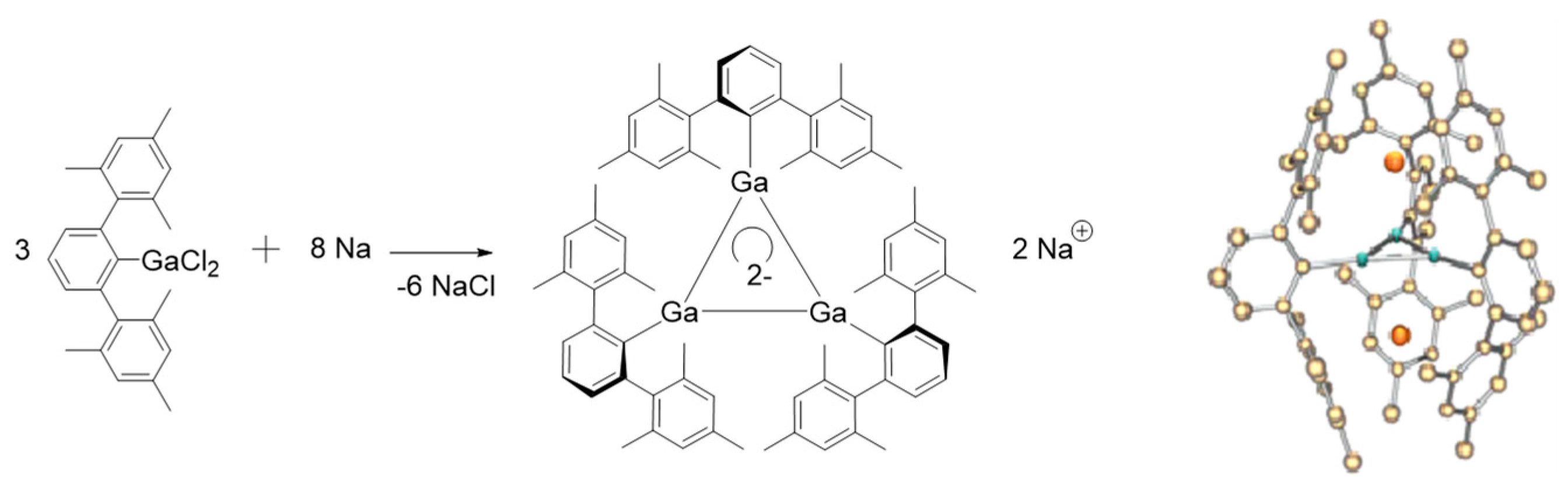
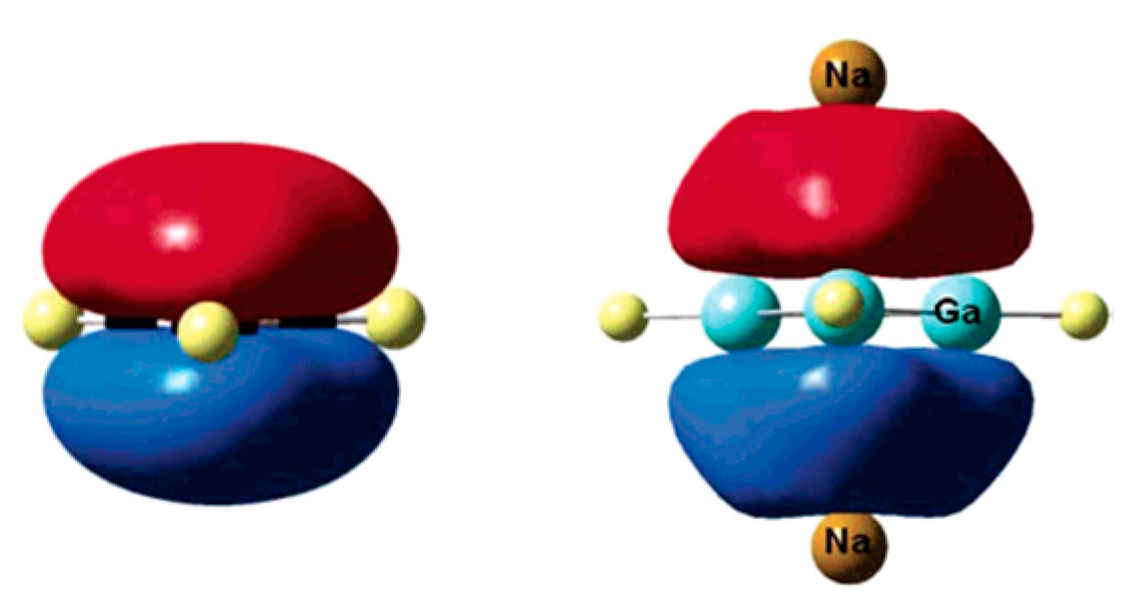
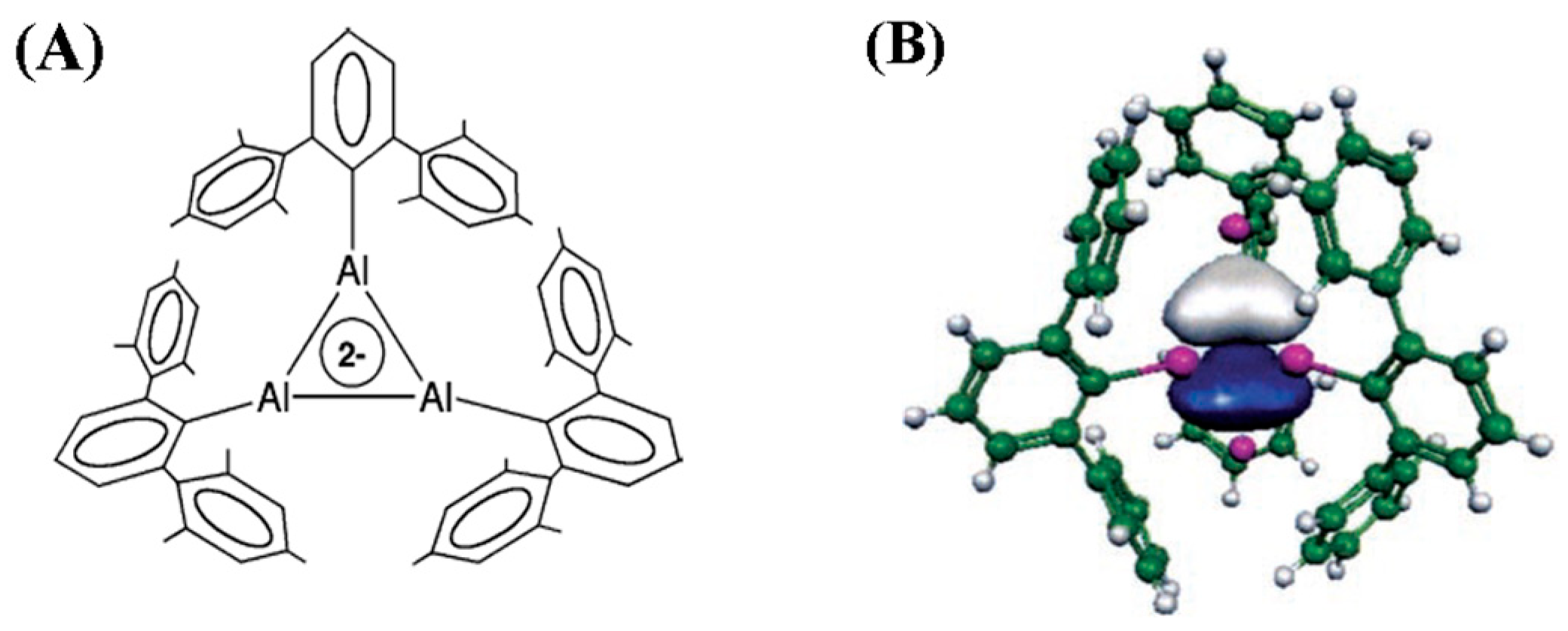
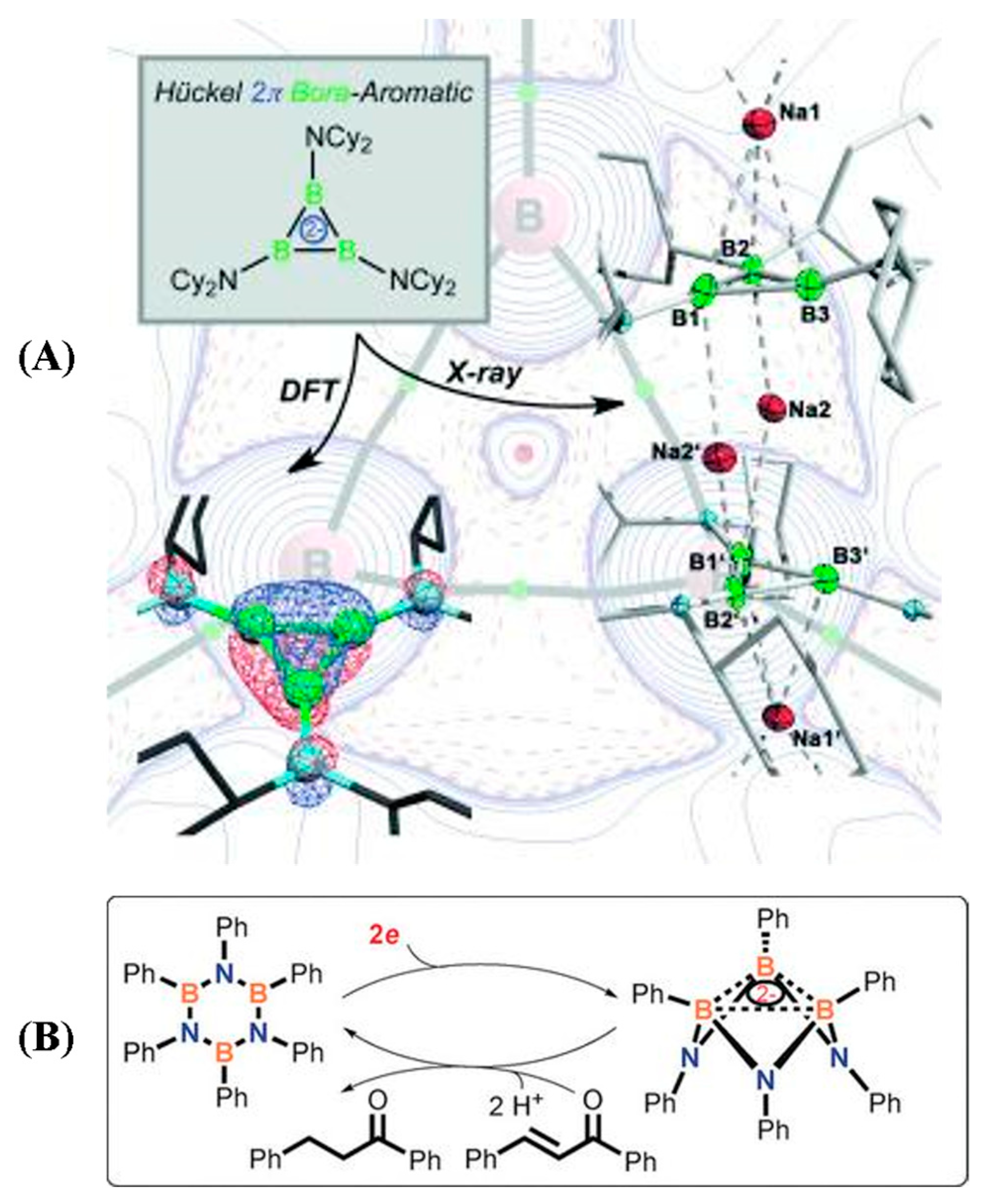
2.1. Triangular π-Aromatic [M3]2− (M = B, Al, Ga)
2.2. Triangular π-Aromatic [Si]3+, [Ge]3+, [Si2C]+
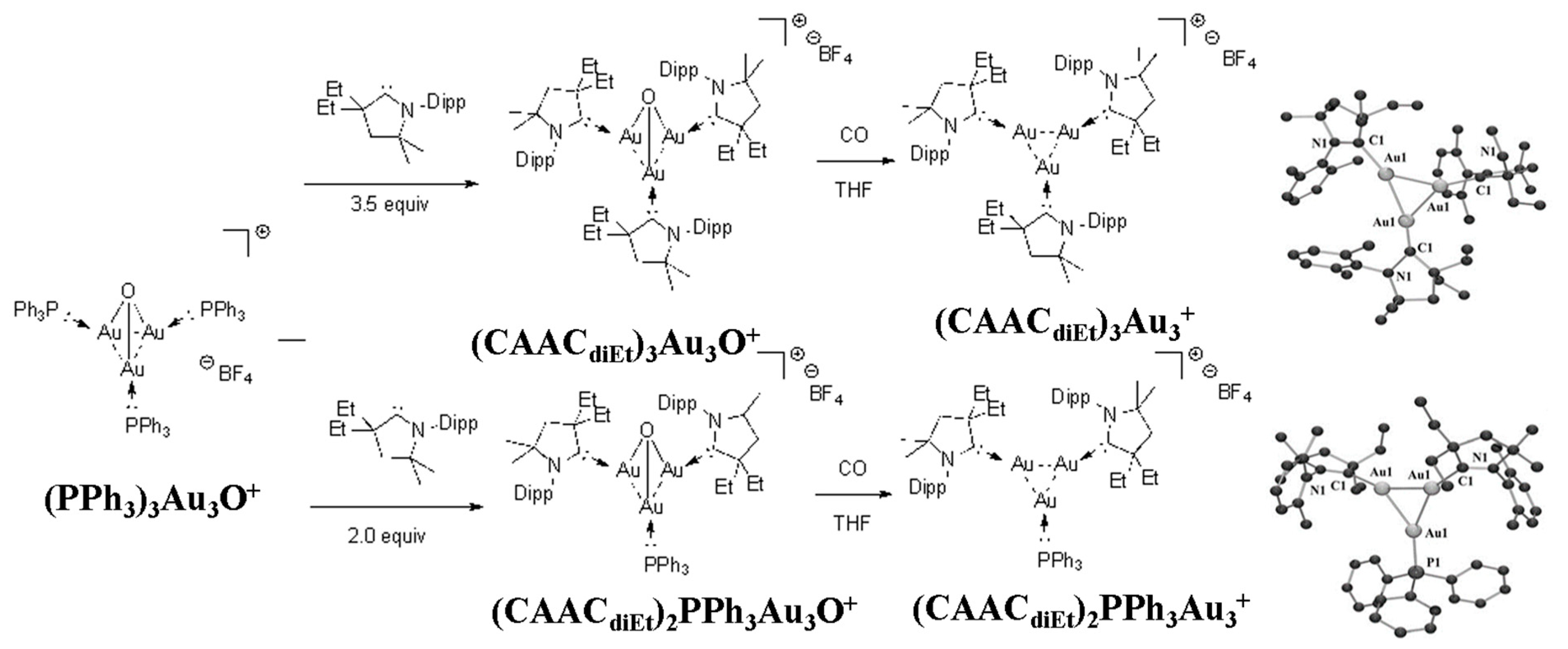
2.3. Triangular σ-Aromatic [Au]3+
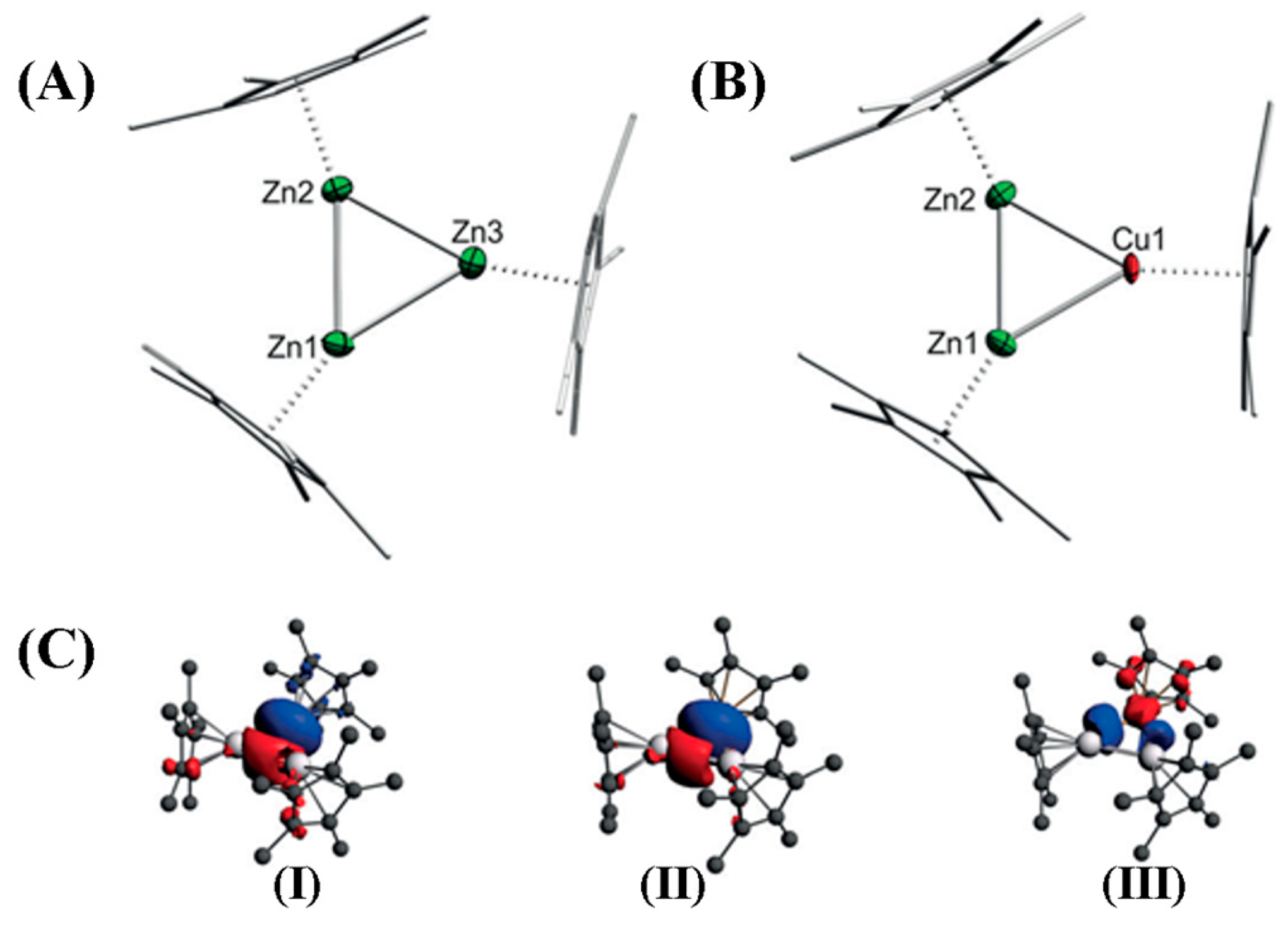
2.4. Triangular σ-Aromatic [Zn3]+, [Zn2Cu]
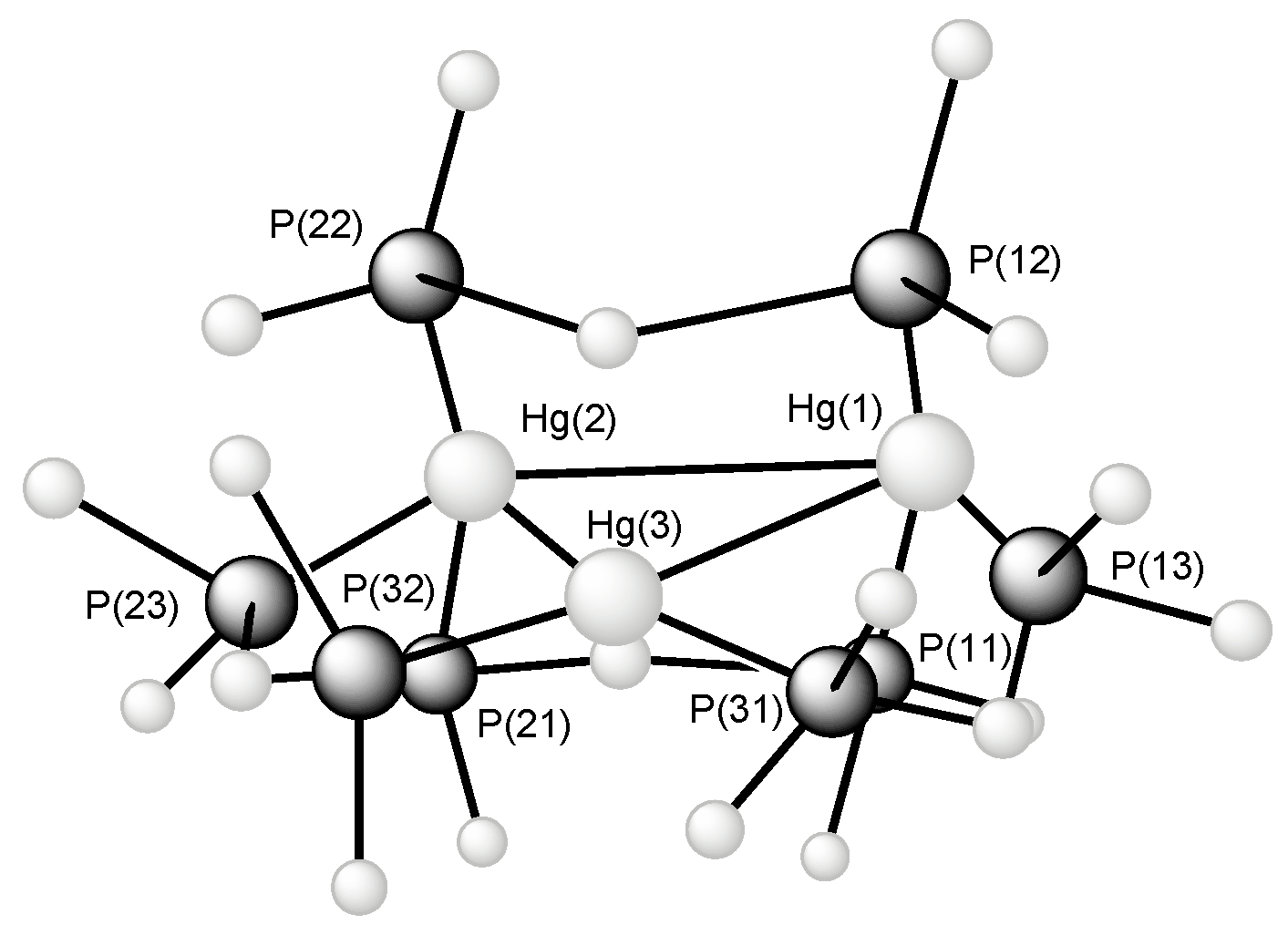
2.5. Triangular Homoleptic [Hg3]4+
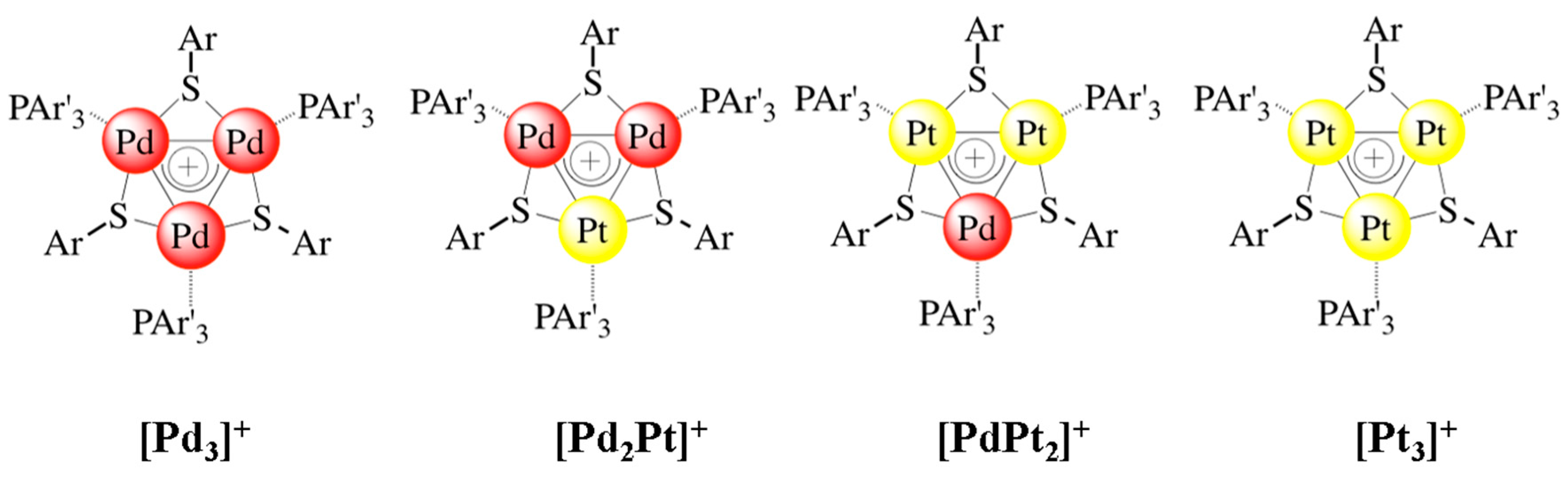
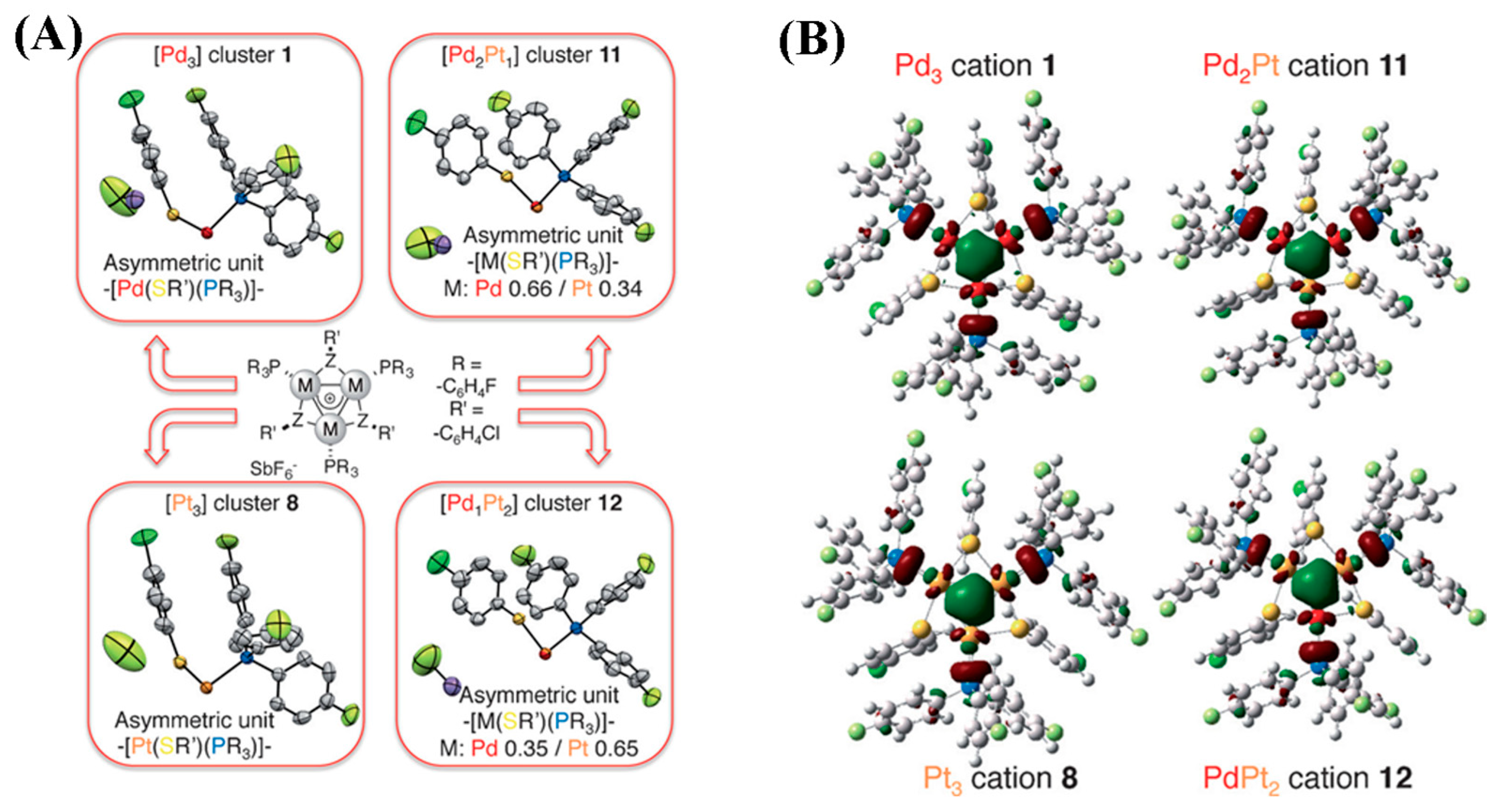
2.6. Triangular δ-Aromatic [Pd3]+, [Pt3]+, [Pd2Pt]+, [PdPt2]+
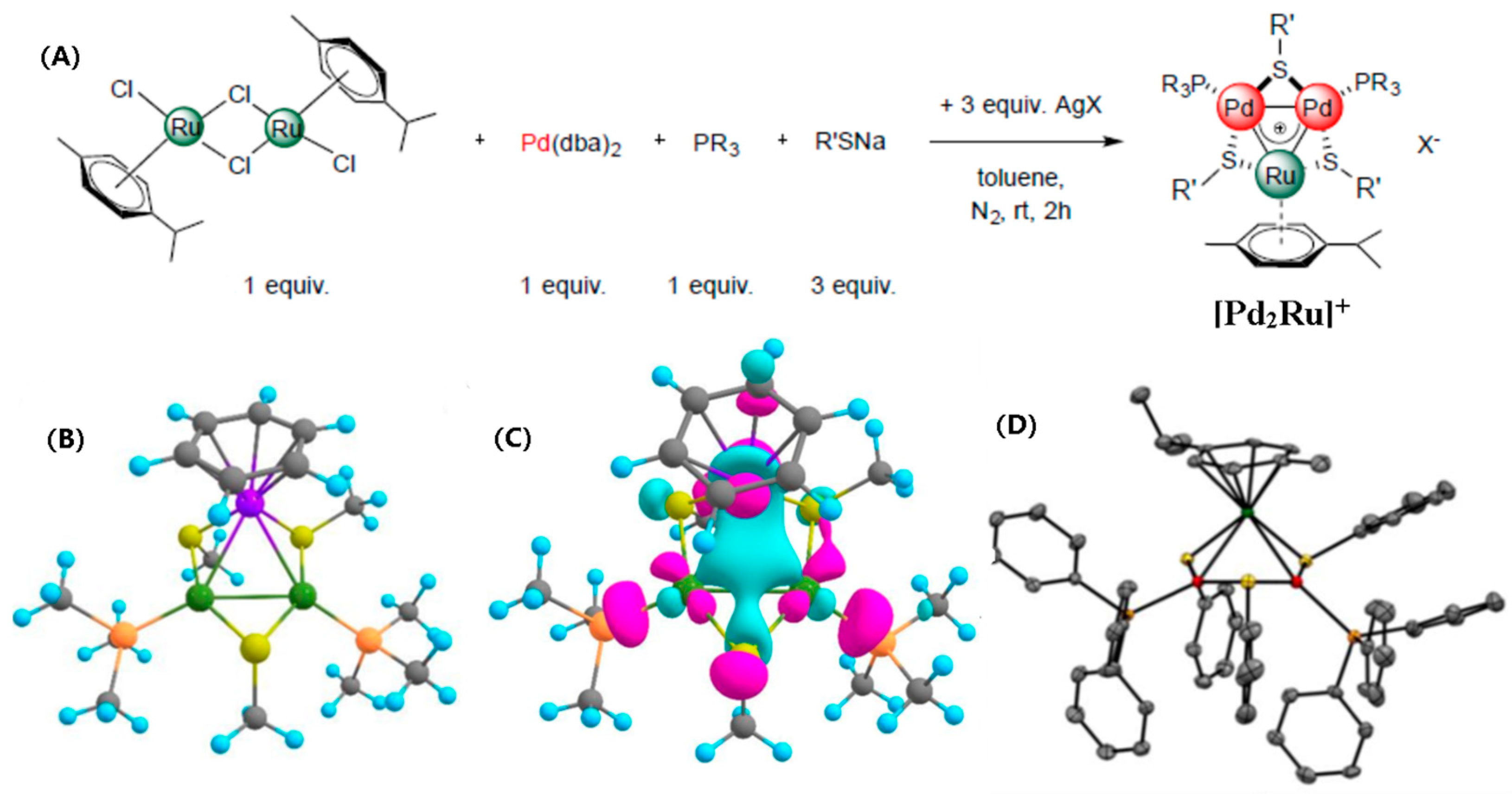
2.7. Triangular Heteroaromatic [Pd2Ru]+
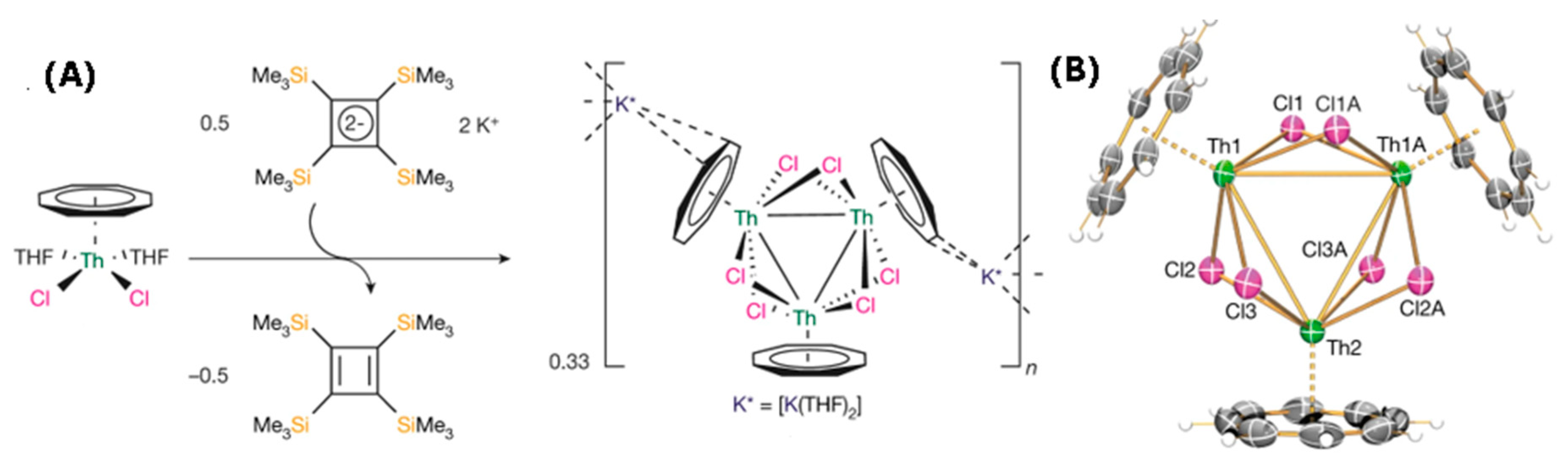
2.8. Triangular σ-Aromatic [Th3]2−
3. Theoretical Predictions of Aromaticity for Triangular All-Metal Clusters
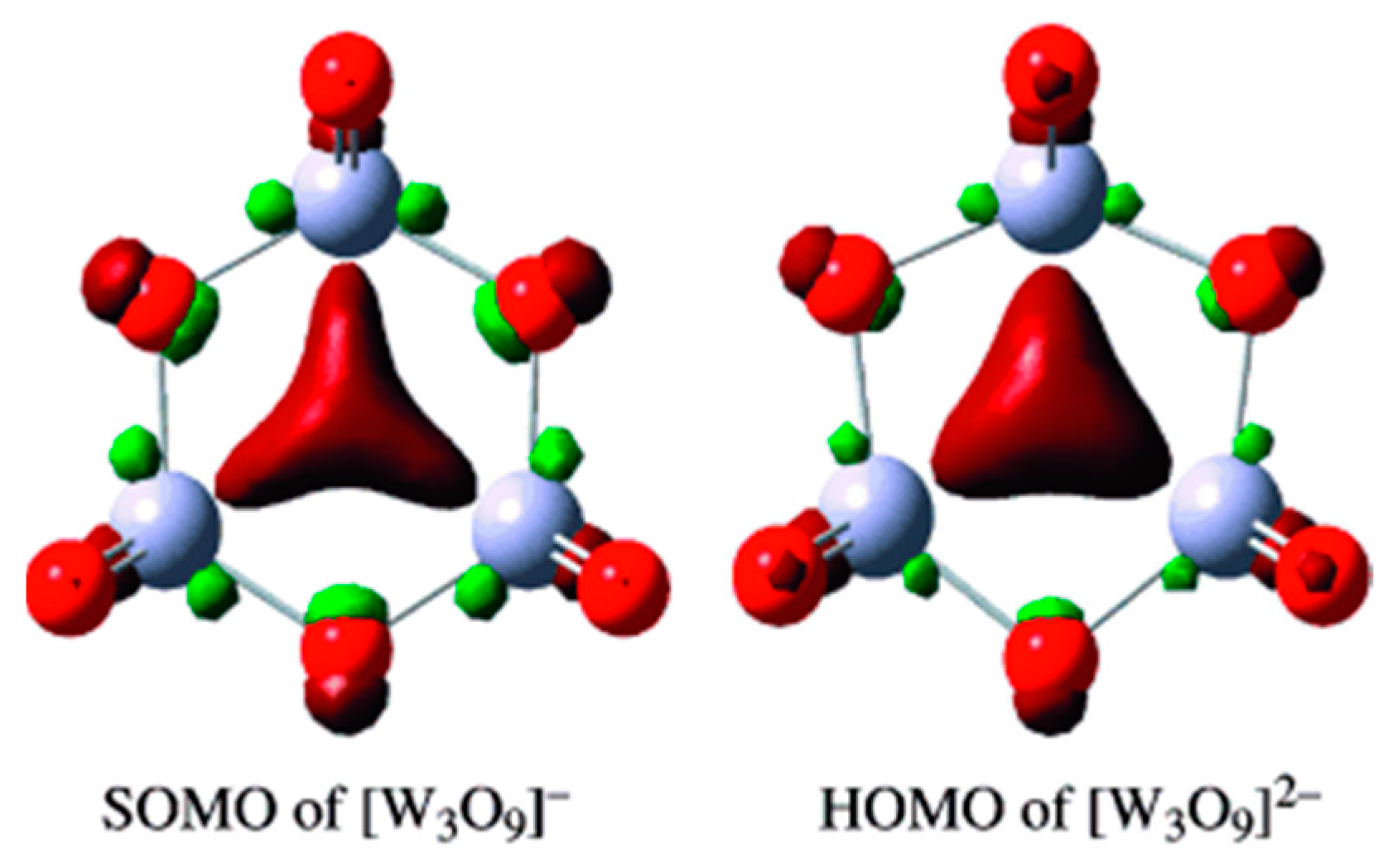
3.1. d-Orbital Aromaticity in Triangular [M3O9]−, [M3O9]2−(M = W, Mo)
3.2. δ-Aromaticity in Triangular [Ta3O3]−
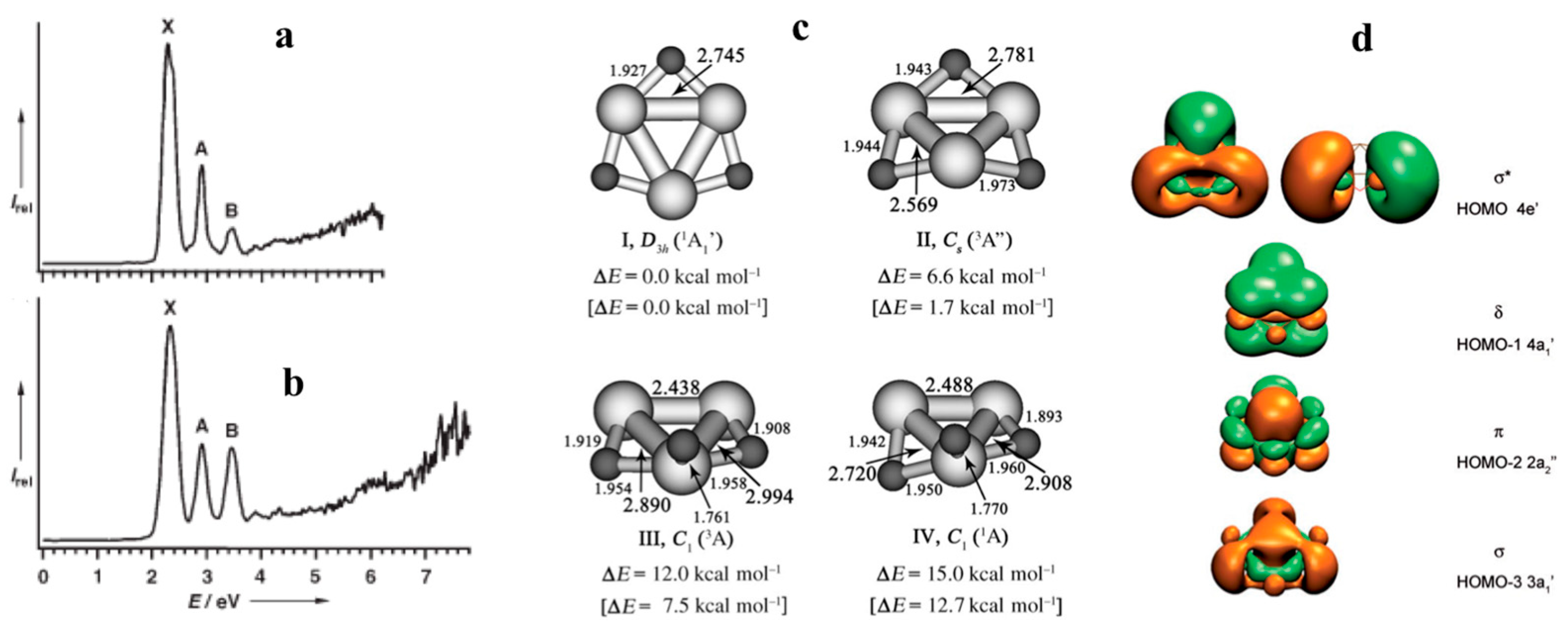
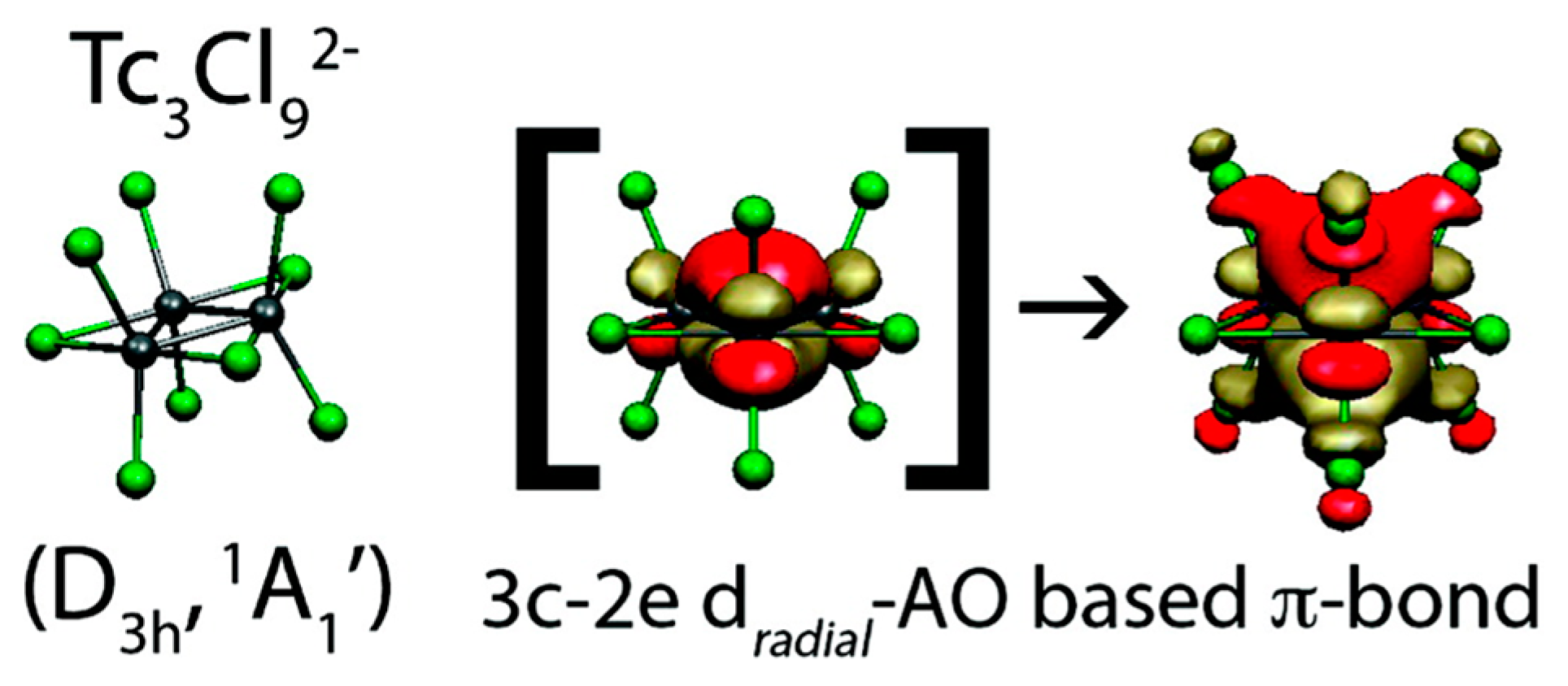
3.3. d-Orbital Aromaticity in Triangular [Tc3X9]2−
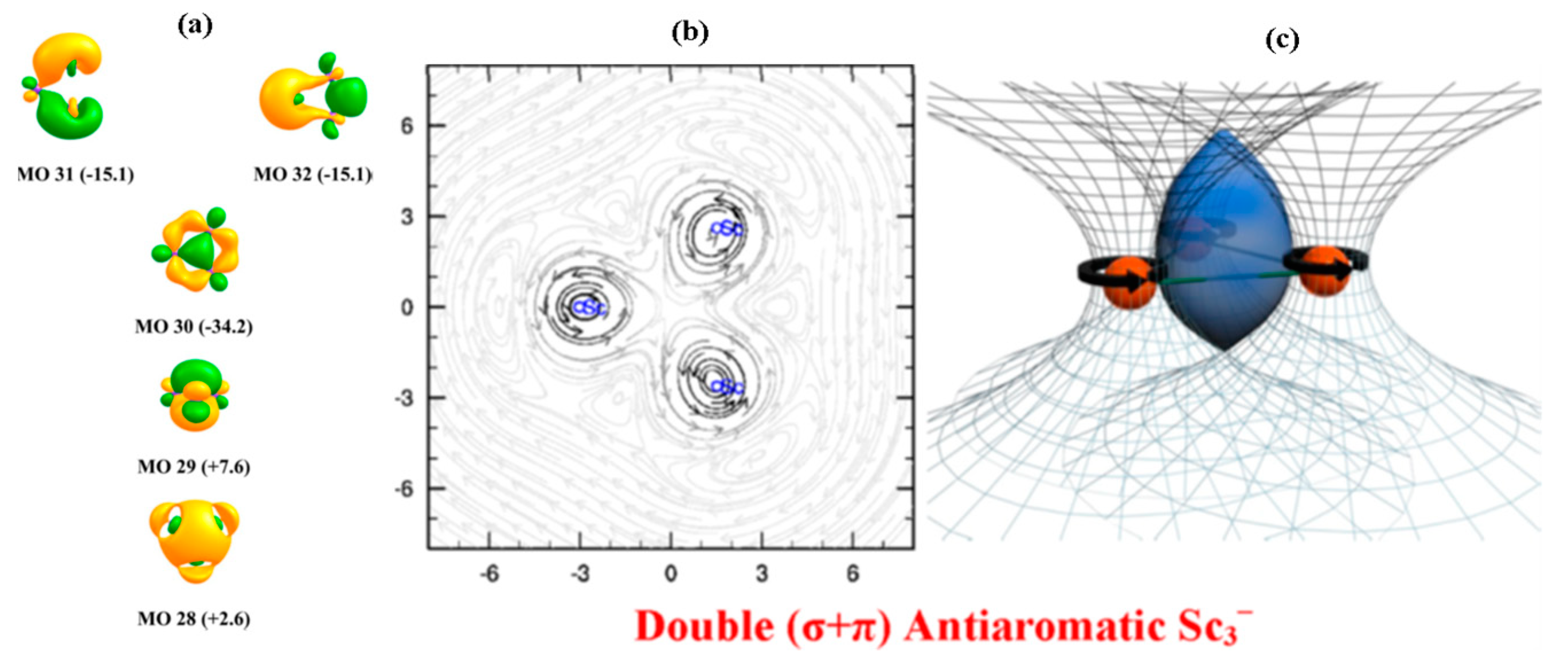
3.4. Aromaticity/Antiaromaticity of Triangular [Sc3]−
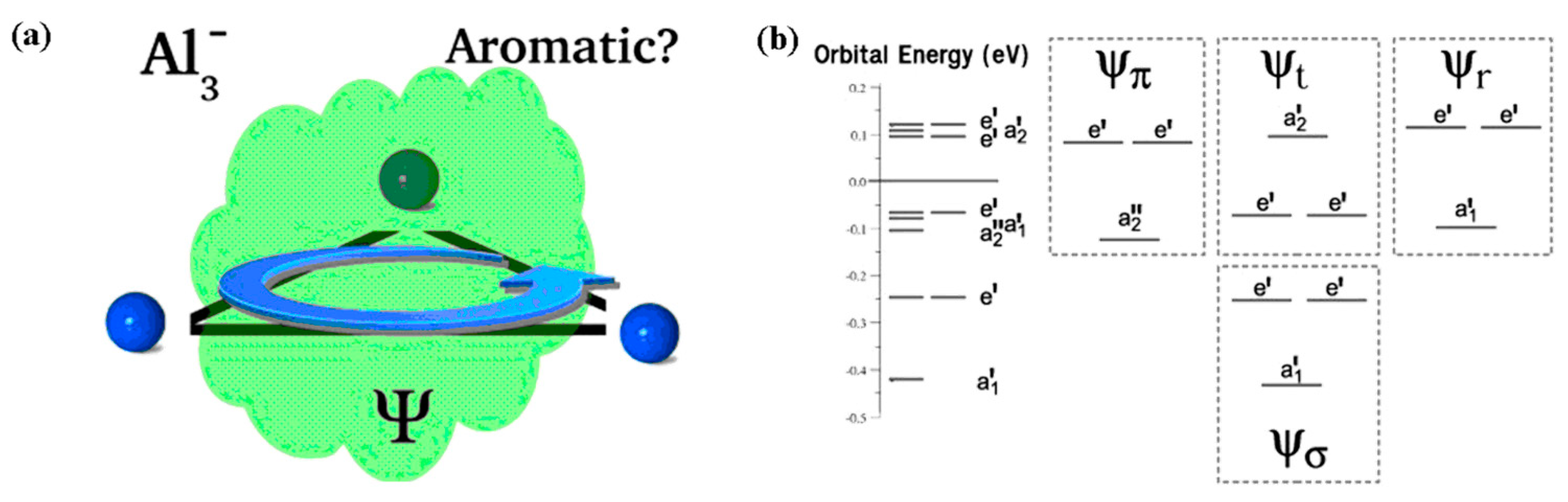
3.5. Aromaticity of Triangular [Al3]−
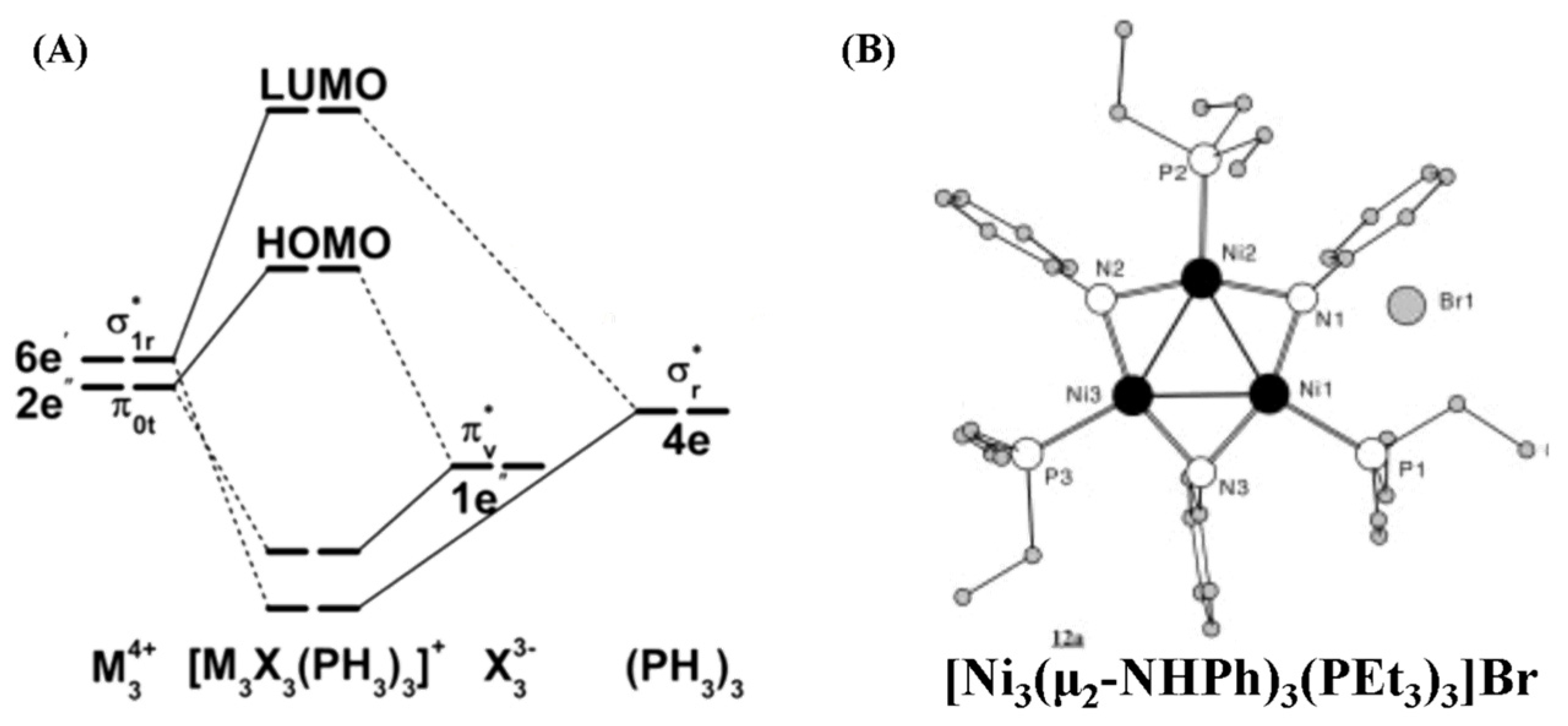
3.6. Aromatic in Triangular [M3]4+ (M = Ni, Pd, Pt)
3.7. Triangular Aromatic Heterobimetallic [Hf3]+
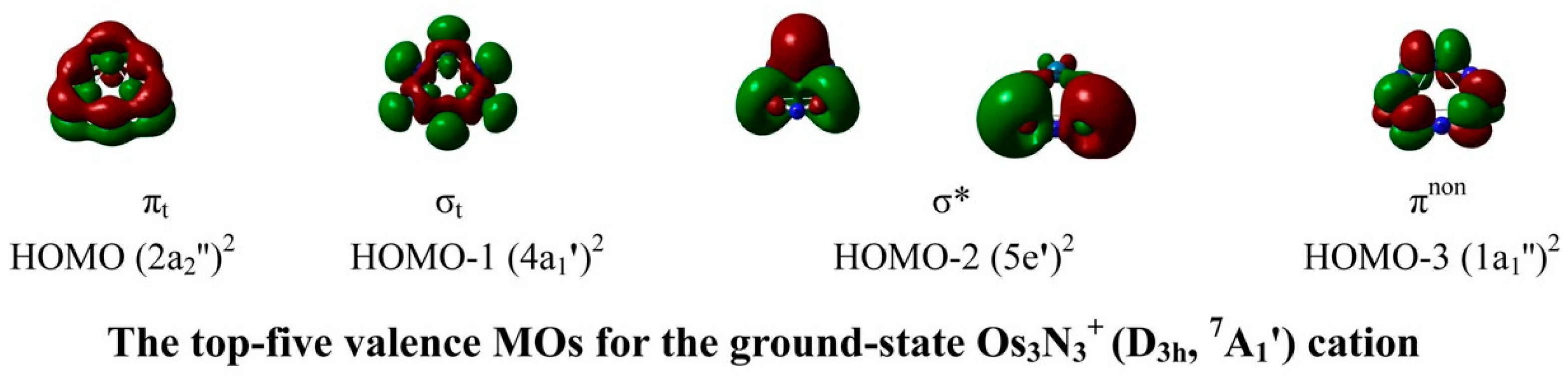
3.8. Triangular Aromatic Os3N3+/−
3.9. Aromaticity in Triangular Ir3N3+/−
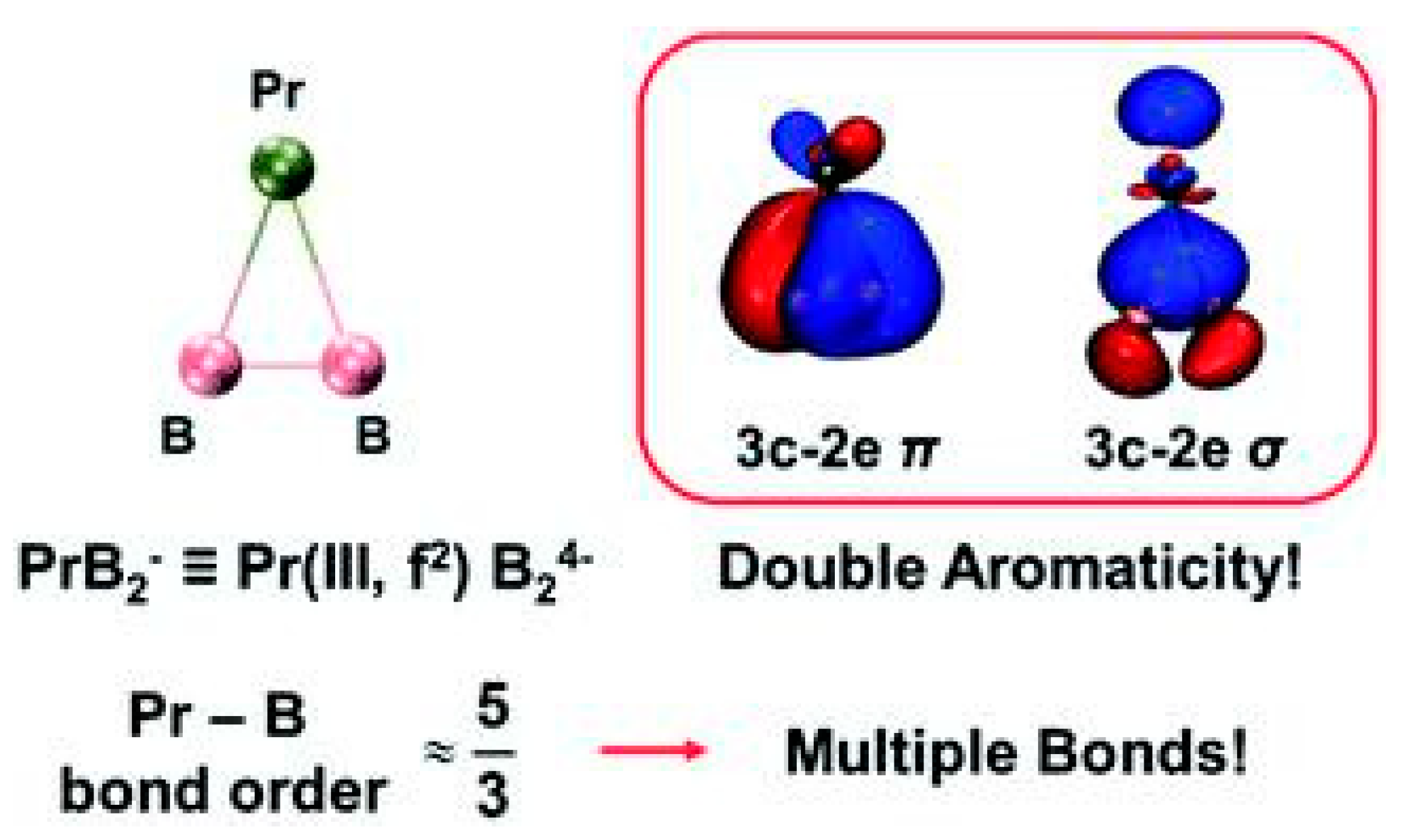
3.10. Aromaticity in Triangular PrB2−
4. Catalytic Applications of Triangular All-Metal Aromatic Clusters
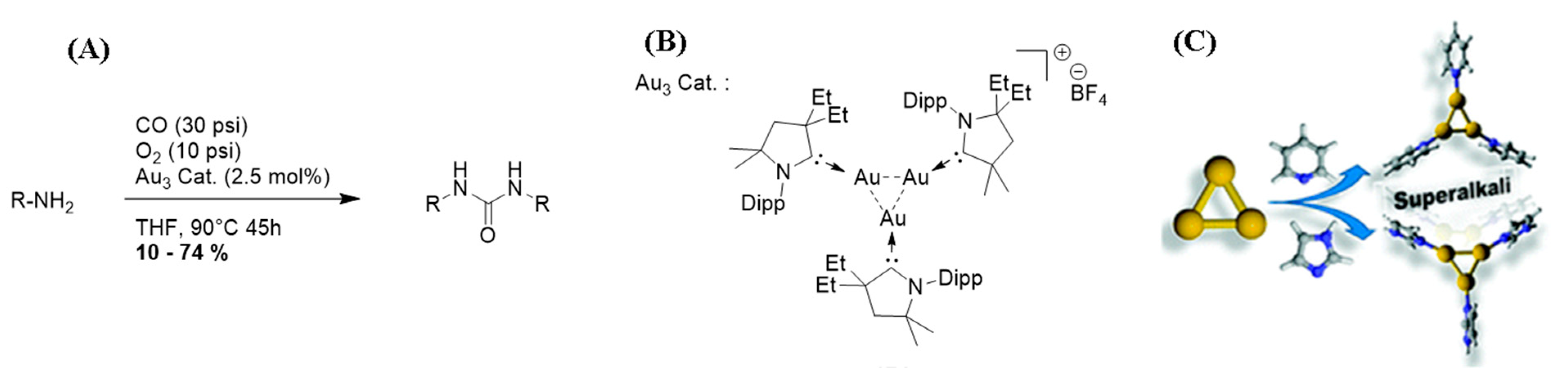
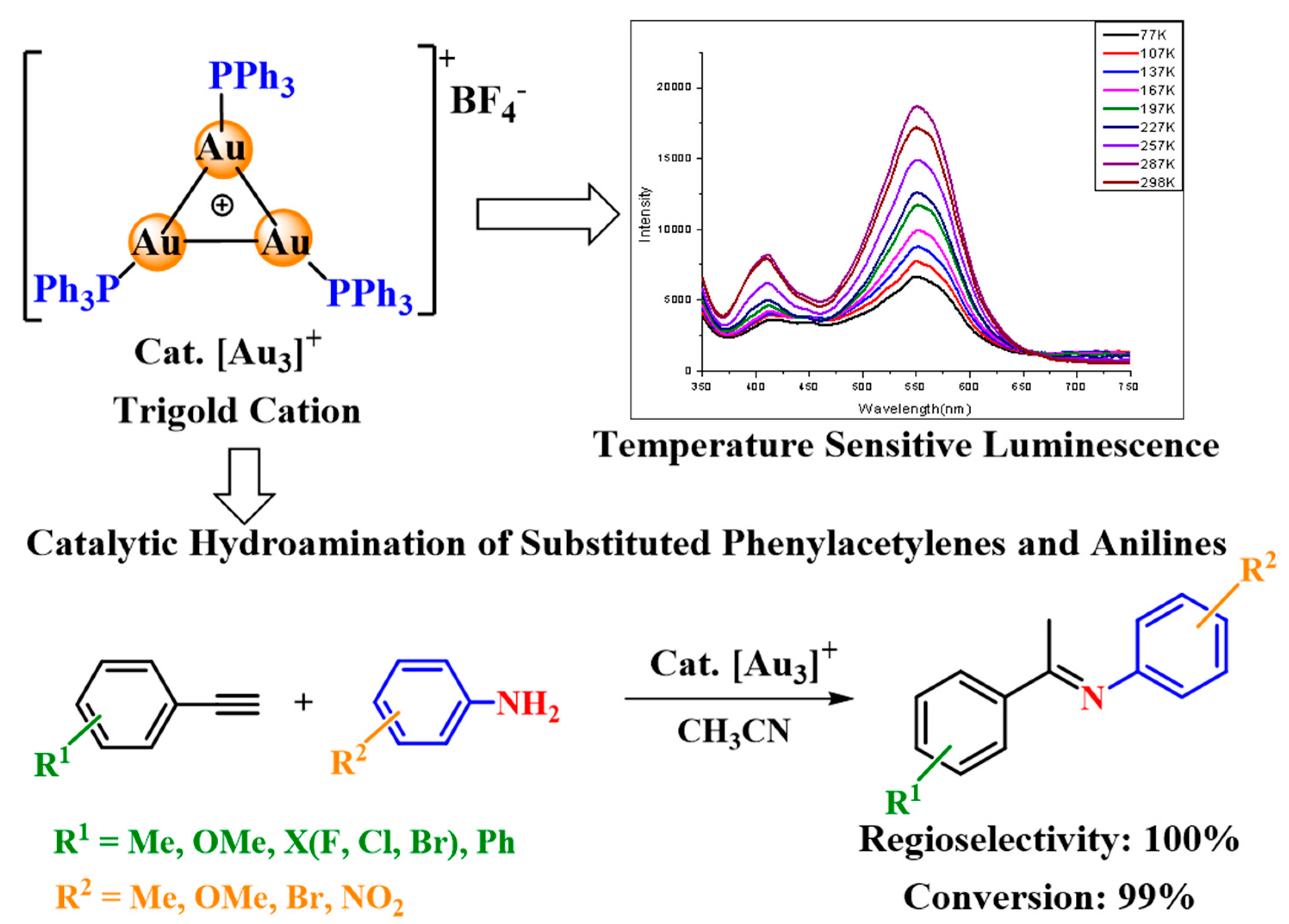
4.1. σ-Aromatic Tri-Gold Cation [Au3]+ Catalyzed Amine Carbonylation
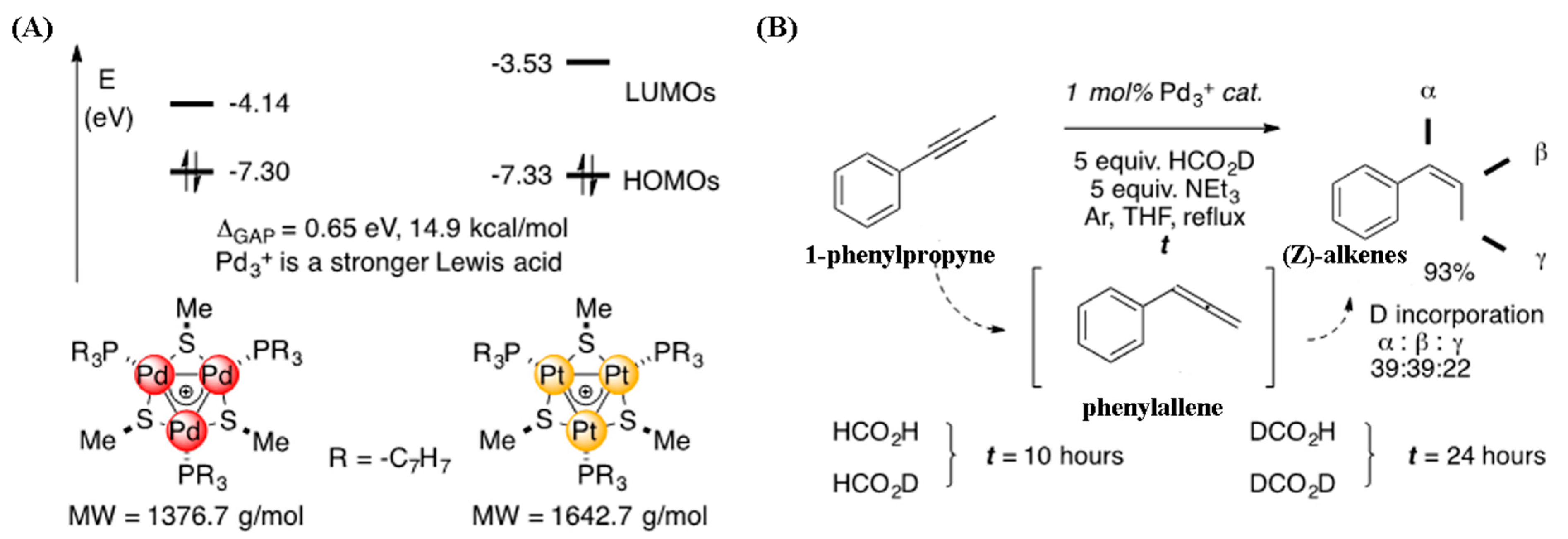
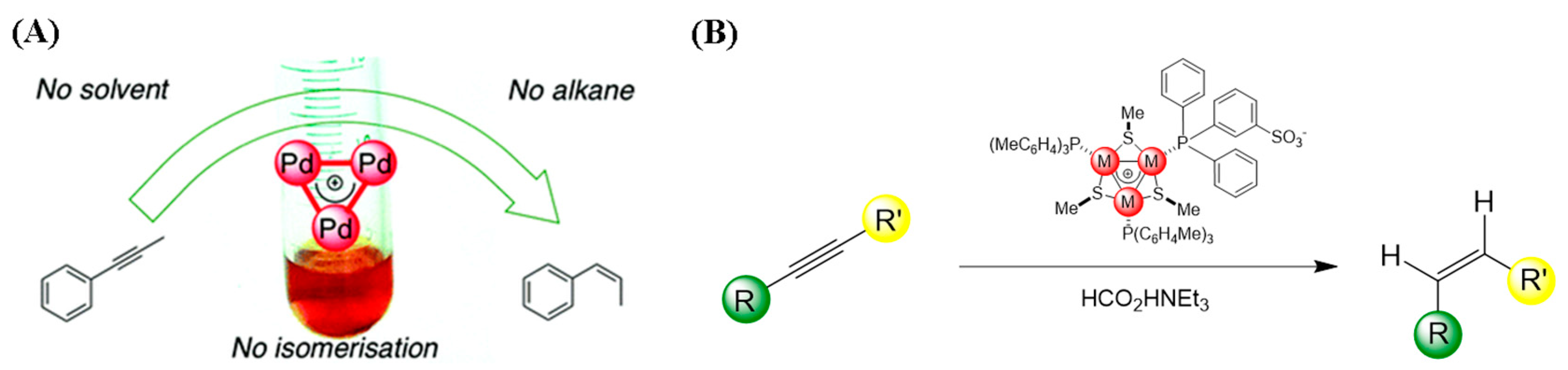
4.2. Aromatic [Pd3]+ Catalyzed Semi-Reduction of Internal Alkynes
4.3. Aromatic [Pd3]+ Catalyzed Cycloisomerization of 1,6-Enynes and Dienynes
4.4. Aromatic [Pd3]+ Catalyzed Coupling Reactions
5. Cation–π Interactions of Triangular All-Metal Aromatics
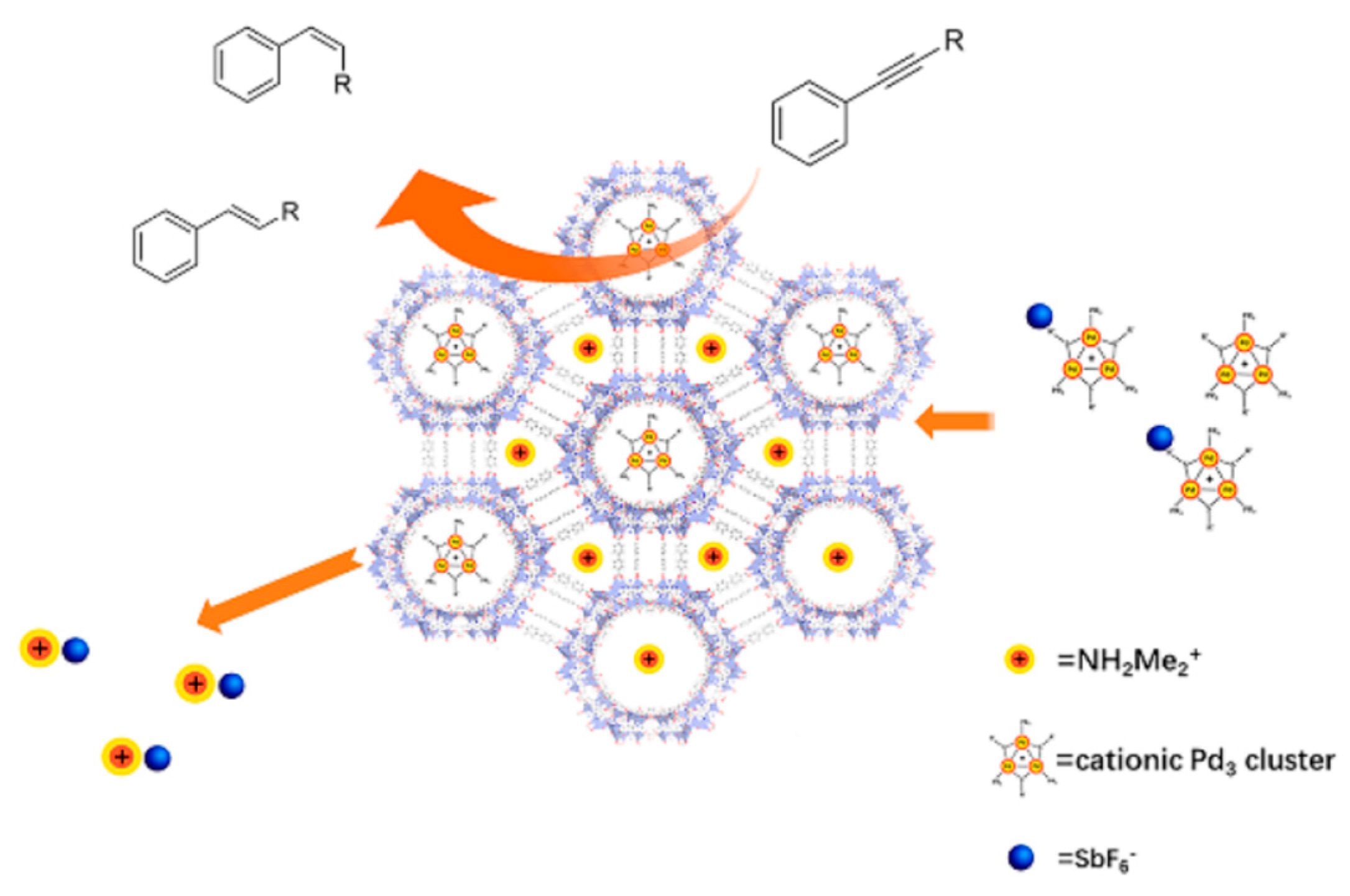
5.1. Coordination of Aromatic [Pd3]+ to Lewis Acids
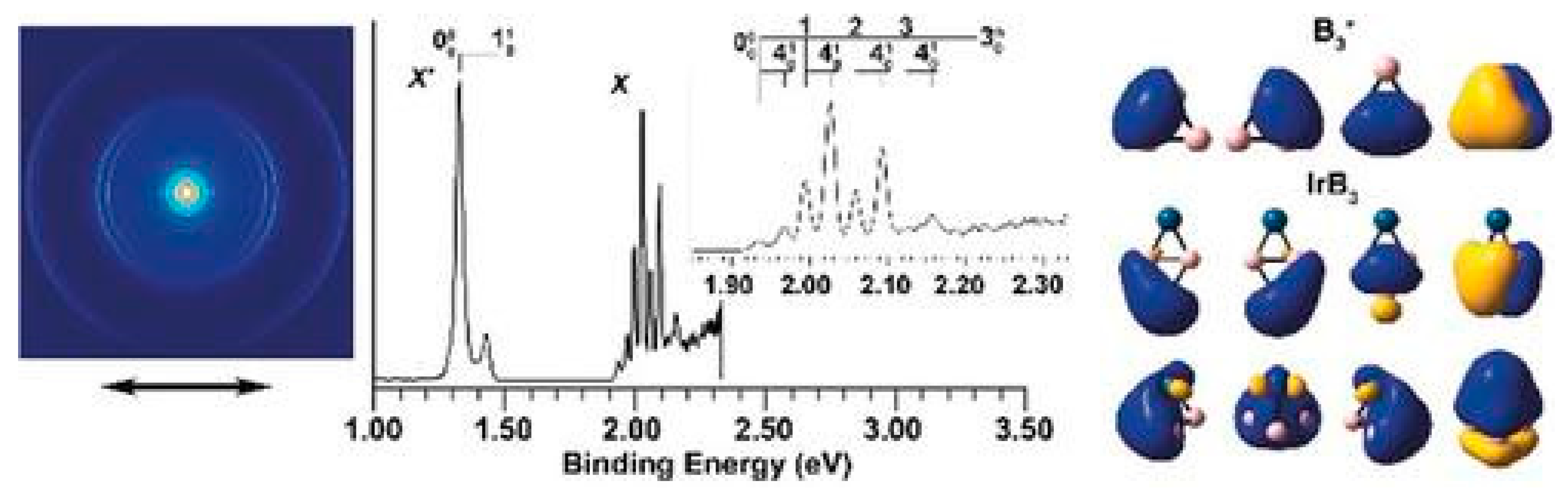
5.2. Coordination of π-Aromatic B3+ to Transition Metals
6. Experimental and Theoretical Developments of Triangular All-Metal Aromatic Sandwiches
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Proft, F.D.; Geerlings, P. Conceptual and Computational DFT in the Study of Aromaticity. Chem. Rev. 2001, 101, 1451–1464. [Google Scholar] [CrossRef]
- Kékule, A. Sur la constitution des substances aromatiques. Bull. Soc. Chim. Fr. 1865, 3, 98–110. [Google Scholar]
- Popov, I.A.; Starikova, A.A.; Steglenko, D.V.; Boldyrev, A.I. Usefulness of the σ-Aromaticity and σ-Antiaromaticity Concepts for Clusters and Solid-State Compounds. Chem. Eur. J. 2018, 24, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Clark, E. Polycyclic Hydrocarbons; Academic Press: New York, NY, USA, 1964. [Google Scholar] [CrossRef]
- Thorn, D.L.; Hoffmann, R. Delocalization in metallocycles. Nouv. J. Chim. 1979, 3, 39–45. [Google Scholar]
- He, X.; Yu, D.; Wu, J.; Wang, B.; Rong, C.; Chattaraj, P.; Liu, S. Towards Understanding Metal Aromaticity in Different Spin States: A Density Functional Theory and Information-Theoretic Approach Analysis. Chem. Phy. Lett. 2020, 761, 138065. [Google Scholar] [CrossRef]
- Hu, H.C.; Zhao, B. Metal-Organic Frameworks Based on Multicenter-Bonded [MI]8 (M = Mn, Zn) Clusters with Cubic Aromaticity. Chem. Eur. J. 2018, 24, 16702–16707. [Google Scholar] [CrossRef]
- Bleeke, J.R. Metallabenzenes. Chem. Rev. 2001, 101, 1205–1228. [Google Scholar] [CrossRef]
- Fernández, I.; Frenking, G.; Merino, G. Aromaticity of Metallabenzenes and Related Compounds. Chem. Soc. Rev. 2015, 44, 6452–6463. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, N.V.; Popov, I.A.; Kulichenko, M.; Fedik, N.; Sun, Z.M.; Muñoz-Castro, A.; Boldyrev, A.I. Bridging Aromatic/Antiaromatic Units: Recent Advances in Aromaticity and Antiaromaticity in Main-Group and Transition-Metal Clusters from Bonding and Magnetic Analyses. Eur. J. Inorg. Chem. 2021, 41, 4239–4250. [Google Scholar] [CrossRef]
- Bleeke, J.R. Aromatic iridacycles. Acc. Chem. Res. 2007, 40, 1035–1047. [Google Scholar] [CrossRef]
- Wright, L.J. Metallabenzenes and metallabenzenoids. Dalton Trans. 2006, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Lanford, C.W.; Haley, M.M. Recent advances in metallabenzene chemistry. Angew. Chem. Int. Ed. 2006, 45, 3914–3936. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.S. All-metal aromaticity and antiaromaticity. Chem. Rev. 2005, 105, 3716–3757. [Google Scholar] [CrossRef] [PubMed]
- You, X.R.; Zhai, H.J. Can Synthetic All-Metal Cluster Compound Support Multifold (π and σ) Aromaticity and d-Orbital Aromaticity? Chin. J. Chem. 2019, 37, 126–130. [Google Scholar] [CrossRef]
- Chen, Z.; Wannere, C.S.; Corminboent, C.; Puchta, R.; von R. Schleyer, P. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P.; King, R.B. Aromaticity of Tri-and Tetranuclear Metal–Carbonyl Clusters Based on Magnetic Criteria. Chem. Eur. J. 2007, 13, 978–984. [Google Scholar] [CrossRef]
