
New Copper Complexes with N,O-Donor Ligands Based on Pyrazole Moieties Supported by 3-Substituted Acetylacetone Scaffolds

 ,
,  , ,
, , 
Abstract

1. Introduction
2. Results and Discussion
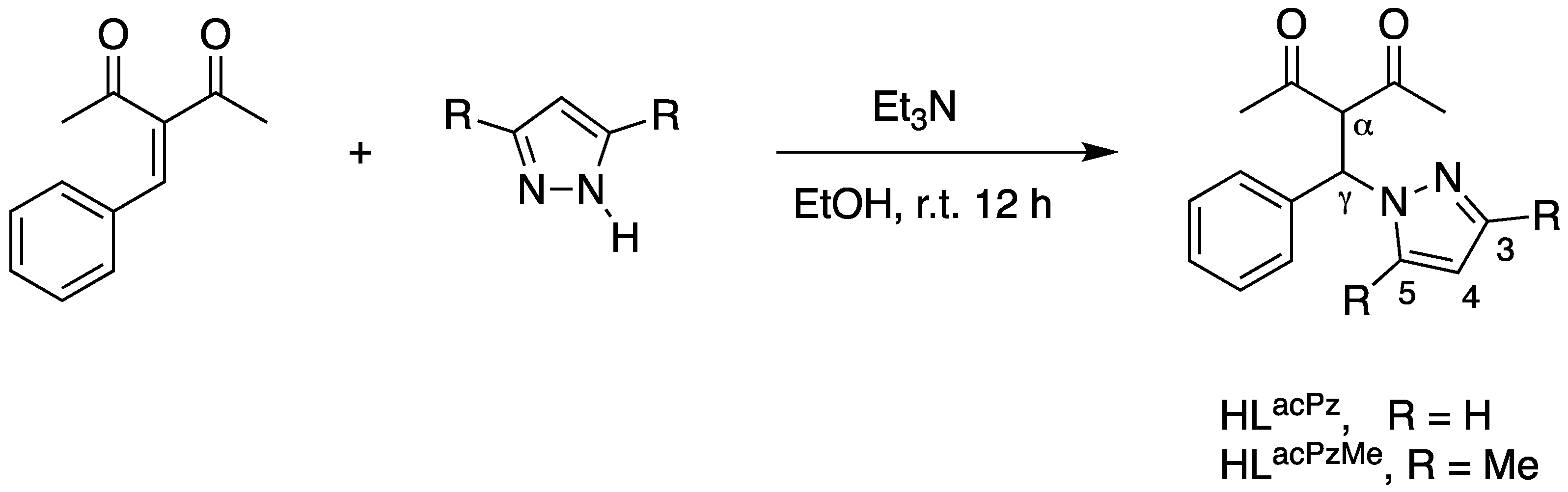
2.1. Synthesis and Characterization
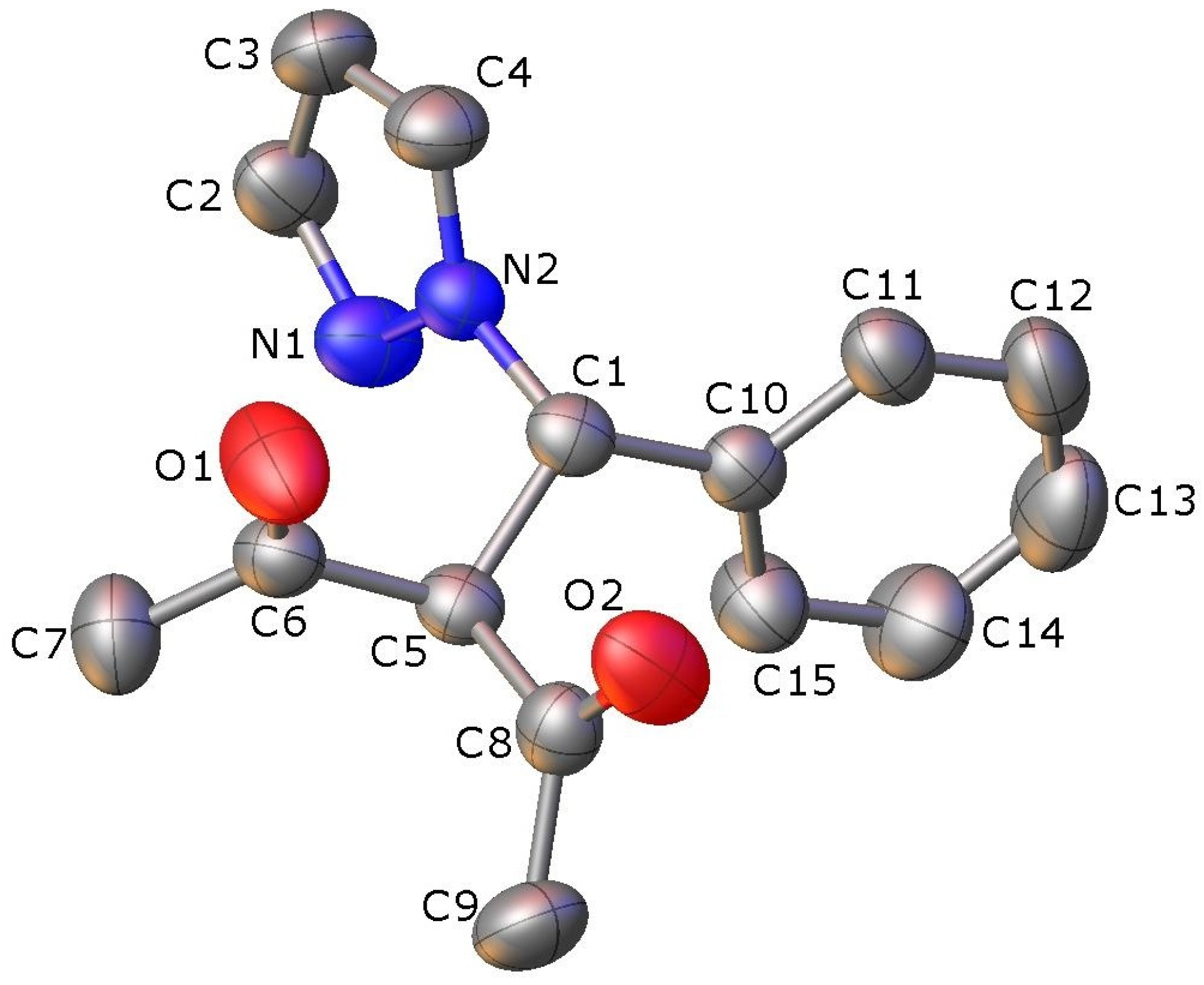
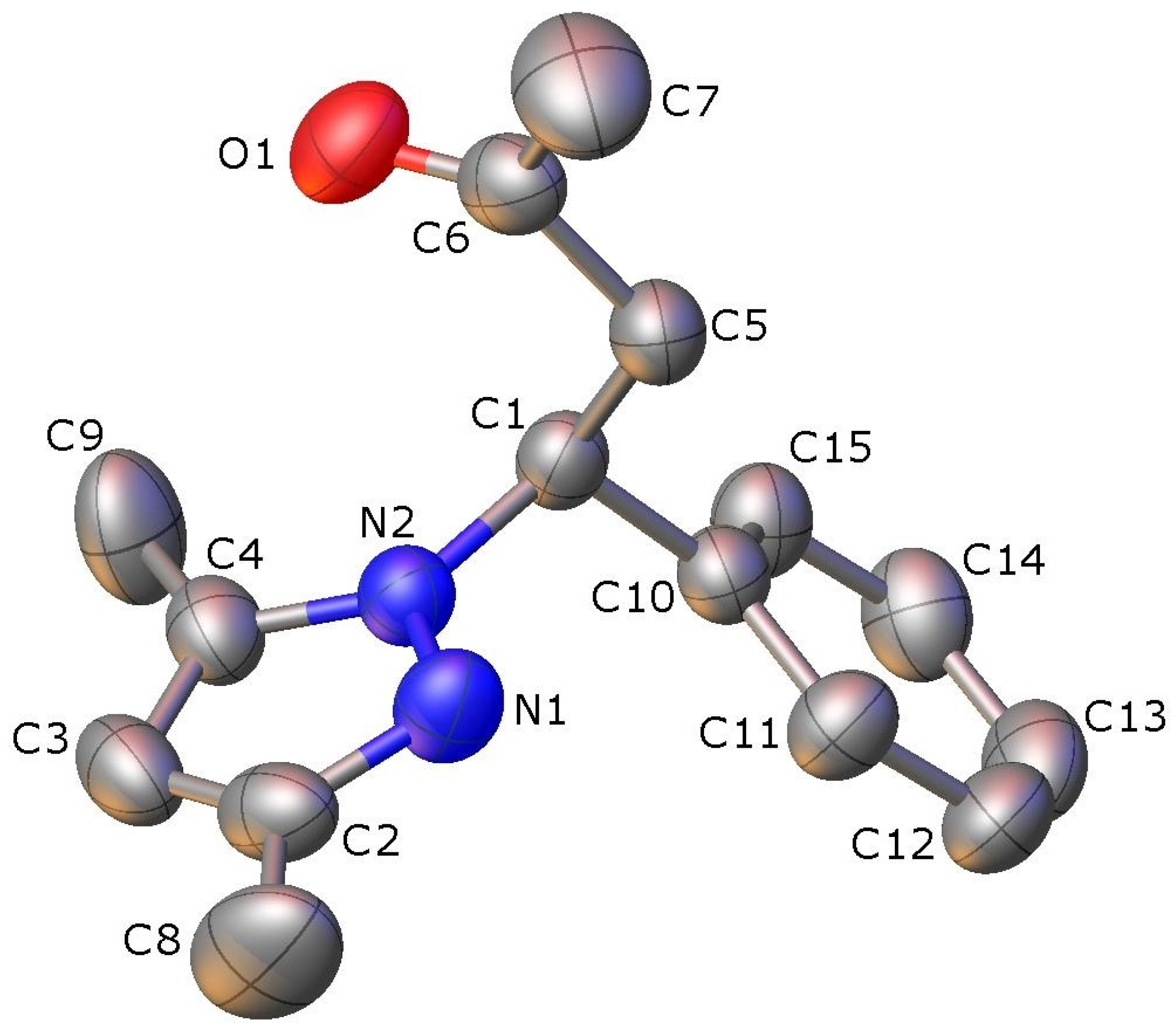
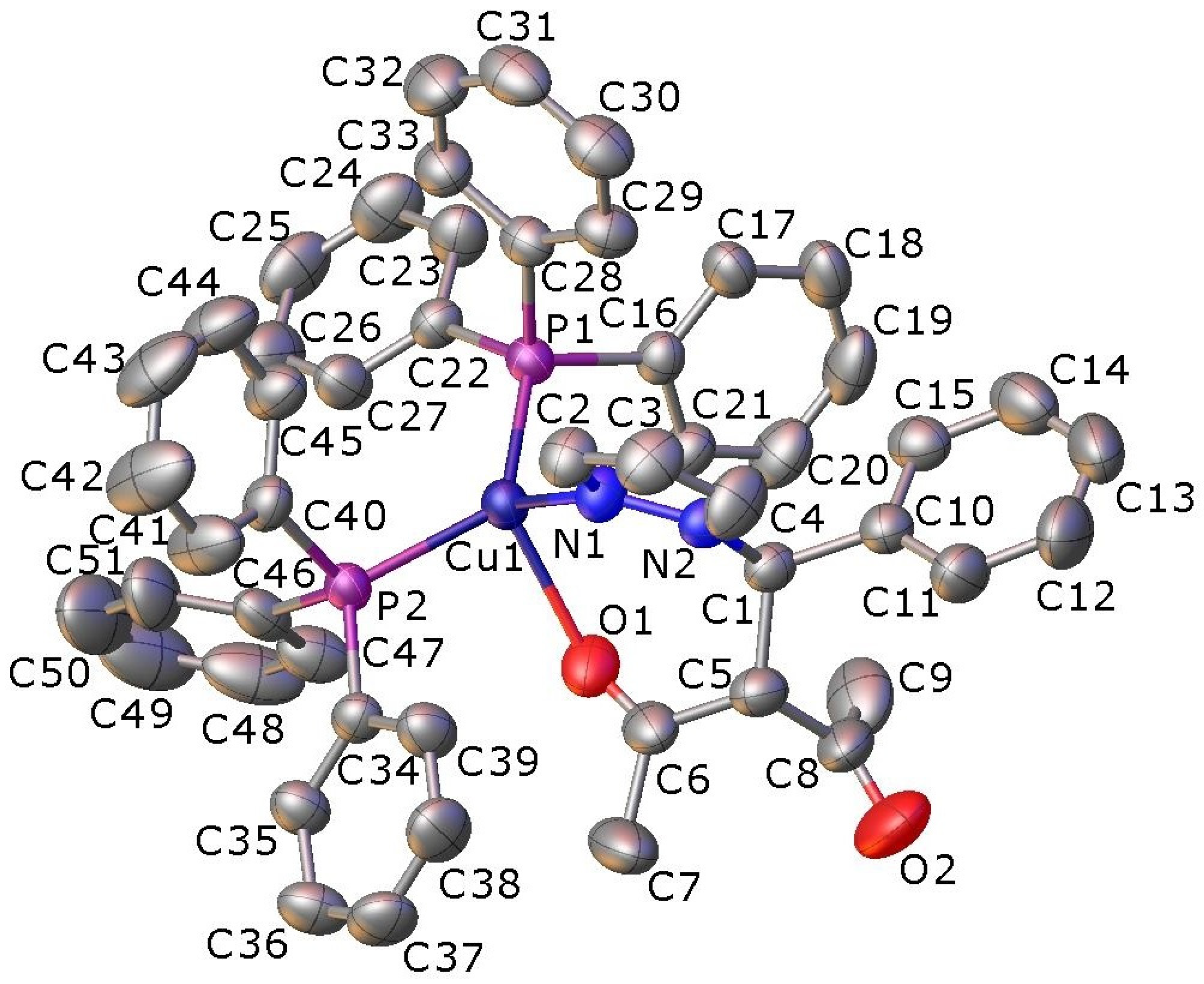
2.2. X-ray Crystallography
3. Experimental Section
3.1. Materials and Instruments
3.2. Synthesis
3.2.1. Synthesis of HLacPz (1)
3.2.2. Synthesis of HLacPzMe (2)
3.2.3. Synthesis of [Cu(HLacPz)(PPh3)2]PF6 (3)
3.2.4. Synthesis of [Cu(HLacPz)2(LacPz)2] (4)
3.2.5. Synthesis of [Cu(HLacPzMe)(PPh3)2]PF6.2CH3CN (5)
3.2.6. Synthesis of [Cu(HLacPzMe)2(LacPzMe)2] (6)
3.2.7. Synthesis of 4-Phenyl-4-(1H-pyrazol-1-yl)butan-2-one, PhPzMEK (7)
3.2.8. Synthesis of 4-(3,5-Dimethyl-1H-pyrazol-1-yl)-4-phenylbutan-2-one, PhPzMe2MEK (8)
3.3. Crystallographic Data Collection and Refinement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Katritzky, A.R.; Wang, Z.; Wang, M.; Wilkerson, C.R.; Hall, C.D.; Akhmedov, N.G. Preparation of β-keto esters and β-diketones by C-acylation/deacetylation of acetoacetic esters and acetonyl ketones with 1-acylbenzotriazoles. J. Org. Chem. 2004, 69, 6617–6622. [Google Scholar] [CrossRef] [PubMed]
- Fargeas, V.; Baalouch, M.; Metay, E.; Baffreau, J.; Menard, D.; Gosselin, P.; Berge, J.P.; Barthomeuf, C.; Lebreton, J. New access to 1,3-diketones from aldehydes. Tetrahedron 2004, 60, 10359–10364. [Google Scholar] [CrossRef]
- Sharma, Y.; Ansari, A. Diketones as building block in organic synthesis with versatile applications and medicinal properties. Mater. Today Proc. 2022; in press. [Google Scholar] [CrossRef]
- Kaur, N.; Bhardwaj, P.; Gupta, M. Recent Developments in the Synthesis of Five- and Six-membered N-heterocycles from Dicarbonyl Compounds. Curr. Org. Chem. 2021, 25, 2765–2790. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; Wiley: Chichester, UK, 2010. [Google Scholar]
- Hansen, P.A.-O. Structural Studies of β-Diketones and Their Implications on Biological Effects. Pharmaceuticals 2021, 14, 1189. [Google Scholar] [CrossRef] [PubMed]
- Grogan, G. Emergent mechanistic diversity of enzyme-catalysed β-diketone cleavage. Biochem. J. 2005, 388, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Grogan, G. β-diketone hydrolases. J. Mol. Catal. B Enzym. 2002, 19, 73–82. [Google Scholar] [CrossRef]
- Pokorny, D.; Steiner, W.; Ribbons, D.W. β-ketolases-Forgotten hydrolytic enzymes? Trends Biotechnol. 1997, 15, 291–296. [Google Scholar] [CrossRef]
- Hozakova, L.; Vokata, B.; Ruml, T.; Ulbrich, P. Targeting the Virus Capsid as a Tool to Fight RNA Viruses. Viruses 2022, 14, 174. [Google Scholar] [CrossRef]
- Reina, M.; Talavera-Contreras, L.G.; Figueroa-DePaz, Y.; Ruiz-Azuara, L.; Hernández-Ayala, L.F. Casiopeinas® as SARS-CoV-2 main protease (Mpro) inhibitors: A combined DFT, molecular docking and ONIOM approach. New J. Chem. 2022, 46, 12500–12511. [Google Scholar] [CrossRef]
- Masui, H. Metalloaromaticity. Coord. Chem. Rev. 2001, 219–221, 957–992. [Google Scholar] [CrossRef]
- Siedle, A.R. Diketones and Related Ligands. In Comprehensive Coordination Chemistry; Wilkinson, G., Gillard, R.D., McCleverty, J.A., Eds.; Pergamon: Oxford, UK, 1987; pp. 365–412. [Google Scholar]
- Kawaguchi, S. Variety in the coordination modes of β-dicarbonyl compounds in metal complexes. Coord. Chem. Rev. 1986, 70, 51–84. [Google Scholar] [CrossRef]
- Mehrotra, R.C.; Bohra, R.; Gaur, D.P. Metal [Beta]-Diketonates and Allied Derivatives; Academic Press: London, UK, 1978. [Google Scholar]
- Crossman, A.S.; Marshak, M.P. 1.11-β-Diketones: Coordination and Application. In Comprehensive Coordination Chemistry III; Constable, E.C., Parkin, G., Que, L., Jr., Eds.; Elsevier: Oxford, UK, 2021; pp. 331–365. [Google Scholar]