- Paul, S.; Misra, A. Interplay among Aromaticity, Magnetism, and Nonlinear Optical Response in All-Metal Aromatic Systems. Inorg. Chem. 2011, 50, 3234–3246. [Google Scholar] [CrossRef]
- Mercero, J.M.; Boldyrev, A.I.; Merino, G.J.M.; Ugalde, J.M. Recent developments and future prospects of all-metal aromatic compounds. Chem. Soc. Rev. 2015, 44, 6519–6534. [Google Scholar] [CrossRef]
- Chen, D.; Hua, Y.; Xia, H. Metallaaromatic Chemistry: History and Development. Chem. Rev. 2020, 120, 12994–13086. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M. Open-Shell Jellium Aromaticity in Metal Clusters. Chem. Commun. 2019, 55, 5559–5562. [Google Scholar] [CrossRef]
- Chen, D.; Xie, Q.; Zhu, J. Unconventional Aromaticity in Organometallics: The Power of Transition Metals. Acc. Chem. Res. 2019, 52, 1449. [Google Scholar] [CrossRef]
- Jr, J.H.D.; Rabelo, J.N.T.; Candido, L. Electron Correlation Effects in All-Metal Aromatic Clusters: A Quantum Monte Carlo Study. Inorg. Chem. 2016, 55, 7442–7447. [Google Scholar] [CrossRef]
- Bigi, F.; Cera, G.; Maggi, R.; Wang, Y.; Malacria, M.; Maestri, G. Is Aromaticity a Driving Force in Catalytic Cycles? A Case from the Cycloisomerization of Enynes Catalyzed by All-Metal Aromatic Pd3+ Clusters and Carboxylic Acids. J. Phys. Chem. A 2021, 125, 10035–10043. [Google Scholar] [CrossRef]
- Bigi, F.; Cauzzi, D.; Della Ca, N.; Malacria, M.; Maggi, R.; Motti, E.; Wang, Y.; Maestri, G. Evolution of Triangular All-Metal Aromatic Complexes from Bonding Quandaries to Powerful Catalytic Platforms. ACS Org. Inorg. Au 2022, 2, 373–385. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I. σ-Aromaticity and σ-antiaromaticity in alkali metal and alkaline earth metal small clusters. J. Phys. Chem. A 2003, 107, 554–560. [Google Scholar] [CrossRef]
- Tsipis, C.A. DFT study of “all-metal” aromatic compounds. Coord. Chem. Rev. 2005, 249, 2740–2762. [Google Scholar] [CrossRef]
- Huang, X.; Zhai, H.J.; Kiran, B.; Wang, L.-S. Observation of d-orbital aromaticity. Angew. Chem. Int. Ed. 2005, 44, 7251–7254. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Soto, L.; Ramírez-Tagle, R.; Arratia-Pérez, R. Spin–orbit effects on the aromaticity of the Re3Cl9 and Re3Br9 clusters. Chem. Phys. Lett. 2008, 467, 94–96. [Google Scholar] [CrossRef]
- Zhai, H.J.; Wang, B.; Huang, X.; Wang, L.S. Probing the Electronic and Structural Properties of the Niobium Trimer Cluster and Its Mono-and Dioxides: Nb3On− and Nb3On (n = 0–2). J. Phys. Chem. A 2009, 113, 3866–3875. [Google Scholar] [CrossRef]
- Alvarado-Soto, L.; Ramírez-Tagle, R.; Arratia-Pérez, R. Spin-Orbit Effects on the Aromaticity of the Re3X92−(X = Cl, Br) Cluster Ions. J. Phys. Chem. A 2009, 113, 1671–1673. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Boldyrev, A.I. The Chemical bonding of Re3Cl9 and revealed by the adaptive natural density partitioning analyses. Inorg. Chem. 2010, 31, 2–12. [Google Scholar] [CrossRef]
- Hirschfelder, J.O. The energy of the triatomic hydrogen molecule and ion. J. Chem. Phys. 1938, 6, 795–806. [Google Scholar] [CrossRef]
- Radom, L.; Hariharan, P.C.; Pople, J.A.; Schleyer, P.v.R. Molecular orbital theory of the electronic structure of organic compounds. XXII. Structures and stabilities of C3H3+ and C3H+ cations. J. Am. Chem. Soc. 1976, 98, 10–14. [Google Scholar] [CrossRef]
- Twamley, B.; Power, P.P. Synthesis of the Square-Planar Gallium Species K2[Ga4(C6H3-2, 6-Trip2)2](Trip=C6H2-2, 4, 6-iPr3): The Role of Aryl–Alkali Metal Ion Interactions in the Structure of Gallium Clusters. Angew. Chem. Int. Ed. 2000, 39, 3500–3502. [Google Scholar] [CrossRef]
- Wiberg, N.; Blank, T.; Westerhausen, M.; Schneiderbauer, S.; Schnöckel, H.; Krossing, I.; Schnepf, A. Disodium Tetrasupersilyltetragallanediide Na2Ga4R*4· 2THF (R*= SitBu3)—Preparation of a Novel Gallium Cluster Compound via Dichlorodisupersilyldigallane R*2Ga2Cl2. Eur. J. Inorg. Chem. 2002, 2002, 351–356. [Google Scholar] [CrossRef]
- Hoffmann, R. The many guises of aromaticity. Am. Sci. 2015, 103, 18–22. [Google Scholar] [CrossRef]
- Li, X.W.; Pennington, W.T.; Robinson, G.H. Metallic system with aromatic character. synthesis and molecular structure of Na2[[(2, 4, 6-Me3C6H2)2C6H3]Ga]3 the first cyclogallane. J. Am. Chem. Soc. 1995, 117, 7578–7579. [Google Scholar] [CrossRef]
- Lichtenthaler, M.; Stahl, F.; Kratzert, D.; Heidinger, L.; Schleicher, E.; Hamann, J.; Himmel, D.; Weber, S.; Krossing, I. Cationic cluster formation versus disproportionation of low-valent indium and gallium complexes of 2,2′-bipyridine. Nat. Commun. 2015, 6, 8288. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Robinson, G.H. Organometallics of the Group 13 M–M Bond (M = Al, Ga, In) and the Concept of Metalloaromaticity. Organometallics 2007, 26, 2–11. [Google Scholar] [CrossRef]
- Li, X.W.; Xie, Y.; Schreiner, P.R.; Gripper, K.D.; Crittendon, R.C.; Campana, C.F.; Schaefer, H.F.; Robinson, G.H. Cyclogallanes and metalloaromaticity. Synthesis and molecular structure of dipotassium tris ((2,6-dimesitylphenyl) cyclogallene), K2[(Mes2C6H3)Ga]3 (Mes = 2,4,6-Me3C6H2): A structural and theoretical examination. Organometallics 1996, 15, 3798–3803. [Google Scholar] [CrossRef]
- Xie, Y.; Schreiner, P.R.; Schaefer, H.F.; Li, X.W.; Robinson, G.H. Are Cyclogallenes [M2 (GaH)3](M = Li, Na, K) Aromatic? J. Am. Chem. Soc. 1996, 118, 10635–10639. [Google Scholar] [CrossRef]
- Xie, Y.; Schreiner, P.R.; Schaefer, H.F.; Li, X.W.; Robinson, G.H. Are Heterocyclic 2π-Electron Aromatic Systems HC–Ga(H)–CH, M[HGa–C(H)–GaH], [HGa–C(H)–GaH]−, HSi–Ga(H)–SiH, M[HGa–Si(H)–GaH](M = Li, Na, and K), and [HGa–Si(H)–GaH]− Stable? Organometallics 1998, 17, 114–122. [Google Scholar] [CrossRef]
- Robinson, G.H. Gallanes, gallenes, cyclogallenes, and gallynes: Organometallic chemistry about the gallium–gallium bond. Acc. Chem. Res. 1999, 32, 773–782. [Google Scholar] [CrossRef]
- Wright, R.J.; Brynda, M.; Power, P.P. Synthesis and Structure of the “Dialuminyne” Na2[Ar′ AlAlAr′] and Na2[(Ar′′ Al)3]: Al-Al Bonding in Al2Na2 and Al3Na2 Clusters. Angew. Chem. Int. Ed. 2006, 45, 5953–5956, Version Angew. Chem. 2006, 118, 6099–6102. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, T.; Braunschweig, H.; Radacki, K. The Triboracyclopropenyl Dianion: The Lightest Possible Main-Group-Element Hückel π Aromatic. Angew. Chem. Int. Ed. 2015, 54, 15084–15088. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wu, B.; Yu, C.; Li, T.; Zhang, W.; Xi, Z. Trishomoaromatic (B3N3Ph6)-Dianion: Characterization and Two-Electron Reduction. Angew. Chem. Int. Ed. 2020, 59, 8868–8872. [Google Scholar] [CrossRef] [PubMed]
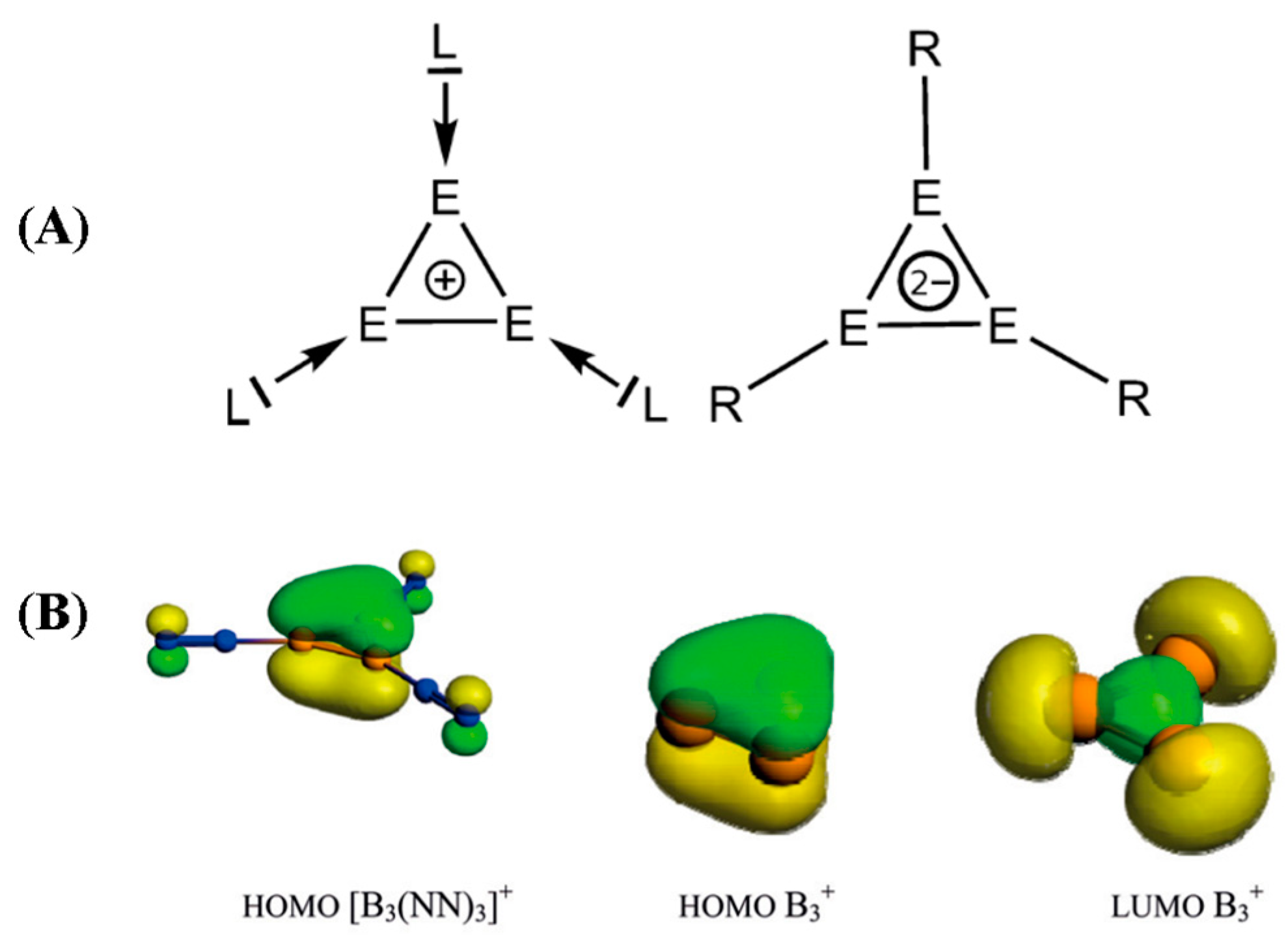
- Jin, J.; Wang, G.; Zhou, M.; Andrada, D.; Hermann, M.; Frenking, G. The [B3(NN)3]+ and [B3(CO)3]+ Complexes Featuring the Smallest π-Aromatic Species B3+. Angew. Chem. 2016, 128, 2118–2122. [Google Scholar] [CrossRef]
- Saha, R.; Pan, S.; Mandal, S.; Orozco, M.; Merino, G.; Chattaraj, P. Noble gas supported B3+ cluster: Formation of strong covalent noble gas–boron bonds. RSC Adv. 2016, 6, 78611–78620. [Google Scholar] [CrossRef]
- Zhang, R.; Li, A.; Li, Z. Circular cationic compounds B3Rgn+ of triangular ion B3+ trapping rare gases. Chem. Res. Chin. Univ. 2017, 33, 958–964. [Google Scholar] [CrossRef]
- Kuznetsov, A.E.; Boldyrev, A.I. Theoretical Evidence of Aromaticity in X3− (X = B, Al, Ga) Species. Struct. Chem. 2002, 13, 141–148. [Google Scholar] [CrossRef]
- Ullah, S.; Mazumder, L.; Kaushik, S.; Das, N.; Brahma, M.; Sharma, P.; Guha, A. Electronic Structure, Stability, and Aromaticity of H2B2XH (X = N, P) molecules: A Theoretical Study. Comput. Theor. Chem. 2017, 1113, 120–125. [Google Scholar] [CrossRef]
- Cheung, L.F.; Kocheril, G.S.; Czekner, J.; Wang, L.S. Observation of Möbius Aromatic Planar Metallaborocycles. J. Am. Chem. Soc. 2020, 142, 3356–3360. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.J.; Wang, L.S. All-Boron Aromatic Clusters as Potential New Inorganic Ligands and Building Blocks in Chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Jemmis, E.D.; Srinivas, G.N.; Leszczynski, J.; Kapp, J.; Korkin, A.A.; von R. Schleyer, P. Group 14 analogs of the cyclopropenium ion: Do they favor classical aromatic structures? J. Am. Chem. Soc. 1995, 117, 11361–11362. [Google Scholar] [CrossRef]
- Cheng, N.; Liu, Y.; Zhang, C. Theoretical studies of traditional and halogen-shared halogen bonds: The doped all-metal aromatic clusters MAl3− (M = Si, Ge, Sn, Pb) as halogen bond acceptors. Theor. Chem. Acc. 2015, 134, 140. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, J.; Bu, Y.; Cheng, S. Observation of “Outlaw” Dual Aromaticity in Unexpectedly Stable Open-Shell Metal Clusters Caused by Near-Degenerate Molecular Orbital Coupling. CCS Chem. 2020, 2, 1913–1920. [Google Scholar] [CrossRef]
- Lee, V.Y.; Sekiguchi, A. Aromaticity of group 14 organometallics: Experimental aspects. Angew. Chem. Int. Ed. 2007, 46, 6596–6620. [Google Scholar] [CrossRef]
- Kar, S.; Chatterjee, D.; Halet, J.; Ghosh, S. Trimetallic Chalcogenide Species: Synthesis, Structures, and Bonding. Molecules 2022, 27, 7473. [Google Scholar] [CrossRef]
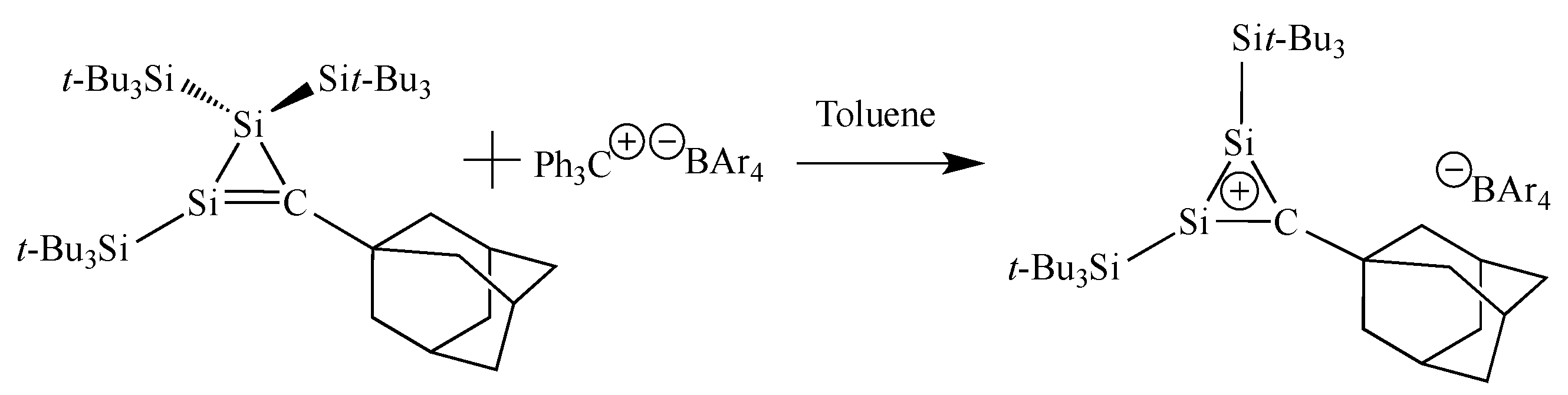
- Sekiguchi, A.; Tsukamoto, M.; Ichinohe, M.A. A free cyclotrigermenium cation with a 2π-electron system. Science 1997, 275, 60–61. [Google Scholar] [CrossRef]
- Ichinohe, M.; Igarashi, M.; Sanuki, K.; Sekiguchi, A. Cyclotrisilenylium ion: The persilaaromatic compound. J. Am. Chem. Soc. 2005, 127, 9978–9979. [Google Scholar] [CrossRef]
- Igarashi, M.; Ichinohe, M.; Sekiguchi, A. Air-Stable Disilacyclopropene with a Si-C Bond and Its Conversion to Disilacyclopropenylium Ion: Silicon–Carbon Hybrid 2π-Electron Systems. J. Am. Chem. Soc. 2007, 129, 12660–12661. [Google Scholar] [CrossRef]
- Kuwabara, T.; Guo, H.D.; Nagase, S.; Saito, M. Diversity of the Structures in a Distannene Complex and its Reduction to Generate a Six-Membered Ti2Sn4 Ring Complex. Angew. Chem. Int. Ed. 2014, 53, 434–438, Version Angew. Chem. 2014, 126, 444–448. [Google Scholar] [CrossRef]
- Pan, F.; Xu, C.; Li, L.; Min, X.; Wang, J.; Li, J.; Zhai, H.; Sun, Z. A niobium-necked cluster [As3Nb(As3Sn3)]3− with aromatic Sn32−. Dalton Trans. 2016, 45, 3874–3879. [Google Scholar] [CrossRef]
- Robilotto, T.J.; Bacsa, J.; Gray, T.G.; Sadighi, J.P. Synthesis of a trigold monocation: An isolobal analogue of [H3]+. Angew. Chem. Int. Ed. 2012, 51, 12077–12080, Version Angew. Chem. 2012, 124, 12243–12246. [Google Scholar] [CrossRef]
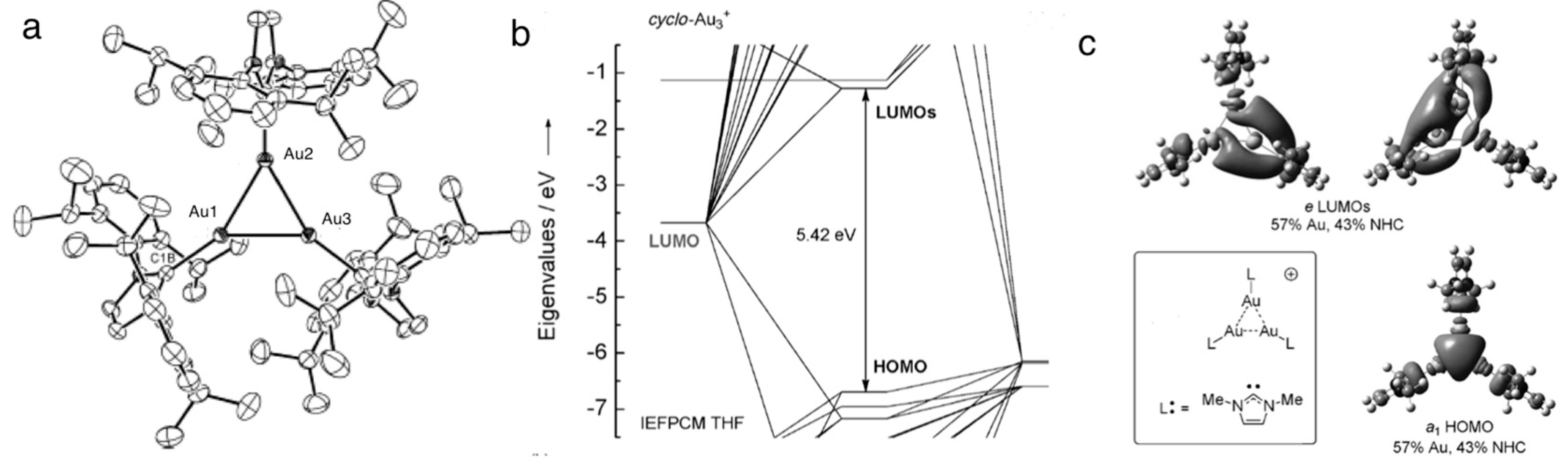
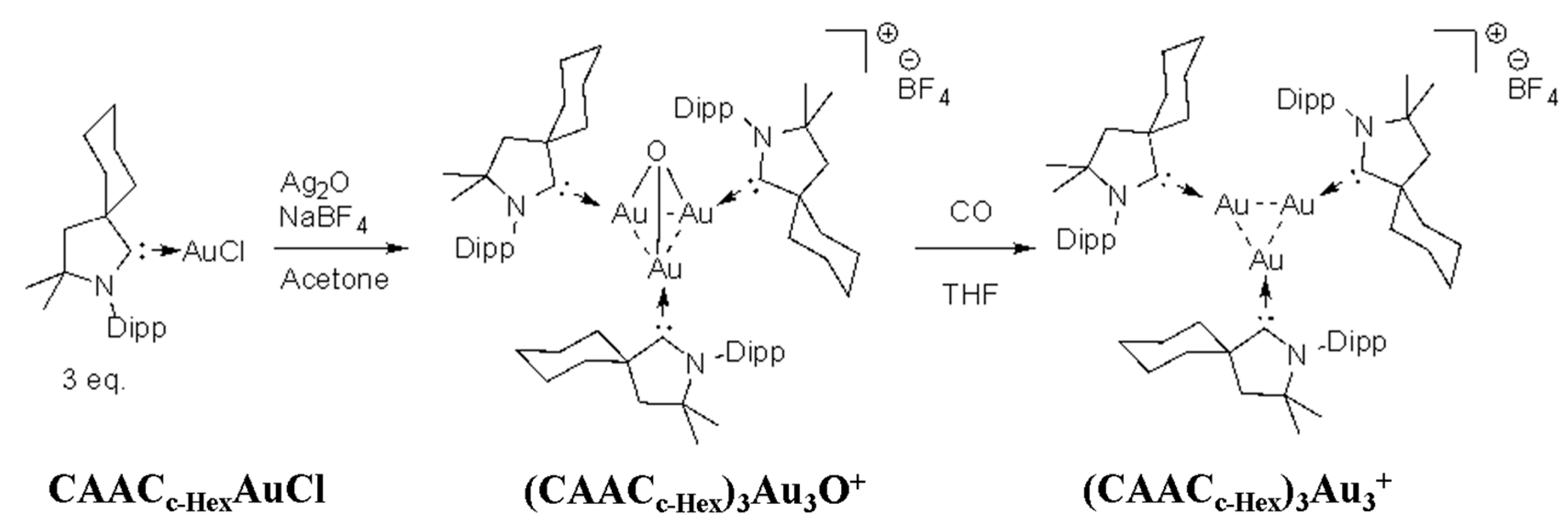