- Primer, D.N.; Molander, G.A. Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc. 2017, 139, 9847–9850. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.C.; Gui, J.; Yabe, Y.; Pan, C.M.; Baran, P.S. Functionalized olefin cross-coupling to construct carbon-carbon bonds. Nature 2014, 516, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Shafir, A.; Buchwald, S.L. Highly selective room-temperature copper-catalyzed C-N coupling reactions. J. Am. Chem. Soc. 2006, 128, 8742–8743. [Google Scholar] [CrossRef] [PubMed]
- Baik, T.G.; Luis, A.L.; Wang, L.C.; Krische, M.J. Diastereoselective cobalt-catalyzed Aldol and Michael cycloreductions. J. Am. Chem. Soc. 2001, 123, 5112–5113. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.K.; Lee, S.H.; Lee, D. Copper-catalyzed N-arylation of amines with hypervalent iodonium salts. Synlett 2000, 2000, 1022–1024. [Google Scholar]
- Isayama, S.; Mukaiyama, T. Hydration of Olefins with Molecular Oxygen and Triethylsilane Catalyzed by Bis(trifluoroacetylacetonato)cobalt(II). Chem. Lett. 1989, 18, 569–572. [Google Scholar] [CrossRef]
- Stalpaert, M.; De Vos, D. Stabilizing Effect of Bulky β-Diketones on Homogeneous Mo Catalysts for Deoxydehydration. ACS Sustain. Chem. Eng. 2018, 6, 12197–12204. [Google Scholar] [CrossRef]
- Krajewski, S.M.; Crossman, A.S.; Akturk, E.S.; Suhrbier, T.; Scappaticci, S.J.; Staab, M.W.; Marshak, M.P. Sterically encumbered β-diketonates and base metal catalysis. Dalton Trans. 2019, 48, 10714–10722. [Google Scholar] [CrossRef]
- Crossman, A.S.; Larson, A.T.; Shi, J.X.; Krajewski, S.M.; Akturk, E.S.; Marshak, M.P. Synthesis of Sterically Hindered β-Diketones via Condensation of Acid Chlorides with Enolates. J. Org. Chem. 2019, 84, 7434–7442. [Google Scholar] [CrossRef] [PubMed]
- Akturk, E.S.; Scappaticci, S.J.; Seals, R.N.; Marshak, M.P. Bulky β-Diketones Enabling New Lewis Acidic Ligand Platforms. Inorg. Chem. 2017, 56, 11466–11469. [Google Scholar] [CrossRef] [PubMed]
- Gromilov, S.A.; Baidina, I.A. Regularities of crystal structures of Cu(II) β-diketonates. J. Struct. Chem. 2004, 45, 1031–1081. [Google Scholar] [CrossRef]
- Bulusheva, L.G.; Okotrub, A.V.; Liskovskaya, T.I.; Krupoder, S.A.; Guselnikov, A.V.; Manaev, A.V.; Traven, V.F. Electronic Structure of 1,5-Cyclooctadiene-copper(I)-hexafluoroacetylacetonate. J. Phys. Chem. A 2001, 105, 8200–8205. [Google Scholar] [CrossRef]
- Chi, K.M.; Shin, H.K.; Hampden-Smith, M.J.; Duesler, E.N.; Kodas, T.T. The chemistry of β-diketonate copper(I) compounds—III. The synthesis of (β-diketonate) Cu(1,5-COD) compounds, the solid state structure and disproportionation of hexafluoroacetylacetonato (1,5-cyclooctadiene)copper(I), (hfac)Cu(1,5-COD). Polyhedron 1991, 10, 2293–2299. [Google Scholar] [CrossRef]
- Nast, R.; Mohr, R.; Schultze, C. Zur Kenntnis von Kupfer(I)-acetylacetonat. Chem. Berichte 1963, 96, 2127–2131. [Google Scholar] [CrossRef]
- Larson, A.T.; Crossman, A.S.; Krajewski, S.M.; Marshak, M.P. Copper(II) as a Platform for Probing the Steric Demand of Bulky β-Diketonates. Inorg. Chem. 2020, 59, 423–432. [Google Scholar] [CrossRef]
- Jakob, A.; Joubert, C.C.; Rüffer, T.; Swarts, J.C.; Lang, H. Chemical and electrochemical oxidation studies on new copper(I) ferrocenyl-functionalised β-diketonates. Inorg. Chim. Acta 2014, 411, 48–55. [Google Scholar] [CrossRef]
- Lang, H.; Leschke, M.; Melter, M.; Walfort, B.; Kohler, K.; Schulz, S.E.; Gessner, T. Mono- and bimetallic copper(I)- and silver(I)-phosphane complexes with β-diketonate units. Z. Anorg. Allg. Chem. 2003, 629, 2371–2380. [Google Scholar] [CrossRef]
- Yang, R.-N.; Wang, D.-M.; Liu, Y.-F.; Jin, D.-M. Synthesis of copper(I) β-diketone complexes. Polyhedron 2001, 20, 585–590. [Google Scholar] [CrossRef]
- Shin, H.K.; Chi, K.M.; Farkas, J.; Hampden-Smith, M.J.; Kodas, T.T.; Duesler, E.N. Chemistry of copper(I).beta.-diketonate complexes. 2. Synthesis, characterization, and physical properties of (.beta.-diketonato)copper(I) trimethylphosphine and bis(trimethylphosphine) complexes. Inorg. Chem. 1992, 31, 424–431. [Google Scholar] [CrossRef]
- Chi, K.-M.; Farkas, J.; Hampden-Smith, M.J.; Kodas, T.T.; Duesler, E.N. The chemistry of copper(I)β-diketonate compounds. Part 4. Syntheses and characterization of CuXL(X = β-diketonate or Cl, L = PMe3, n = 2 or 4; L = PEt3, n = 2). Dalton Trans. 1992, 3111–3117. [Google Scholar] [CrossRef]
- Shin, H.K.; Hampden-Smith, M.J.; Duesler, E.N.; Kodas, T.T. The chemistry of copper(I) β-diketonate compounds. Part V. Syntheses and characterization of (β-diketonate)CuLn species where L = PBu3, PPh3, and PCy3; n = 1 and 2; crystal and molecular structures of (acac)Cu(PCy3), (tfac)Cu(PCy3), (hfac)Cu(PCy3), and (hfac)Cu(PCy3)2. Can. J. Chem. 1992, 70, 2954–2966. [Google Scholar]
- Baum, T.H.; Larson, C.E. A novel copper complex and CVD precursor: (.eta.2-butyne)copper(I) hexafluoroacetylacetonate. Chem. Mater. 1992, 4, 365–369. [Google Scholar] [CrossRef]
- Reynolds, S.K.; Smart, C.J.; Baran, E.F.; Baum, T.H.; Larson, C.E.; Brock, P. Chemical vapor deposition of copper from 1,5-cyclooctadiene copper(I) exafluoroacetylacetonate. J. Appl. Phys. Lett. 1991, 59, 2332. [Google Scholar] [CrossRef]
- Beach, D.B.; LeGoues, F.K.; Hu, C.K. Low-temperature chemical vapor deposition of high purity copper from an organometallic source. Chem. Mater. 1990, 2, 216–219. [Google Scholar] [CrossRef]
- Chow, Y.L.; Buono-Core, G.E. Triplet-state benzophenone-sensitized photoreduction of bis(acetylacetonato)copper(II): The generation and stability of copper(I) complexes. Can. J. Chem. 1983, 61, 795–800. [Google Scholar] [CrossRef]
- Restivo, R.J.; Costin, A.; Ferguson, G.; Carty, A.J. Perchlorato, Nitrato, and Acetylacetonato Complexes of Copper(I). The Crystal and Molecular Structure of Perchloratobis(tricyclohexyiphosphine)Copper(I). Can. J. Chem. 1975, 53, 1949–1957. [Google Scholar] [CrossRef]
- Anderson, W.A.; Carty, A.J.; Palenik, G.J.; Schreiber, G. Nitrato and Acetylacetonato Complexes of Copper(I). Can. J. Chem. 1971, 49, 761–766. [Google Scholar] [CrossRef]
- Khamidullina, L.A.; Puzyrev, I.S.; Burygin, G.L.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Mitrofanova, A.V.; Khrustalev, V.N.; Timofeeva, T.V.; Slepukhin, P.A.; Tobysheva, P.D.; et al. Unsymmetrical Trifluoromethyl Methoxyphenyl β-Diketones: Effect of the Position of Methoxy Group and Coordination at Cu(II) on Biological Activity. Molecules 2021, 26, 6466. [Google Scholar] [CrossRef]
- Pellei, M.; Santini, C.; Bagnarelli, L.; Caviglia, M.; Sgarbossa, P.; De Franco, M.; Zancato, M.; Marzano, C.; Gandin, V. Novel Silver Complexes Based on Phosphanes and Ester Derivatives of Bis(pyrazol-1-yl)acetate Ligands Targeting TrxR: New Promising Chemotherapeutic Tools Relevant to SCLC Management. Int. J. Mol. Sci. 2023, 24, 4091. [Google Scholar] [CrossRef] [PubMed]
- Del Bello, F.; Pellei, M.; Bagnarelli, L.; Santini, C.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; Battocchio, C.; Iucci, G.; Schiesaro, I.; et al. Cu(I) and Cu(II) Complexes Based on Lonidamine-Conjugated Ligands Designed to Promote Synergistic Antitumor Effects. Inorg. Chem. 2022, 61, 4919–4937. [Google Scholar] [CrossRef] [PubMed]
- Pellei, M.; Bagnarelli, L.; Luciani, L.; Del Bello, F.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; De Franco, M.; Gandin, V.; Marzano, C.; et al. Synthesis and Cytotoxic Activity Evaluation of New Cu(I) Complexes of Bis(pyrazol-1-yl) Acetate Ligands Functionalized with an NMDA Receptor Antagonist. Int. J. Mol. Sci. 2020, 21, 2616. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Amantini, C.; Santoni, G.; Pellei, M.; Santini, C.; Cimarelli, C.; Marcantoni, E.; Petrini, M.; Del Bello, F.; Giorgioni, G.; et al. Novel antitumor copper(ii) complexes designed to act through synergistic mechanisms of action, due to the presence of an NMDA receptor ligand and copper in the same chemical entity. New J. Chem. 2018, 42, 11878–11887. [Google Scholar] [CrossRef]
- Gandin, V.; Ceresa, C.; Esposito, G.; Indraccolo, S.; Porchia, M.; Tisato, F.; Santini, C.; Pellei, M.; Marzano, C. Therapeutic potential of the phosphino Cu(I) complex (HydroCuP) in the treatment of solid tumors. Sci. Rep. 2017, 7, 13936. [Google Scholar] [CrossRef] [PubMed]