- Jin, L.; Weinberger, D.S.; Melaimi, M.; Moore, C.E.; Rheingold, A.L.; Bertrand, G. Trinuclear gold clusters supported by cyclic (alkyl)(amino) carbene ligands: Mimics for gold heterogeneous catalysts. Angew. Chem. Int. Ed. 2014, 53, 9059–9063. [Google Scholar] [CrossRef]
- Freitag, K.; Gemel, C.; Jerabek, P.; Oppel, M.I.; Seidel, R.W.; Frenking, G.; Banh, H.; Dilchert, K.; Fischer, R.A. The σ-Aromatic Clusters [Zn3]+ and [Zn2Cu]: Embryonic Brass; Version. Angew. Chem. Int. Ed. 2015, 54, 4370–4374, Version Angew. Chem. 2015, 127, 4445–4449. [Google Scholar] [CrossRef]
- Banh, H.; Hornung, J.; Kratz, T.; Gemel, C.; Pöthig, A.; Gam, F.; Kahlal, S.; Saillard, J.Y.; Fischer, R.A. Embryonic Brass: Pseudo Two Electron Cu/Zn Clusters. Chem. Sci. 2018, 9, 8906–8913. [Google Scholar] [CrossRef]
- Resa, I.; Carmona, E.; Gutierrez-Puebla, E.; Monge, A. Decamethyldizincocene, a stable compound of Zn (I) with a Zn-Zn bond. Science 2004, 305, 1136–1138. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101–194117. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R.; Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Mühlecker-Knoepfler, A.; Ellmerer-Muller, E.; Konrat, R.; Ongania, K.H.; Wurst, K.; Peringer, P.J. Synthesis and crystal structure of the subvalent mercury cluster [triangulo-Hg3(µ-dmpm)4][O3SCF 3]4 (dmpm = Me2PCH2PMe2). Chem. Soc. Dalton Trans. 1997, 1607–1610. [Google Scholar] [CrossRef]
- Hämmerle, B.; Müller, E.P.; Wilkinson, D.L.; Muüller, G.; Peringer, P. Synthesis and structure of bis[sulphato]tris[µ-bis(diphenylphosphino) methane]-triangulo-trimercury. J. Chem. Soc. Chem. Commun. 1989, 1527–1528. [Google Scholar] [CrossRef]
- Blanchard, S.; Fensterbank, L.; Gontard, G.; Lacôte, E.; Maestri, G.; Malacria, M. Synthesis of triangular tripalladium cations as noble-metal analogues of the cyclopropenyl cation. Angew. Chem. Int. Ed. 2014, 53, 1987–1991, Version Angew. Chem. 2014, 126, 2018–2022. [Google Scholar] [CrossRef]
- Wang, Y.; Deyris, P.A.; Caneque, T.; Blanchard, F.; Li, Y.; Bigi, F.; Maggi, R.; Blanchard, S.; Maestri, G.; Malacria, M. A Simple Synthesis of Triangular All-Metal Aromatics Allowing Access to Isolobal All-Metal Heteroaromatics. Chem. Eur. J. 2015, 21, 12271–12274. [Google Scholar] [CrossRef]
- Albrecht, C.; Schwieger, S.; Bruhn, C.; Wagner, C.; Kluge, R.; Schmidt, H.; Steinborn, D.A. Alkylthio bridged 44 cve triangular platinum clusters: Synthesis, oxidation, degradation, ligand substitution, and quantum chemical calculations. J. Am. Chem. Soc. 2007, 129, 4551–4566. [Google Scholar] [CrossRef]
- Murahashi, T.; Usui, K.; Tachibana, Y.; Kimura, S.; Ogoshi, S. Selective Construction of Pd2Pt and PdPt2 Triangles in a Sandwich Framework: Carbocyclic Ligands as Scaffolds for a Mixed-Metal System. Chem. Eur. J. 2012, 18, 8886–8890. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Natural bond orbitals and extensions of localized bonding concepts. Chem. Educ. Res. Pract. Eur. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef]
- Aihara, J. Circuit resonance energy: A key quantity that links energetic and magnetic criteria of aromaticity. J. Am. Chem. Soc. 2006, 128, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Proft, F.; von R. Schleyer, P.; Lenthe, J.H.; Stahl, F.; Geerlings, P. Magnetic Properties and Aromaticity of o-, m-, and p-Benzyne. Chem. Eur. J. 2002, 8, 3402–3410. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Barczynski, P.; Musumarra, G.; Pisano, D.; Szafran, M. Aromaticity as a quantitative concept. 1. A statistical demonstration of the orthogonality of classical and magnetic aromaticity in five-and six-membered heterocycles. J. Am. Chem. Soc. 1989, 111, 7–15. [Google Scholar] [CrossRef]
- Kulichenko, M.; Fedik, N.; Monfredini, A.; Muñoz-Castro, A.; Balestri, D.; Boldyrev, A.I.; Maestri, G. “Bottled” spiro-doubly aromatic trinuclear [Pd2Ru]+ complexes. Chem. Sci. 2021, 12, 477–486. [Google Scholar] [CrossRef]
- Boronski, J.T.; Seed, J.A.; Hunger, D.; Woodward, A.W.; van Slageren, J.; Wooles, A.J.; Natrajan, L.S.; Kaltsoyannis, N.; Liddle, S.T. A Crystalline Tri-Thorium Cluster with σ-Aromatic Metal–Metal Bonding. Nature 2021, 598, 75. [Google Scholar] [CrossRef]
- Cuyacot, B.J.R.; Foroutan-Nejad, C. [{Th(C8H8)Cl2}3]2− is stable but not aromatic. Nature 2022, 603, E18. [Google Scholar] [CrossRef]
- Chen, W.J.; Zhai, H.J.; Zhang, Y.F.; Huang, X.; Wang, L.S. On the Electronic and Structural Properties of Tri-Niobium Oxide Clusters Nb3On− (n = 3–8): Photoelectron Spectroscopy and Density Functional Calculations. J. Phys. Chem. A 2010, 114, 5958–5966. [Google Scholar] [CrossRef]
- Weck, P.F.; Sergeeva, A.P.; Kim, E.; Boldyrev, A.I.; Czerwinski, K.R. Chemical bonding and aromaticity in trinuclear transition-metal halide clusters. Inorg. Chem. 2011, 50, 1039–1046. [Google Scholar] [CrossRef]
- Galeev, T.R.; Boldyrev, A.I. Recent advances in aromaticity and antiaromaticity in transition-metal systems. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 2011, 107, 124–147. [Google Scholar] [CrossRef]
- Ivanov, A.S.; Zhang, X.; Wang, H.; Boldyrev, A.I.; Gantefoer, G.; Bowen, K.H.; Cernusak, I. Anion photoelectron spectroscopy and CASSCF/CASPT2/RASSI study of Lan− (n = 1, 3–7). J. Phys. Chem. A 2015, 119, 11293–11303. [Google Scholar] [CrossRef]
- Vásquez-Espinal, A.; Pino-Rios, R.; Alvarez-Thon, L.; Rabanal-León, W.A.; Torres-Vega, J.J.; Arratia-Perez, R.; Tiznado, W. New Insights into Re3(μ-Cl)3Cl6 Aromaticity. Evidence of σ-and π-Diatropicity. J. Phys. Chem. Lett. 2015, 6, 4326–4330. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.S. Beyond organic chemistry: Aromaticity in atomic clusters. Phys. Chem. Chem. Phys. 2016, 18, 11589–11605. [Google Scholar] [CrossRef]
- von R. Schleyer, P.; Maerker, C.; Dransfeld, A.; Jiao, H.; Eikema Hommes, N.J.R. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Li, X.; Kuznetsov, A.E.; Zhang, H.F.; Boldyrev, A.I.; Wang, L.S. Observation of all-metal aromatic molecules. Science 2001, 291, 859–861. [Google Scholar] [CrossRef]
- Frenking, G. Building a quintuple bond. Science 2005, 310, 796–797. [Google Scholar] [CrossRef]
- Radius, U.; Breher, F. To boldly pass the metal–metal quadruple bond. Angew. Chem. Int. Ed. 2006, 45, 3006–3010, Version Angew. Chem. 2006, 118, 3072–3077. [Google Scholar] [CrossRef]
- Brynda, M.; Gagliardi, L.; Widmark, P.O.; Power, P.P.; Roos, B.O. A Quantum Chemical Study of the Quintuple Bond between Two Chromium Centers in [PhCrCrPh]: Trans-Bent versus Linear Geometry. Angew. Chem. 2006, 118, 3888–3891, Version Angew. Chem. Int. Ed. 2006, 45, 3804–3807. [Google Scholar] [CrossRef]
- Zhai, H.; Averkiev, B.B.; Zubarev, D.Y.; Wang, L.S.; Boldyrev, A.I. δ Aromaticity in [Ta3O3]−. Angew. Chem. 2007, 119, 4355–4358. [Google Scholar] [CrossRef]
- Wang, L.S.; Wu, H. Advances in Metaland Semiconductor Clusters, IV; Cluster, Materials; Duncan, M.A., Ed.; JAI: Greenwich, CT, USA, 1998; pp. 299–343. [Google Scholar]
- Badri, Z.; Pathak, S.; Fliegl, H.; Rashidi-Ranjbar, P.; Bast, R.; Marek, R.; Foroutan-Nejad, C.; Ruud, K. All-metal aromaticity: Revisiting the ring current model among transition metal clusters. J. Chem. Theory Comput. 2013, 9, 4789–4796. [Google Scholar] [CrossRef]
- Mercero, J.M.; Matito, E.; Ruiperez, F.; Infante, I.; Lopez, X.; Ugalde, J.M. The Electronic Structure of the Al3− Anion: Is it Aromatic? Chem. Eur. J. 2015, 21, 9610–9614. [Google Scholar] [CrossRef]
- Chen, J.; Yang, H.; Wang, J.; Cheng, S. Theoretical investigations on the d-p hybridized aromaticity, photoelectron spectroscopy and neutral salts of the LaX2− (X = Al, Ga, In) clusters. Spectrochim. Acta Part A 2018, 203, 132–138. [Google Scholar] [CrossRef]
- Yang, H.; Wu, D.; He, H.; Yu, D.; Li, Y.; Li, Z. The behavior of the aluminum trimer when combining with different superatom clusters. RSC Adv. 2018, 8, 6667–6674. [Google Scholar] [CrossRef]
- Xu, C.Q.; Xing, D.H.; Xiao, H.; Li, J. Manipulating stabilities and catalytic properties of trinuclear metal clusters through tuning the chemical bonding: H2 adsorption and activation. J. Phys. Chem. C 2017, 121, 10992–11001. [Google Scholar] [CrossRef]
- Reid, S.; Hernández, H. Characterization of the Effects of Ligands on Bonding and σ-Aromaticity of Small Pt Nanoclusters. J. Phys. Chem. A 2023, 127, 4237–4244. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Kimura, S.; Takase, K.; Yamamoto, K.; Kurashige, Y.; Yanai, T.; Murahashi, T. Modulation of Benzene or Naphthalene Binding to Palladium Cluster Sites by the Backside-Ligand Effect. Angew. Chem. Int. Ed. 2015, 54, 2482–2486. [Google Scholar] [CrossRef]
- Link, H.; Reiss, P.; Chitsaz, S.; Pfistner, H.; Fenske, D. Synthese und Molekülstrukturen von amido-und imidoverbrückten Clustern elektronenreicher Übergangsmetalle. Z. Für Anorg. Und Allg. Chem. 2003, 629, 755–768. [Google Scholar] [CrossRef]
- Lee, S.W.; Trogler, W.C. Synthesis and structure of the trinuclear palladium cluster [Pd3(PEt3)3 (μ2-NPh)2(μ2-NHPh)]Cl, containing bridging imido and amido ligands. Inorg. Chem. 1990, 29, 1099–1102. [Google Scholar] [CrossRef]
- Averkiev, B.B.; Boldyrev, A.I. Hf3 Cluster Is Triply (σ-, π-, and δ-) Aromatic in the Lowest D3h, 1A1′ State. J. Phys. Chem. A 2007, 111, 12864–12866. [Google Scholar] [CrossRef]
- Jin, Q.; Jin, B.; Jin, F.-K.; Li., J.-P. Theoretical evidence of triple (σ-, π-, and δ-) aromaticity in the Os3N3+/− clusters. Comput. Thero. Chem. 2017, 1101, 127–131. [Google Scholar] [CrossRef]
- Jin, Q.; Jin, B.; Gong, L.; Jin, F. Aromaticity of the bare osmium trimers and the bindings to group IA/IIA all-metal series. Comput. Theor. Chem. 2017, 1102, 74–79. [Google Scholar] [CrossRef]
- Jin, Q.; Jin, B.; Jin, Y.; Ding, H. On the electronic structures and multiple aromaticity in the Ir3N32+/0/2− clusters and the Ir3N3M−/0 or Ir3N3M2 complexes with group IA/IIA metals. Comput. Theor. Chem. 2017, 1118, 75–80. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, T.; Chen, W.; Li, W.; Zhao, J.; Jiang, X.; Li, J.; Wang, L.; Hu, H. The smallest 4f-metalla-aromatic molecule of cyclo-PrB2− with Pr-B multiple bonds. Chem. Sci. 2022, 13, 10082–10094. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; Proft, F.; Fowler, P.W.; Geerlings, P. σ-Aromaticity in H3+ and Li3+: Insights from ring-current maps. Chem. Phys. Lett. 2005, 407, 391–396. [Google Scholar] [CrossRef]
- Park, J.; Yoon, J.; Chung, Y. Cobalt/Rhodium Heterobimetallic Nanoparticle-Catalyzed Oxidative Carbonylation of Amines in the Presence of Carbon Monoxide and Molecular Oxygen to Ureas. Adv. Synth. Catal. 2009, 351, 1233–1237. [Google Scholar] [CrossRef]