- Tisato, F.; Marzano, C.; Peruzzo, V.; Tegoni, M.; Giorgetti, M.; Damjanovic, M.; Trapananti, A.; Bagno, A.; Santini, C.; Pellei, M.; et al. Insights into the cytotoxic activity of the phosphane copper(I) complex Cu(thp)4PF6. J. Inorg. Biochem. 2016, 165, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Papini, G.; Bandoli, G.; Dolmella, A.; Gioia Lobbia, G.; Pellei, M.; Santini, C. New homoleptic carbene transfer ligands and related coinage metal complexes. Inorg. Chem. Commun. 2008, 11, 1103–1106. [Google Scholar] [CrossRef]
- Pellei, M.; Del Gobbo, J.; Caviglia, M.; Karade, D.V.; Gandin, V.; Marzano, C.; Noonikara Poyil, A.; Dias, H.V.R.; Santini, C. Synthesis and cytotoxicity studies of Cu(I) and Ag(I) complexes based on sterically hindered β-diketonates with different degrees of fluorination. Dalton Trans. 2023, 52, 12098–12111. [Google Scholar] [CrossRef]
- Emsley, J. The composition, structure and hydrogen bonding of the β-diketones. In Complex Chemistry; Emsley, J., Ernst, R.D., Hathaway, B.J., Warren, K.D., Eds.; Springer: Berlin/Heidelberg, Germany, 1984; Volume 57, pp. 147–191. [Google Scholar]
- Liu, J.-H.; Lv, X.-J.; Liu, Y.-K. Asymmetric Retro-Claisen Reaction Catalyzed by Chiral Aza-Bisoxazoline–Zn(II) Complex: Enantioselective Synthesis of α-Arylated Ketones. Org. Lett. 2023, 25, 1706–1710. [Google Scholar] [CrossRef]
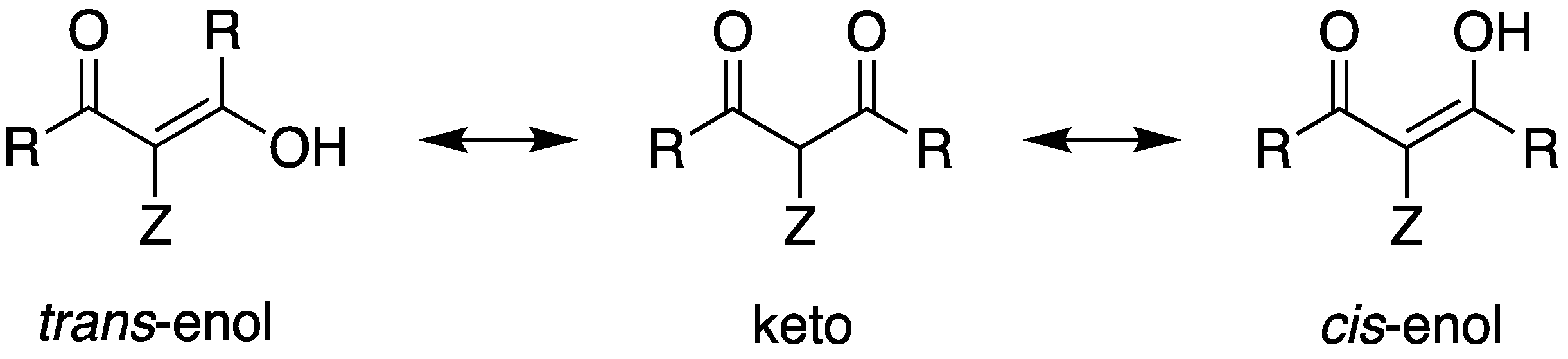
- Kol’tsov, A.I.; Kheifets, G.M. Investigation of Keto–Enol Tautomerism by Nuclear Magnetic Resonance Spectroscopy. Russ. Chem. Rev. 1971, 40, 773–788. [Google Scholar] [CrossRef]
- Tanaka, M.; Shono, T.; Shinra, K. Tautomerism in 3-Substituted-2,4-pentanediones and Their Copper Chelates. Bull. Chem. Soc. Jpn. 1969, 42, 3190–3194. [Google Scholar] [CrossRef]
- Tong, J.; Jia, L.-M.; Shang, P.; Yu, S.-Y. Controlled Synthesis of Supramolecular Architectures of Homo- and Heterometallic Complexes by Programmable Self-Assembly. Cryst. Growth Des. 2019, 19, 30–39. [Google Scholar] [CrossRef]
- Lee, E.; Seo, S.J.; Lee, S.S.; Lindoy, L.F. Assembling latter d-block heterometal coordination polymers: Synthetic strategies and structural outcomes. Coord. Chem. Rev. 2017, 348, 121–170. [Google Scholar] [CrossRef]
- Cook, T.R.; Stang, P.J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Z.J.; Liu, D.; Jin, G.X. Multi-component coordination-driven self-assembly toward heterometallic macrocycles and cages. Coord. Chem. Rev. 2015, 293–294, 139–157. [Google Scholar] [CrossRef]
- Ward, M.D.; Raithby, P.R. Functional behaviour from controlled self-assembly: Challenges and prospects. Chem. Soc. Rev. 2013, 42, 1619–1636. [Google Scholar] [CrossRef] [PubMed]
- Silvernail, C.M.; Yap, G.; Sommer, R.D.; Rheingold, A.L.; Day, V.W.; Belot, J.A. An effective synthesis of alkyl β-cyano-α,γ-diketones using chlorosulfonylisocyanate and a representative Cu(II) complex. Polyhedron 2001, 20, 3113–3117. [Google Scholar] [CrossRef]
- Nieuwenhuyzen, M.; Schobert, R.; Hampel, F.; Hoops, S. Reactions of dicarbonyltitanocenes with 2-diazo-1,3-diketones: O-,N-versus O-,O-chelation and self-assembly of a novel heteroleptic Ti5O6-cage compound. Inorg. Chim. Acta 2000, 304, 118–121. [Google Scholar] [CrossRef]
- Turner, S.S.; Collison, D.; Mabbs, F.E.; Halliwell, M. Preparation, magnetic properties and crystal structure of bis 3-(4-pyridyl)pentane-2,4-dionato copper(II). J. Chem. Soc. Dalton Trans. 1997, 1117–1118. [Google Scholar] [CrossRef]
- Isakova, V.G.; Baidina, I.A.; Morozova, N.B.; Igumenov, I.K. gamma-Halogenated iridium(III) acetylacetonates. Polyhedron 2000, 19, 1097–1103. [Google Scholar] [CrossRef]
- Gildenast, H.; Hempelmann, G.; Gruszien, L.; Englert, U. A Rigid Linker for Site-Selective Coordination of Transition Metal Cations: Combining an Acetylacetone with a Caged Phosphine. Inorg. Chem. 2023, 62, 3178–3185. [Google Scholar] [CrossRef]
- Gildenast, H.; Nölke, S.; Englert, U. 3-(4-Methylthiophenyl)acetylacetone–ups and downs of flexibility in the synthesis of mixed metal–organic frameworks. Ditopic bridging of hard and soft cations and site-specific desolvation. CrystEngComm 2020, 22, 1041–1049. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Englert, U. An Acetylacetonate or a Pyrazole? Both! 3-(3,5-Dimethyl-pyrazol-4-yl)pentane-2,4-dione as a Ditopic Ligand. Cryst. Growth Des. 2016, 16, 5127–5135. [Google Scholar] [CrossRef]
- van Terwingen, S.; Nachtigall, N.; Ebel, B.; Englert, U. N-Donor-Functionalized Acetylacetones for Heterobimetallic Coordination Polymers, the Next Episode: Trimethylpyrazoles. Cryst. Growth Des. 2021, 21, 2962–2969. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Merkens, C.; Si, R.Z.; Englert, U. Crosslinking of the Pd(acacCN)2 building unit with Ag(I) salts: Dynamic 1D polymers and an extended 3D network. Crystengcomm 2015, 17, 4383–4393. [Google Scholar] [CrossRef]
- Merkens, C.; Truong, K.N.; Englert, U. 3-(4-Pyridyl)-acetylacetone-a fully featured substituted pyridine and a flexible linker for complex materials. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Merkens, C.; Pecher, O.; Steuber, F.; Eisenhut, S.; Gorne, A.; Haarmann, F.; Englert, U. Crystal-to-Crystal Transformations in a Seven-Coordinated Scandium Complex. Z. Anorg. Allg. Chem. 2013, 639, 340–346. [Google Scholar] [CrossRef]
- Merkens, C.; Pan, F.F.; Englert, U. 3-(4-Pyridyl)-2,4-pentanedione-a bridge between coordinative, halogen, and hydrogen bonds. Crystengcomm 2013, 15, 8153–8158. [Google Scholar] [CrossRef]
- Merkens, C.; Englert, U. Ordered bimetallic coordination networks featuring rare earth and silver cations. Dalton Trans. 2012, 41, 4664–4673. [Google Scholar] [CrossRef]
- Merkens, C.; Becker, N.; Lamberts, K.; Englert, U. Bimetallic coordination networks based on Al(acacCN)3: A building block between inertness and lability. Dalton Trans. 2012, 41, 8594–8599. [Google Scholar] [CrossRef]
- Kondracka, M.; Englert, U. Bimetallic Coordination Polymers via Combination of Substitution-inert Building Blocks and Labile Connectors. Inorg. Chem. 2008, 47, 10246–10257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Chen, B.L.; Fronczek, F.R.; Maverick, A.W. A nanoporous Ag-Fe mixed-metal-organic framework exhibiting single-crystal-to-single-crystal transformations upon guest exchange. Inorg. Chem. 2008, 47, 4433–4435. [Google Scholar] [CrossRef] [PubMed]
- Burrows, A.D.; Cassar, K.; Mahon, M.F.; Warren, J.E. The stepwise formation of mixed-metal coordination networks using complexes of 3-cyanoacetylacetonate. Dalton Trans. 2007, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Vreshch, V.D.; Lysenko, A.B.; Chernega, A.N.; Sieler, J.; Domasevitch, K.V. Heterobimetallic Cd(Zn)/Be coordination polymers involving pyridyl functionalized beryllium diketonates. Polyhedron 2005, 24, 917–926. [Google Scholar] [CrossRef]
- Chen, B.L.; Fronczek, F.R.; Maverick, A.W. Porous Cu-Cd mixed-metal-organic frameworks constructed from Cu(PyaC)2 {Bis 3-(4-pyridyl)pentane-2,4-dionato copper(II)}. Inorg. Chem. 2004, 43, 8209–8211. [Google Scholar] [CrossRef] [PubMed]
- Vreshch, V.D.; Lysenko, A.B.; Chernega, A.N.; Howard, J.A.K.; Krautscheid, H.; Sieler, J.; Domasevitch, K.V. Extended coordination frameworks incorporating heterobimetallic squares. Dalton Trans. 2004, 2899–2903. [Google Scholar] [CrossRef] [PubMed]
- Vreshch, V.D.; Chernega, A.N.; Howard, J.A.K.; Sieler, J.; Domasevitch, K.V. Two-step construction of molecular and polymeric mixed-metal Cu(Co)/Be complexes employing functionality of a pyridyl substituted acetylacetonate. Dalton Trans. 2003, 1707–1711. [Google Scholar] [CrossRef]
- Boldog, I.; Rusanov, E.B.; Chernega, A.N.; Sieler, J.; Domasevitch, K.V. Acentric extended solids by self assembly of 4,4′-bipyrazolyls. Angew. Chem. Int. Ed. Engl. 2001, 40, 3435–3438. [Google Scholar] [CrossRef]
- Mackay, L.G.; Anderson, H.L.; Sanders, J.K.M. A platinum-linked cyclic porphyrin trimer. J. Chem. Soc. Chem. Commun. 1992, 43–44. [Google Scholar] [CrossRef]
- Kremer, M.; Englert, U. N Donor substituted acetylacetones–versatile ditopic ligands. Z. Für Krist. Cryst. Mater. 2018, 233, 437–452. [Google Scholar] [CrossRef]