- Shelton, P.; Zhang, Y.; Nguyena, T.; McElwee-White, L. NaIO4-oxidized carbonylation of amines to ureas. Chem. Commun. 2009, 947–949. [Google Scholar] [CrossRef]
- Díaz, D.; Darko, A.; McElwee-White, L. Transition Metal-Catalyzed Oxidative Carbonylation of Amines to Ureas. Eur. J. Org. Chem. 2007, 2007, 4453–4465. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M. Reviews. In Catalytic Carbonylation Reactions; Beller, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 239–272. [Google Scholar]
- Ragaini, F. Away from phosgene: Reductive carbonylation of nitroarenes and oxidative carbonylation of amines, understanding the mechanism to improve performance. Dalton Trans. 2009, 32, 6251–6266. [Google Scholar] [CrossRef]
- Hashmi, A.S.K. Sub-nanosized gold catalysts. Science 2012, 338, 1434. [Google Scholar] [CrossRef]
- Gramage-Doria, R.; Reek, J.N.H. Neues aus der Goldkatalyse–die Größe zählt. New Endeavors in Gold Catalysis—Size Matters. Angew. Chem. 2013, 125, 13384–13386, New Endeavors in Gold Catalysis—Size Matters. Angew. Chem. Int. Ed. 2013, 52, 13146–13148. [Google Scholar] [CrossRef]
- Blanco Jaimes, M.C.; Bçhling, C.R.N.; Serrano-Becerra, J.M.; Hashmi, A.S.K. Hochaktive einkernige NAC-Gold (I)-Katalysatoren. Angew. Chem. 2013, 125, 8121–8124, Highly active mononuclear NAC–gold (I) catalysts. Angew. Chem. Int. Ed. 2013, 52,7963–7966. [Google Scholar] [CrossRef]
- Blanco Jaimes, M.C.; Rominger, F.; Pereira, M.M.; Carrilho, R.M.B.; Carabineiroc, S.A.C.; Hashmi, A.S.K. Highly active phosphite gold (I) catalysts for intramolecular hydroalkoxylation, enyne cyclization and furanyne cyclization. Chem. Commun. 2014, 50, 4937–4940. [Google Scholar] [CrossRef]
- Pan, S.; Saha, R.; Mandal, S.; Chattaraj, P. σ-Aromatic cyclic M3+ (M = Cu, Ag, Au) clusters and their complexation with dimethyl imidazol-2-ylidene, pyridine, isoxazole, furan, noble gases and carbon monoxide. Phys. Chem. Chem. Phys. 2016, 18, 11661–11676. [Google Scholar] [CrossRef]
- Wang, L.; Xu, J.; Kira, M.; Yan, L.; Xiao, X.; Li, Z. A Stable Cyclic (R2SnAu)3 Anion Having In-Plane σ-Möbius Aromaticity. Angew. Chem. Int. Ed. 2020, 132, 1996–2000. [Google Scholar] [CrossRef]
- Parida, R.; Reddy, G.N.; Ganguly, A.; Roymahapatra, G.; Chakraborty, A.; Giri, S. On the making of aromatic organometallic superalkali complexes. Chem. Commun. 2018, 54, 3903–3906. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Sun, L.; Wang, X.; Yuan, L.; Wu, L.; Liu, X.; Wang, Y. Syntheses of Triangular Gold Complexes and Their Applications in Hydroamination Reaction. Eur. J. Inorg. Chem. 2021, 40, 4230–4237. [Google Scholar] [CrossRef]
- Zeng, M.S.; Li, L.; Herzon, S.B. A highly active and air-stable ruthenium complex for the ambient temperature anti-Markovnikov reductive hydration of terminal alkynes. J. Am. Chem. Soc. 2014, 136, 7058–7067. [Google Scholar] [CrossRef]
- Li, G.; Jin, R.-C. Gold nanocluster-catalyzed semihydrogenation: A unique activation pathway for terminal alkynes. J. Am. Chem. Soc. 2014, 136, 11347–11354. [Google Scholar] [CrossRef]
- Radkowski, K.; Sundararaju, B.; Furstner, A. A Functional-Group-Tolerant Catalytic trans Hydrogenation of Alkynes. Angew. Chem. Int. Ed. 2013, 52, 355–360, Version Angew. Chem. 2013, 125, 373–378. [Google Scholar] [CrossRef]
- Jeddi, N.; Scott, N.; Fairlamb, I. Well-Defined Pdn Clusters for Cross-Coupling and Hydrogenation Catalysis: New Opportunities for Catalyst Design. ACS Catal. 2022, 12, 11615–11638. [Google Scholar] [CrossRef]
- Karunananda, M.; Mankad, N. E-Selective Semi-Hydrogenation of Alkynes by Heterobimetallic Catalysis. J. Am. Chem. Soc. 2015, 137, 14598–14601. [Google Scholar] [CrossRef]
- Deyris, P.A.; Caneque, T.; Wang, Y.; Retailleau, P.; Bigi, F.; Maggi, R.; Maestri, G.; Malacria, M. Catalytic Semireduction of Internal Alkynes with All-Metal Aromatic Complexes. ChemCatChem. 2015, 7, 3266–3269. [Google Scholar] [CrossRef]
- Fürstner, A. Gold and platinum catalysis—A convenient tool for generating molecular complexity. Chem. Soc. Rev. 2009, 38, 3208–3221. [Google Scholar] [CrossRef]
- Fürstner, A.; Davies, P.W. Catalytic carbophilic activation: Catalysis by platinum and gold π acids. Angew. Chem. Int. Ed. 2007, 46, 3410–3449. [Google Scholar] [CrossRef]
- Fürstner, A. From understanding to prediction: Gold-and platinum-based π-acid catalysis for target oriented synthesis. Acc. Chem. Res. 2014, 47, 925–938. [Google Scholar] [CrossRef]
- Guo, H.; Zheng, Z.L.; Yu, F.; Ma, S.M.; Holuigue, A.; Tromp, D.S.; Elsevier, C.J.; Yu, Y.H. [Pd(Ar-BIAN)(alkene)]-Catalyzed Highly Chemo-, Regio-, and Stereoselective Semihydrogenation of 1, 2-Allenyl Phosphonates and Related Compounds. Angew. Chem. Int. Ed. 2006, 45, 4997–5000, Version Angew. Chem. 2006, 118, 5119–5122. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gallego, M.; Sierra, M.A. Kinetic isotope effects in the study of organometallic reaction mechanisms. Chem. Rev. 2011, 111, 4857–4963. [Google Scholar] [CrossRef]
- Monfredini, A.; Santacroce, V.; Deyris, P.A.; Maggi, R.; Bigi, F.; Maestri, G.; Malacria, M. Boosting catalyst activity in cis-selective semi-reduction of internal alkynes by tailoring the assembly of all-metal aromatic tri-palladium complexes. Dalton Trans. 2016, 45, 15786–15790. [Google Scholar] [CrossRef] [PubMed]