- Pellei, M.; Papini, G.; Trasatti, A.; Giorgetti, M.; Tonelli, D.; Minicucci, M.; Marzano, C.; Gandin, V.; Aquilanti, G.; Dolmella, A.; et al. Nitroimidazole and glucosamine conjugated heteroscorpionate ligands and related copper(II) complexes. Syntheses, biological activity and XAS studies. Dalton Trans. 2011, 40, 9877–9888. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Chemical Hardness; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1997. [Google Scholar]
- Maier, T.; Mildenberger, H. β-Azolyl-α,α-dicarbonyl compounds. Angew. Chem. Int. Ed. Engl. 1980, 19, 137–138. [Google Scholar] [CrossRef]
- Raman, N.; Pravin, N. Lasing the DNA fragments through β-diketimine framed Knoevenagel condensed Cu(II) and Zn(II) complexes–An in vitro and in vivo approach. Spectrochim. Acta Part A 2014, 118, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Watanabe, Y.; Mitsudo, T.-a.; Takegami, Y. The Reductive Deacylation of the Knoevenagel Condensates by the Use of the Tetracarbonylhydridoferrate(0) Anion. A New Synthetic Route of Ketones from Aldehydes. Bull. Chem. Soc. Jpn. 1978, 51, 835–838. [Google Scholar] [CrossRef]
- Cheng, B.; Yi, H.; He, C.; Liu, C.; Lei, A. Revealing the Ligand Effect on Copper(I) Disproportionation via Operando IR Spectra. Organometallics 2014, 34, 206–211. [Google Scholar] [CrossRef]
- Hussein, M.A.; Huynh, V.T.; Hommelsheim, R.; Koenigs, R.M.; Nguyen, T.V. An efficient method for retro-Claisen-type C-C bond cleavage of diketones with tropylium catalyst. Chem. Commun. 2018, 54, 12970–12973. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.C.; Kotadiya, G.M.; Vaja, D.V. Synthesis and biological evaluation of some novel quinoline based pyrimidine derivatives. Indian J. Chem. Sect. B Org. Chem. Incl. Med. Chem. 2018, 57, 965–973. [Google Scholar]
- Kudyakova, Y.S.; Bazhin, D.N.; Slepukhin, P.A.; Burgart, Y.V.; Saloutin, V.I.; Charushin, V.N. Unexpected formation of diethyl 2-ethoxy-6-CF3-2H-pyran-3,5-dicarboxylate from the condensation of ethyl 4,4,4-trifluoroacetoacetate with CH(OEt)3. Tetrahedron Lett. 2017, 58, 744–747. [Google Scholar] [CrossRef]
- Bazhin, D.N.; Kudyakova, Y.S.; Nemytova, N.A.; Burgart, Y.V.; Saloutin, V.I. Detrifluoroacetylation of 4,4,4-trifluoro-3,3-dihydroxy-2-(hydroxyimino)butan-1-ones as a convenient synthetic strategy for acyl cyanides. J. Fluor. Chem. 2016, 186, 28–32. [Google Scholar] [CrossRef]
- Yang, D.M.; Zhou, Y.H.; Xue, N.; Qu, J.P. Synthesis of Trifluoromethyl Ketones via Tandem Claisen Condensation and Retro-Claisen C-C Bond-Cleavage Reaction. J. Org. Chem. 2013, 78, 4171–4176. [Google Scholar] [CrossRef]
- Biswas, S.; Maiti, S.; Jana, U. An Efficient Iron-Catalyzed Carbon-Carbon Single-Bond Cleavage via Retro-Claisen Condensation: A Mild and Convenient Approach to Synthesize a Variety of Esters or Ketones. Eur. J. Org. Chem. 2010, 2010, 2861–2866. [Google Scholar] [CrossRef]
- Siirola, E.; Frank, A.; Grogan, G.; Kroutil, W. C-C Hydrolases for Biocatalysis. Adv. Synth. Catal. 2013, 355, 1677–1691. [Google Scholar] [CrossRef]
- Xie, F.; Yan, F.X.; Chen, M.M.; Zhang, M. Base-catalyzed retro-Claisen condensation: A convenient esterification of alcohols via C-C bond cleavage of ketones to afford acylating sources. Rsc Adv. 2014, 4, 29502–29508. [Google Scholar] [CrossRef]
- Jukic, M.; Sterk, D.; Casar, Z. Recent Advances in the Retro-Claisen Reaction and Its Synthetic Applications. Curr. Org. Synth. 2012, 9, 488–512. [Google Scholar] [CrossRef]
- Rao, C.B.; Rao, D.C.; Babu, D.C.; Venkateswarlu, Y. Retro-Claisen Condensation with Fe-III as Catalyst under Solvent-Free Conditions. Eur. J. Org. Chem. 2010, 2010, 2855–2859. [Google Scholar] [CrossRef]
- Kawata, A.; Takata, K.; Kuninobu, Y.; Takai, K. Indium-catalyzed retro-claisen condensation. Angew. Chem. Int. Ed. Engl. 2007, 46, 7793–7795. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.K. ORTEP, Report ORNL–5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976.
- New and Improved CSD–The 2023 November Data Update. Cambridge Structural Database (CSD). 2023. Available online: https://www.ccdc.cam.ac.uk/discover/blog/new-and-improved-csd-the-2023-november-data-update/ (accessed on 27 December 2023).
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Volov, A.N.; Zamilatskov, I.A. Ethyl N-(2-acetyl-3-oxo-1-phenylbutyl)carbamate. Acta Crystallogr. Sect. E Crystallogr. Commun. 2013, 69, o1529. [Google Scholar]
- Gotsko, M.D.; Saliy, I.V.; Vashchenko, A.V.; Trofimov, B.A. 1-Phenyl-3,3-di(1H-pyrazol-1-yl)propan-1-one. Molbank 2022, 2022, M1472. [Google Scholar] [CrossRef]
- Peters, L.; Hübner, E.; Haas, T.; Heinemann, F.W.; Burzlaff, N. 3,3-Bis(3,5-dimethylpyrazol-1-yl)propionic acid: A tripodal N,N,O ligand for manganese and rhenium complexes-Syntheses and structures. J. Organomet. Chem. 2009, 694, 2319–2327. [Google Scholar] [CrossRef]
- Kunz, P.C.; Berghahn, M.; Brückmann, N.E.; Dickmeis, M.; Kettel, M.; Spingler, B. Functionalised Tris(pyrazolyl)methane Ligands and Re(CO)3 Complexes Thereof. Z. Anorg. Allg. Chem. 2009, 635, 471–478. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Malecki, J.G. (Department of Crystallography, Institute of Chemistry, University of Silesia, Szkolna 9 str., 40–006 Katowice, Poland). Personal communication, 2016.
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 955–964. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro. Versions 1.171.41.123a and 1.171.42.49 (Rigaku Oxford Diffraction). 2022.
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | 8 |
|---|---|---|---|---|
| Radiation (all experiments) | Cu Kα (λ = 1.54184) | |||
| Empirical formula | C15H16N2O2 | C17H20N2O2 | C51H46N2O2F6P3Cu | C15H18N2O |
| Formula weight | 256.30 | 284.35 | 989.35 | 242.31 |
| Temperature/K | 298.1(4) | 299.4(8) | 298(1) | 298.8(8) |
| Crystal system | monoclinic | monoclinic | orthorhombic | monoclinic |
| Space group | C2/c | P21/c | Pna21 | P21/c |
| a/Å | 26.9711(5) | 8.3223(2) | 20.9587(2) | 9.1203(7) |
| b/Å | 9.7500(2) | 12.4916(3) | 9.96560(10) | 12.8652(8) |
| c/Å | 10.5745(2) | 30.5285(5) | 22.7132(2) | 12.5423(9) |
| α/° | 90.00 | 90.0 | 90.00 | 90.0 |
| β/° | 95.406(2) | 91.296(2) | 90.00 | 108.296(8) |
| γ/° | 90.00 | 90.0 | 90.00 | 90.0 |
| Volume/Å3 | 2768.39(9) | 3172.90(12) | 4744.02(8) | 1397.25(18) |
| Z | 8 | 8 | 4 | 4 |
| ρcalc Mg/m3 | 1.230 | 1.191 | 1.385 | 1.152 |
| μ/mm−1 | 0.668 | 0.629 | 2.164 | 0.575 |
| F(000) | 1088 | 1216 | 2040 | 520 |
| Crystal size/mm3 | 0.80 × 0.40 × 0.10 | 0.44 × 0.40 × 0.24 | 0.40 × 0.30 × 0.22 | 0.44 × 0.30 × 0.06 |
| Reflections collected | 20,260 | 53,329 | 41,569 | 5370 |
| Independent reflections/Rint | 2658/0.0374 | 6320/0.0276 | 8023/0.0393 | 2484/0.0338 |
| Restraints/parameters | 0/237 | 0/539 | 1/643 | 0/236 |
| Goodness of fit on F2 | 1.049 | 1.049 | 1.038 | 0.984 |
| Final R (R1; wR2) indexes [I > 2σ (I)] | 0.0473, 0.1113 | 0.0468, 0.1307 | 0.0336, 0.0838 | 0.0446, 0.1136 |
| Largest diff. peak/hole/e Å−3 | 0.187/−0.175 | 0.222/−0.233 | 0.317/−0.182 | 0.149/−0.120 |
| 1 | |||
|---|---|---|---|
| Bond | Angle | ||
| N1-N2 | 1.3412(18) | N2-N1-C2 | 103.65(14) |
| N1-C2 | 1.333(2) | N1-N2-C4 | 112.21(14) |
| N2-C1 | 1.4694(19) | N1-N2-C1 | 120.53(12) |
| N2-C4 | 1.338(2) | N2-C1-C5 | 109.98(12) |
| C1-C5 | 1.531(2) | N2-C1-C10 | 110.68(12) |
| C1-C10 | 1.520(2) | C1-C5-C6 | 110.1(9) |
| C2-C3 | 1.371(3) | C1-C5-C8 | 110.32(13) |
| C3-C4 | 1.359(3) | C2-C3-C4 | 104.79(16) |
| C5-C6 | 1.536(2) | C5-C6-C7 | 116.89(15) |
| C5-C8 | 1.532(2) | C6-C5-C8 | 107.35(12) |
| C6-O1 | 1.1953(19) | C5-C6-O1 | 120.18(15) |
| C8-O2 | 1.195(2) | C5-C8-O2 | 120.39(14) |
| 2 * | |||||
|---|---|---|---|---|---|
| Bond | Angle | ||||
| N1-N2 | 1.3606(15) | 1.3645(15) | N2-N1-C2 | 104.76(12) | 104.73(11) |
| N1-C2 | 1.3293(17) | 1.3298(17) | N1-N2-C4 | 112.45(11) | 112.25(11) |
| N2-C1 | 1.4637(15) | 1.4621(15) | N1-N2-C1 | 119.53(10) | 119.45(10) |
| N2-C4 | 1.3513(17) | 1.3496(17) | N2-C1-C5 | 109.65(10) | 109.59(10) |
| C1-C5 | 1.5363(18) | 1.5362(18) | N2-C1-C12 | 111.46(10) | 112.36(10) |
| C1-C12 | 1.5213(18) | 1.5211(18) | C1-C5-C6 | 111.23(10) | 111.70(10) |
| C2-C3 | 1.388(2) | 1.393(2) | C1-C5-C8 | 110.41(10) | 110.27(11) |
| C3-C4 | 1.373(2) | 1.366(2) | C2-C3-C4 | 106.70(13) | 106.73(13) |