- Monfredini, A.; Santacroce, V.; Marchiò, L.; Maggi, R.; Bigi, F.; Maestri, G.; Malacria, M. Semi-Reduction of internal alkynes with prototypical subnanometric metal surfaces: Bridging homogeneous and heterogeneous catalysis with trinuclear all-metal aromatics. ACS Sustain. Chem. Eng. 2017, 5, 8205–8212. [Google Scholar] [CrossRef]
- Serafino, A.; Camedda, N.; Lanzi, M.; Ca’, N.; Cera, G.; Maestri, G. Inter/Intramolecular Cascade of 1,6-Enynes Catalyzed by All-Metal Aromatic Tripalladium Complexes and Carboxylic Acids. J. Org. Chem. 2021, 86, 15433–15452. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Lan, P.C.; Chen, M.; Zhang, W.; Ma, S. Heterogenization of Trinuclear palladium complex into an anionic metal–organic framework through postsynthetic cation exchange. Organometallics 2019, 38, 3460–3465. [Google Scholar] [CrossRef]
- Lanzi, M.; Cañeque, T.; Marchioò, L.; Maggi, R.; Bigi, F.; Malacria, M.; Maestri, G. Alternative Routes to Tricyclic Cyclohexenes with Trinuclear Palladium Complexes. ACS Catal. 2018, 8, 144–147. [Google Scholar] [CrossRef]
- Cecchini, C.; Lanzi, M.; Cera, G.; Malacria, M.; Maestri, G. Complementary Reactivity of 1, 6-Enynes with All-Metal Aromatic Trinuclear Complexes and Carboxylic Acids. Synthesis 2019, 51, 1216–1224. [Google Scholar] [CrossRef]
- Fürstner, A.; Davies, P.; Gress, T. Cyclobutenes by Platinum-Catalyzed Cycloisomerization Reactions of Enynes. J. Am. Chem. Soc. 2005, 127, 8244–8245. [Google Scholar] [CrossRef]
- Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. Platinum- and Gold-Catalyzed Cycloisomerization Reactions of Hydroxylated Enynes. J. Am. Chem. Soc. 2004, 126, 8654–8655. [Google Scholar] [CrossRef] [PubMed]
- Fu, F.; Xiang, J.; Cheng, H.; Cheng, L.; Chong, H.; Wang, S.; Li, P.; Wei, S.; Zhu, M.; Li, Y. A robust and efficient Pd3 cluster catalyst for the Suzuki reaction and its odd mechanism. ACS Catal. 2017, 7, 1860–1867. [Google Scholar] [CrossRef]
- Sain, S.; Jain, S.; Srivastava, M.; Vishwakarma, R.; Dwivedi, J. Application of Palladium-Catalyzed Cross-Coupling Reactions in Organic Synthesis. Curr. Org. Synth. 2019, 16, 1105–1142. [Google Scholar] [CrossRef]
- Pan, C.; Liu, M.; Zhang, L.; Wu, H.; Ding, J.; Cheng, J. Palladium catalyzed ligand-free Suzuki cross-coupling reaction. Catal. Commun. 2008, 9, 508–510. [Google Scholar] [CrossRef]
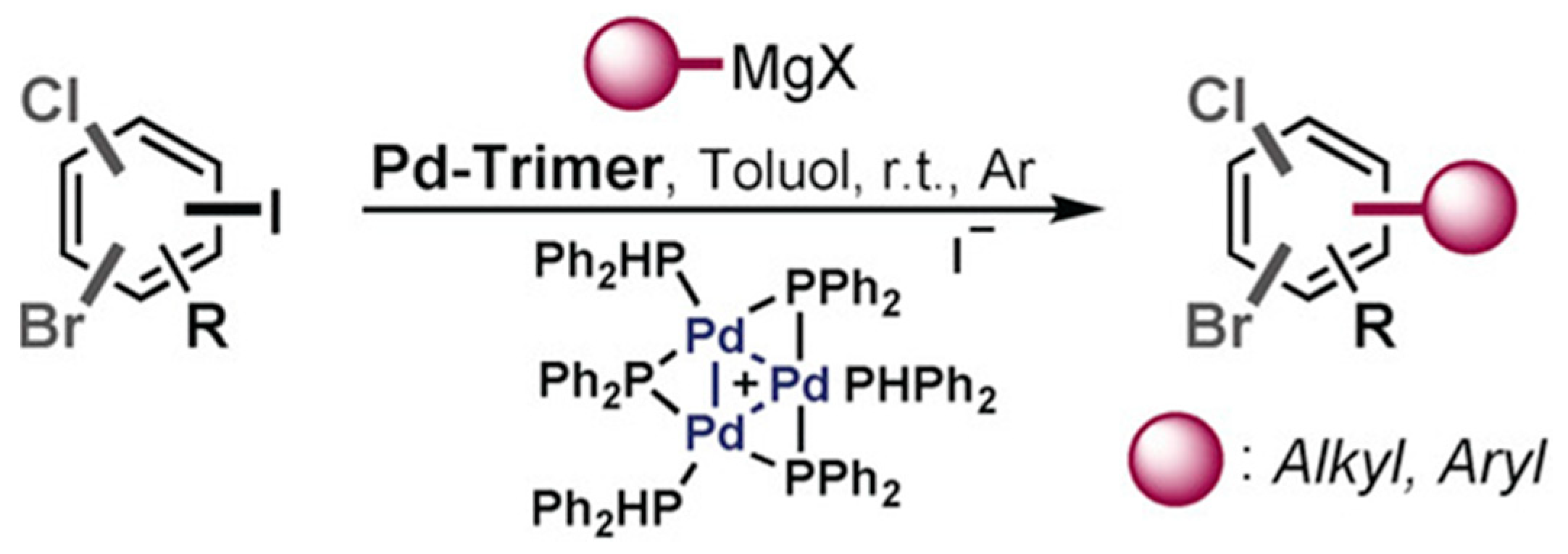
- Diehl, C.J.; Scattolin, T.; Englert, U.; Schoenebeck, F. C–I-Selective Cross-Coupling Enabled by a Cationic Palladium Trimer. Angew. Chem. Int. Ed. 2019, 58, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.W.J.; Ford, M.J.; Schotes, C.; Parker, R.R.; Whitwood, A.C.; Fairlamb, I.J.S. The Ubiquitous Cross-Coupling Catalyst System ’Pd(OAc)2/2PPh3 Forms a Unique Dinuclear PdI Complex: An Important Entry Point into Catalytically Competent Cyclic Pd3 Clusters. Chem. Sci. 2019, 10, 7898–7906. [Google Scholar] [CrossRef]
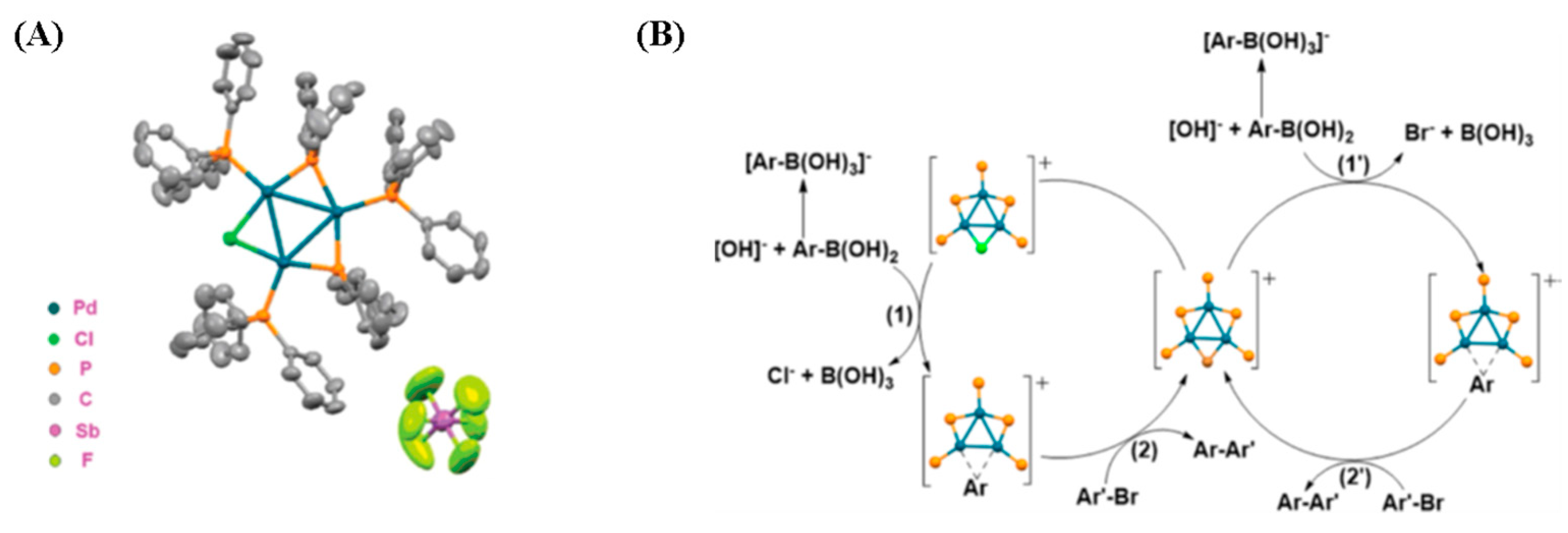
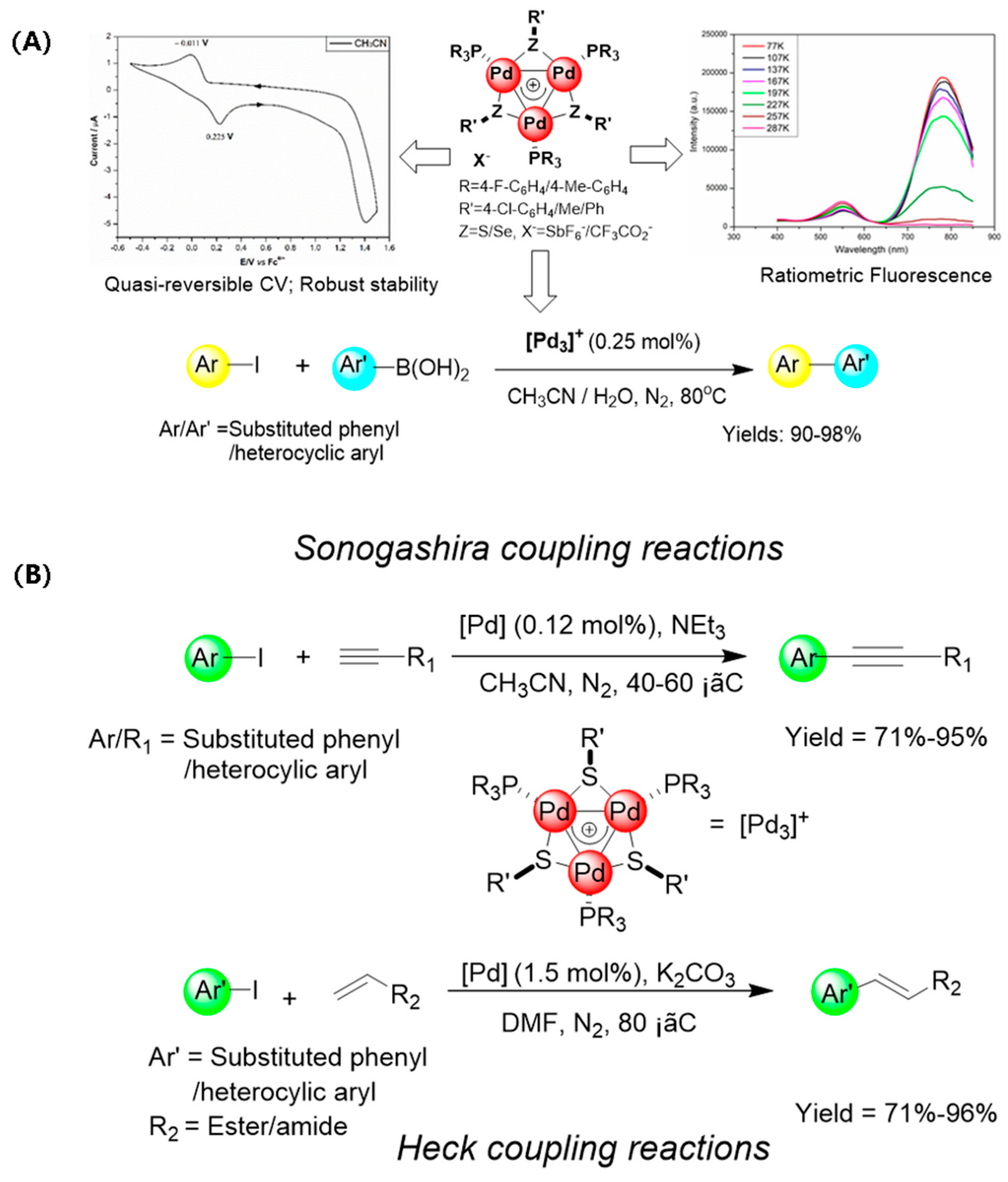
- Li, X.; Li, J.; Wang, X.; Wu, L.; Wang, Y.; Maestri, G.; Malacria, M.; Liu, X. Sulfur and aryl phosphine stabilized aromatic triangular tri-palladium complexes [Pd3]+: Photoelectric properties and their catalytic applications in the Suzuki–Miyaura reaction. Dalton Trans. 2021, 50, 11834–11842. [Google Scholar] [CrossRef]
- Wang, X.; Sun, L.; Wang, M.; Maestri, G.; Malacria, M.; Liu, X.; Wang, Y.; Wu, L. Sonogashira and Heck coupling reactions catalyzed by [Pd3]+. Eur. J. Org. Chem. 2022, 2022, e202200009. [Google Scholar] [CrossRef]
- Ma, J.C.; Dougherty, D.A. The cation–π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Schottel, B.L.; Chifotides, H.T.; Dunbar, K.R. Anion-π interactions. Chem. Soc. Rev. 2008, 37, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Tsipis, A.C.; Tsipis, C.A. Ligand-stabilized aromatic three-membered gold rings and their sandwichlike complexes. J. Am. Chem. Soc. 2005, 127, 10623–10638. [Google Scholar] [CrossRef]
- Jentzsch, A.V.; Hennig, A.; Mareda, J.; Matile, S. Synthetic ion transporters that work with anion–π interactions, halogen bonds, and anion–macrodipole interactions. Acc. Chem. Res. 2013, 46, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Archambault, C.; Bender, R.; Braunstein, P.; Dusausoy, Y.; Welter, R. Reactions of trinuclear platinum clusters with electrophiles: Ionisation isomerism with [Pt3(μ2-I)(μ-PPh2)2(PPh3)3]I and [Pt3 (μ-PPh2)2I2(PPh3)3]. Structures of [Pt3(μ2-Cl)(μ-PPh2)2(PPh3)3] PF6,[Pt3(μ-PPh2)2I2(PPh3)3] and of the Pt–Ag cluster [Pt3{μ3-AgBF4}(μ2-I)(μ-PPh2)2(PPh3)3]BF4. Dalton Trans. 2014, 43, 8609–8619. [Google Scholar] [CrossRef] [PubMed]
- Bender, R.; Welter, R.; Braunstein, P. Phosphanido-bridged triangular platinum clusters as versatile platforms: A personal account. Inorganica Chim. Acta 2015, 424, 20–28. [Google Scholar] [CrossRef]
- Gamrad, W.; Dreier, A.; Goddard, R.; Porschke, K.R. Cation–Cation Pairing by N–C–H···O Hydrogen Bonds. Angew. Chem. Int. Ed. 2015, 54, 4482–4487. [Google Scholar] [CrossRef]
- Bremer, D.C.M.; von R. Schleyer, P.; Schötz, K.; Kausch, M.; Schindler, M. Four-Center Two-Electron Bonding in a Tetrahedral Topology. Experimental Realization of Three-Dimensional Homoaromaticity in the 1, 3-Dehydro-5, 7-adamantanediyl Dication. Angew. Chem. Int. Ed. 1987, 26, 761–763. [Google Scholar] [CrossRef]
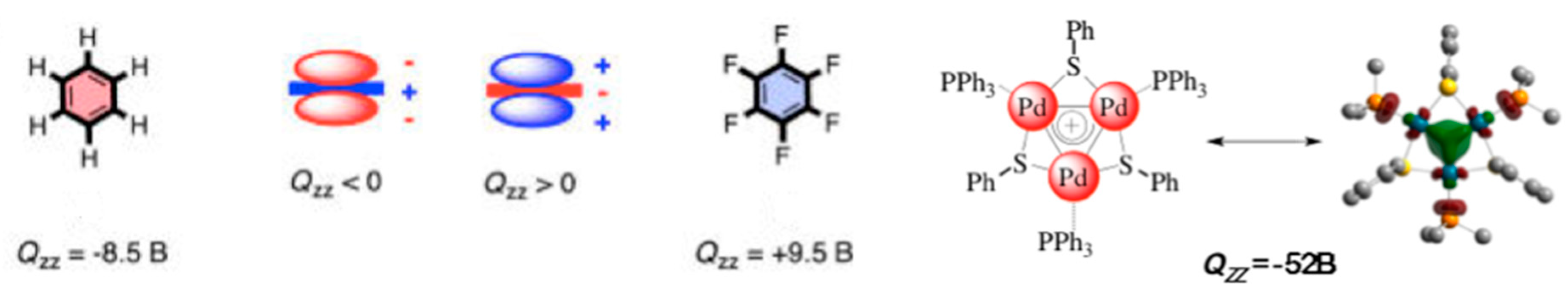