| C5-C6 | 1.5411(17) | 1.5388(17) | C5-C6-C7 | 118.13(11) | 117.63(12) |
| C5-C8 | 1.5380(17) | 1.5342(17) | C6-C5-C8 | 107.02(10) | 106.88(10) |
| C6-O1 | 1.2016(16) | 1.2026(17) | C5-C6-O1 | 119.26(12) | 119.57(13) |
| C8-O2 | 1.1964(18) | 1.1948(19) | C5-C8-O2 | 120.77(13) | 120.66(14) |
| 8 | |||
|---|---|---|---|
| Bond | Angle | ||
| N1-N2 | 1.357(2) | N2-N1-C2 | 104.30(16) |
| N1-C2 | 1.334(3) | N1-N2-C4 | 112.47(15) |
| N2-C1 | 1.465(2) | N1-N2-C1 | 119.84(15) |
| N2-C4 | 1.352(2) | N2-C1-C5 | 109.70(15) |
| C1-C5 | 1.522(3) | N2-C1-C10 | 112.90(15) |
| C1-C10 | 1.518(2) | C1-C5-C6 | 113.97(17) |
| C2-C3 | 1.384(3) | C2-C3-C4 | 106.17(19) |
| C3-C4 | 1.372(3) | C5-C6-C7 | 116.7(2) |
| C5-C6 | 1.504(3) | C5-C6-O1 | 121.29(19) |
| C6-O1 | 1.211(2) |
| 3 | |||
|---|---|---|---|
| Bond | Angle | ||
| Cu1-P1 | 2.2385(9) | N1-Cu1-P1 | 110.40(9) |
| Cu1-P2 | 2.2578(9) | N1-Cu1-P2 | 109.52(10) |
| Cu1-N1 | 2.039(3) | O1-Cu1-P1 | 119.80(10) |
| Cu1-O1 | 2.307(3) | O1-Cu1-P2 | 93.11(9) |
| N1-N2 | 1.347(5) | P1-Cu1-P2 | 128.25(4)) |
| N1-C2 | 1.323(5) | N1-Cu1-O1 | 87.81(13) |
| N2-C4 | 1.348(6) | Cu1-P1-C16 | 114.70(12) |
| N2-C1 | 1.470(5) | Cu1-P2-C34 | 117.08(12) |
| C1-C5 | 1.545(5) | Cu1-N1-N2 | 129.9(3) |
| C5-C6 | 1.525(6) | Cu1-O1-C6 | 136.9(3) |
| C6-O1 | 1.203(6) | N1-N2-C1 | 121.4(3) |
| C5-C8 | 1.551(7) | N2-C1-C5 | 110.9(3) |
| C8-O2 | 1.195(7) | C1-C5-C6 | 112.7(4) |
| C2-C3 | 1.369(6) | C5-C6-O1 | 122.1(4) |
| C3-C4 | 1.351(7) | N1-N2-C4 | 109.4(4) |
| P1-C16 | 1.833(4) | N2-N1-C2 | 105.8(3) |
| P1-C22 | 1.825(4) | C16-P1-C28 | 106.79(17) |
| P1-C28 | 1.828(4) | C28-P1-C22 | 102.83(18) |
| P2-C34 | 1.824(3) | C34-P2-C46 | 103.63(17) |
| P2-C40 | 1.829(4) | C46-P2-C40 | 106.5(2) |
| P2-C46 | 1.820(4) | F-P3-F (av.) | 103.9 |
| P3-F (av.) | 1.540 |
| Molecule | Acceptor Atom (A) Atom | Donor (D) Atom | Parent (P) Atom * | Distance A····D (Å) | Angle A–D–P (°) | Symmetry Op. |
|---|---|---|---|---|---|---|
| 1 | O1 | H15 | C15 | 2.50 | 141.6 | x, 1 − y, −1/2 + z |
| N1 | H11 | C11 | 2.70 | 171.6 | x, 1 − y, 1/2 + z | |
| 2 | O1A | H7B | C7 | 2.53 | 147.0 | x, y, z |
| O1A | H14A | C14A | 2.60 | 143.6 | 1 + x, y, z | |
| O1 | H7AC | C7A | 2.52 | 156.0 | 1 + x, y, z | |
| O1 | H14 | C14 | 2.56 | 152.3 | −1 + x, y, z | |
| O2 | H16 | C16 | 2.62 | 175.0 | 2 − x, 2 − y,1 − z | |
| 8 | O1 | H15 | C15 | 2.55 | 153.4 | 1 − x, 1 − y, −z |
| O1 | H1 | C1 | 2.65 | 157.3 | 1 − x, 1 − y, −z | |
| O1 | H14 | C14 | 2.66 | 136.6 | 1 + x, y, z | |
| 3 | F1A | H14 | C14 | 2.56 | 135.3 | 1/2 + x, 3/2 − y, z |
| F1A | H18 | C18 | 2.44 | 160.7 | 1/2 + x, 3/2 − y, z | |
| F2A | H9B | C9 | 2.51 | 142.8 | x, y, z | |
| F2 | H9B | C9 | 2.56 | 130.7 | x, y, z | |
| F2 | H42 | C42 | 2.44 | 145.6 | 1 − x, 1 − y, 1/2 + z | |
| F3 | H11 | C11 | 2.51 | 158.2 | x, 1 + y, z | |
| F3 | H32 | C32 | 2.63 | 160.6 | 1/2 − x, 1/2 + y, 1/2 + z | |
| F4A | H38 | C38 | 2.56 | 129.9 | x, 1 + y, z | |
| F4 | H5 | C5 | 2.46 | 154.5 | x, 1 + y, z | |
| F5A | H21 | C21 | 2.57 | 148.4 | x, y, z | |
| F6A | H42 | C42 | 2.55 | 146.6 | 1 − x, 1 − y, 1/2 + z | |
| C15 | H37 | C37 | 2.79 | 153.4 | 1 − x, 1/2 − y, 1/2 + z |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Gobbo, J.; Santini, C.; Dolmella, A.; Li, Z.; Caviglia, M.; Pellei, M. New Copper Complexes with N,O-Donor Ligands Based on Pyrazole Moieties Supported by 3-Substituted Acetylacetone Scaffolds. Molecules 2024, 29, 621. https://doi.org/10.3390/molecules29030621
Del Gobbo J, Santini C, Dolmella A, Li Z, Caviglia M, Pellei M. New Copper Complexes with N,O-Donor Ligands Based on Pyrazole Moieties Supported by 3-Substituted Acetylacetone Scaffolds. Molecules. 2024; 29(3):621. https://doi.org/10.3390/molecules29030621
Chicago/Turabian StyleDel Gobbo, Jo’, Carlo Santini, Alessandro Dolmella, Zhenzhen Li, Miriam Caviglia, and Maura Pellei. 2024. "New Copper Complexes with N,O-Donor Ligands Based on Pyrazole Moieties Supported by 3-Substituted Acetylacetone Scaffolds" Molecules 29, no. 3: 621. https://doi.org/10.3390/molecules29030621
APA StyleDel Gobbo, J., Santini, C., Dolmella, A., Li, Z., Caviglia, M., & Pellei, M. (2024). New Copper Complexes with N,O-Donor Ligands Based on Pyrazole Moieties Supported by 3-Substituted Acetylacetone Scaffolds. Molecules, 29(3), 621. https://doi.org/10.3390/molecules29030621