- Wang, Y.; Monfredini, A.; Deyris, P.A.; Blanchard, F.; Derat, E.; Maestri, G.; Malacria, M. All-metal aromatic cationic palladium triangles can mimic aromatic donor ligands with Lewis acidic cations. Chem. Sci. 2017, 8, 7394–7402. [Google Scholar] [CrossRef]
- Czekner, J.; Cheung, L.; Kocheril, G.; Kulichenko, M.; Boldyrev, A.; Wang, L. High-Resolution Photoelectron Imaging of IrB3−: Observation of a π-Aromatic B3+ Ring Coordinated to a Transition Metal. Angew. Chem. Int. Ed. 2019, 58, 8877–8881. [Google Scholar] [CrossRef]
- Cheung, L.; Czekner, J.; Kocheril, G.; Wang, L. High-resolution photoelectron imaging of MnB3−: Probing the bonding between the aromatic B3 cluster and 3d transition metals. J. Chem. Phys. 2020, 152, 244306. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Sheng, H.; Astruc, D.; Zhu, M. Atomically Precise Noble Metal Nanoclusters as Efficient Catalysts: A Bridge between Structure and Properties. Chem. Rev. 2020, 120, 526–622. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Bai, J.; Kang, X.; Zhu, M.; Guo, Y.; Wang, X. Non-directed C–H arylation of electron-deficient arenes by synergistic silver and Pd3 cluster catalysis. Nanoscale 2023, 15, 3560–3565. [Google Scholar] [CrossRef] [PubMed]
- Giner, X.; Najera, C.; Kovacs, G.; Lledos, A.; Ujaqueb, G. Gold versus Silver-Catalyzed Intermolecular Hydroaminations of Alkenes and Dienes. Adv. Synth. Catal. 2011, 353, 3451–3466. [Google Scholar] [CrossRef]
- Bjoremark, P.M.; Olsson, S.; Kokoli, T.; Hakansson, M. Absolute asymmetric synthesis of a tetrahedral silver complex. Chem. Eur. J. 2015, 21, 8750–8753. [Google Scholar] [CrossRef] [PubMed]
- Urnėžius, E.; Brennessel, W.W.; Cramer, C.J.; Ellis, J.E.; Schleyer, P.v.R. A carbon-free sandwich complex [(P5)2Ti]2−. Science 2002, 295, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Osuga, T.; Murase, T.; Ono, K.; Yamauchi, Y.; Fujita, M. [m×n] metal ion arrays templated by coordination cages. J. Am. Chem. Soc. 2010, 132, 15553–15555. [Google Scholar] [CrossRef] [PubMed]
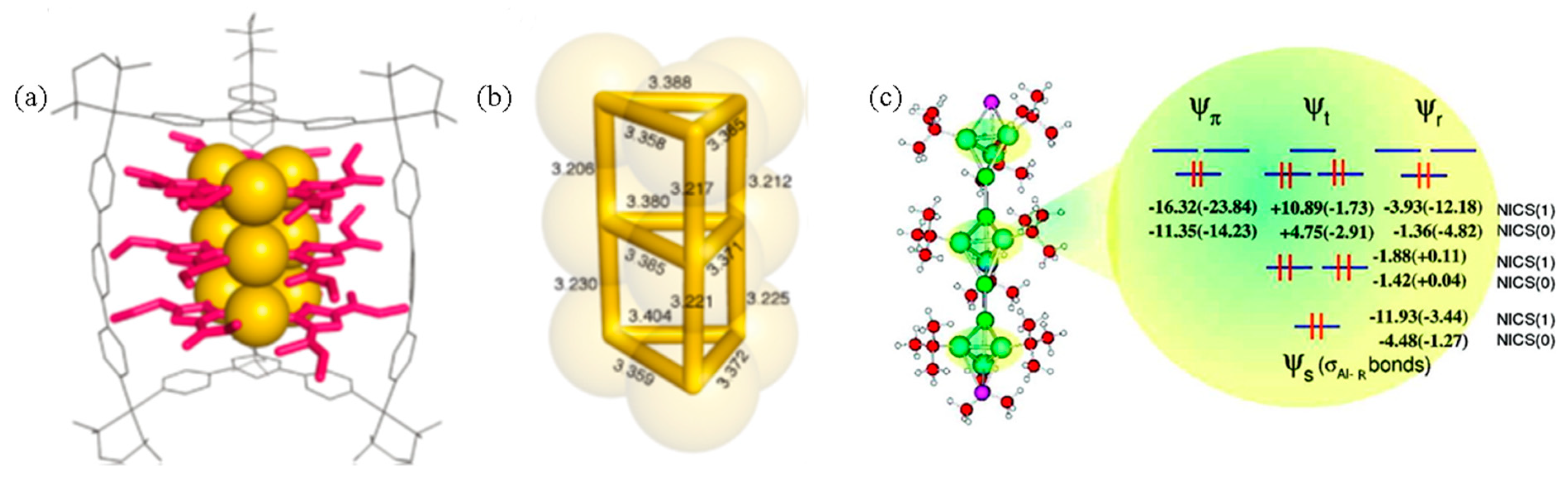
- Mercero, J.M.; Piris, M.; Matxain, J.M.; Lopez, X.; Ugalde, J.M. Sandwich complexes of the metalloaromatic η3-Al3R3 ligand. J. Am. Chem. Soc. 2009, 131, 6949–6951. [Google Scholar] [CrossRef]
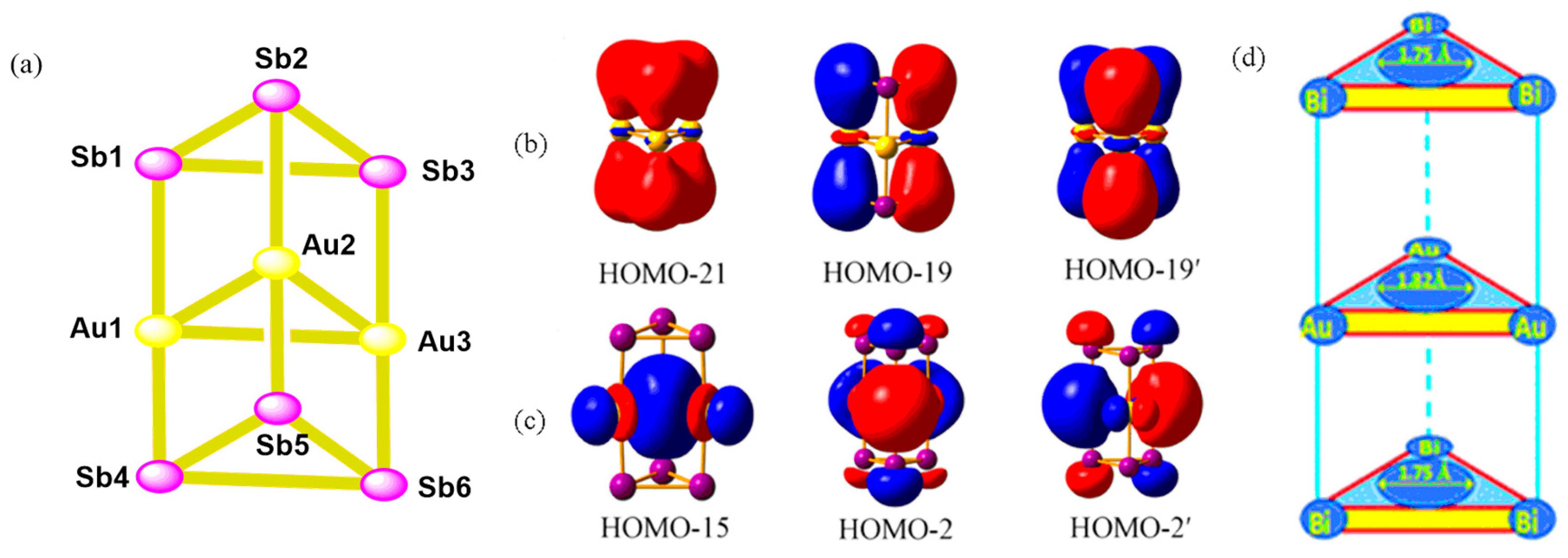
- Pan, F.X.; Li, L.J.; Wang, Y.J.; Guo, J.C.; Zhai, H.J.; Xu, L.; Sun, Z.M. An all-metal aromatic sandwich complex [Sb3Au3Sb3]3−. J. Am. Chem. Soc. 2015, 137, 10954–10957. [Google Scholar] [CrossRef]
- Li, W.; Xu, C.; Hua, S.; Li, J. Theoretical studies on the bonding and electron structures of a [Au3Sb6]3− complex and its oligomers. Dalton Trans. 2016, 45, 11657–11667. [Google Scholar] [CrossRef]
- Tang, L.H.; Zhu, T.T.; Ning, P.; Li, K.; Bao, S.Y.; Jin, X.; Zhang, X.Y. Design and structural characterization of the all-metal aromatic sandwich species [Bi3Au3Bi3]3−: Insight from density functional theory. New J. Chem. 2017, 41, 2321–2327. [Google Scholar] [CrossRef]
- Peerless, B.; Schmidt, A.; Franzke, Y.; Dehnen, S. φ-Aromaticity in prismatic {Bi6}-based clusters. Nat. Chem. 2023, 15, 347–356. [Google Scholar] [CrossRef]
- Zhu, H.; Cao, T.; Zhang, Q.; Liang, X.; Suo, B.; Zou, W.; Han, H.; Huang, Y.; Li, J. All-Metal Aromatic Sandwich Binuclear Complexes: Electronic Structures, Aromaticity and Interactions with Hydrogen via Multicenter Bonds. ChemistrySelect 2017, 2, 6206–6211. [Google Scholar] [CrossRef]
- Zhu, H.; Han, Y.; Suo, B.; Zhai, G.; Wen, Z. All-metal binuclear sandwich complexes Al4Ti2Al4: High capacity hydrogen storage through multicenter bonds. Int. J. Hydrogen Energy 2017, 42, 5440–5446. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, L.; Zhai, H. Sandwich-type Na6B7− and Na8B7+ clusters: Charge-transfer complexes, four-fold π/σ aromaticity, and dynamic fluxionality. Phys. Chem. Chem. Phys. 2019, 21, 18338–18345. [Google Scholar] [CrossRef]
- Yamaura, H.; Yamamoto, K.; Murahashi, T. Selective dimerization of a trinuclear mixed-metal sandwich complex: Construction of an axially chiral metal skeleton. Chem. Commun. 2021, 57, 9120–9123. [Google Scholar] [CrossRef] [PubMed]
- Benmachiche, A.; Zouchoune, B. Coordination and ligands’ effects in trinuclear [Pd3(COT)2(L)]2+(L = H2O, CO, N2, HCN, HNC, NH3, PH3, PCl3, PF3, CS, CH2) sandwich complexes of cyclooctatetraene: Theoretical investigation. Struct. Chem. 2019, 30, 2339–2346. [Google Scholar] [CrossRef]
- Jandl, C.; Pankhurst, J.; Love, J.; Pothig, A. Rational Synthesis and Electronic Structure of Functionalized Trinuclear Pd Metal Sheet Sandwich Complexes. Organometallics 2017, 36, 2772–2783. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Wang, Y. Advances for Triangular and Sandwich-Shaped All-Metal Aromatics. Molecules 2024, 29, 763. https://doi.org/10.3390/molecules29040763
Wang M, Wang Y. Advances for Triangular and Sandwich-Shaped All-Metal Aromatics. Molecules. 2024; 29(4):763. https://doi.org/10.3390/molecules29040763
Chicago/Turabian StyleWang, Miaomiao, and Yanlan Wang. 2024. "Advances for Triangular and Sandwich-Shaped All-Metal Aromatics" Molecules 29, no. 4: 763. https://doi.org/10.3390/molecules29040763
APA StyleWang, M., & Wang, Y. (2024). Advances for Triangular and Sandwich-Shaped All-Metal Aromatics. Molecules, 29(4), 763. https://doi.org/10.3390/molecules29040763