3.2. Synthesis and Characterization of the Compounds
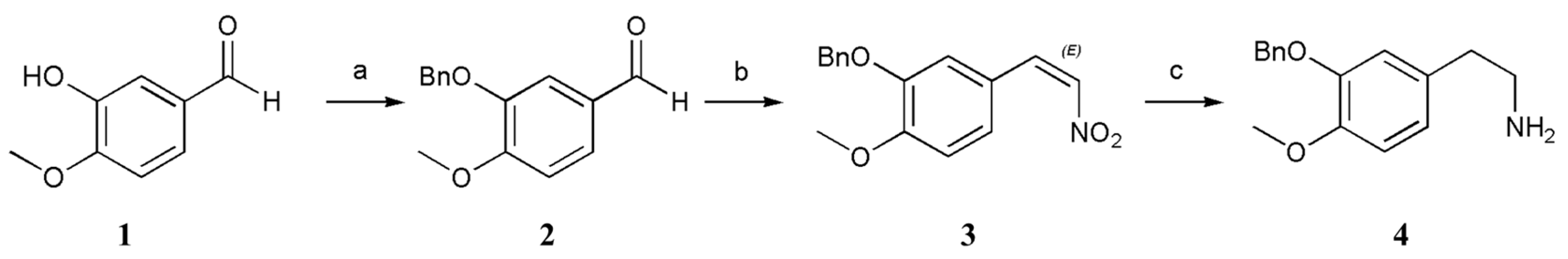
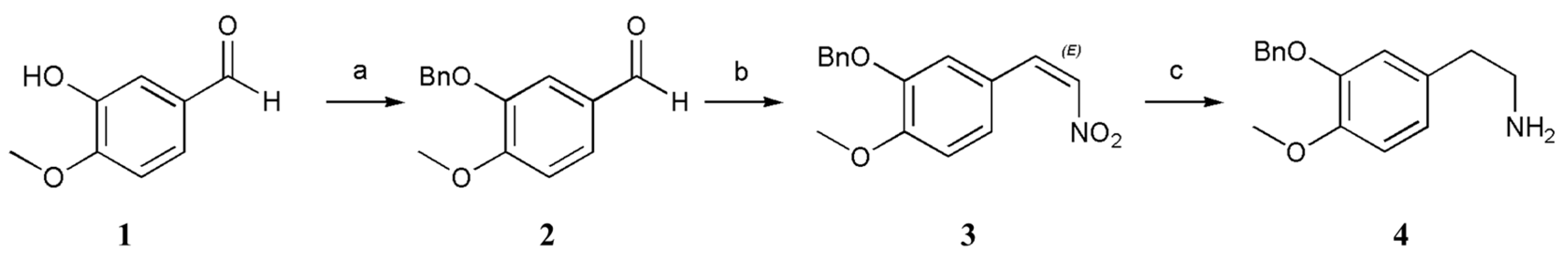
3.2.1. 3-(Benzyloxy)-4-methoxybenzaldehyde (2)
A solution of 3-hydroxy-4-methoxybenzaldehyde (1; 1.52 g, 10.0 mmol, 1 equiv.), BnCl (1.52 g, 12.0 mmol, 1.2 equiv.), K2CO3 (4.15 g, 30.0 mmol, 3 equiv.), and KI (4.15 mg, 0.025 mmol, 0.025 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 2 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (20 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure. The resultant crude products were purified by recrystallization (hexane), which furnished 3-(benzyloxy)-4-methoxybenzaldehyde (2) (83% yield).
3-(Benzoxy)-4-methoxybenzaldehyde (2): white solid; Rf (Hexane/EtOAc 80:20) = 0.62; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 90:10); 1H-NMR (400 MHz, CDCl3): δ = 9.80 (s, 1H), 7.46–7.44 (m, 4H), 7.38–7.34 (m, 2H), 7.32 (m, 1H), 7.00 (d, J = 8.8 Hz, 1H), 5.17 (s, 2H), 3.94 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 190.97, 155.15, 148.79, 136.38, 130.07, 128.75 (2C), 128.23, 127.59 (2C), 127.01, 111.44, 110.88, 70.93, 56.27. HRMS (ESI) calculated for C15H15O3+ [M + H]+ 243.1016, found 243.1013.
3.2.2. 2-(Benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3)
A solution of 3-(benzyloxy)-4-methoxybenzaldehyde (2; 2.42 g, 10.0 mmol, 1 equiv.), NH4OAc (1.00 g, 13.0 mmol, 1.3 equiv.), and CH3NO2 (2.44 g, 40.0 mmol, 4 equiv.) in anhydrous HOAc (10 mL) was stirred at 118 °C for 4 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The precipitate was washed with water to neutral, which furnished 2-(benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3) (89% yield).
2-(Benzoxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3): yellow solid; Rf (Hexane/EtOAc 80:20) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 90:10) 1H-NMR (400 MHz, CDCl3): δ = 7.89 (d, J = 13.6 Hz, 1H), 7.42–7.30 (m, 6H), 7.15–7.13 (m, 1H), 7.01 (s, 1H), 6.91–6.89 (m, 1H), 5.14 (s, 2H), 3.91 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 153.56, 148.68, 139.42, 136.37, 135.20, 128.83 (2C), 128.32, 127.43 (2C), 125.02, 122.74, 113.22, 111.83, 71.25, 56.20. HRMS (ESI) calculated for C16H16NO4+ [M + H]+ 286.1074, found 286.1072.
3.2.3. 2-(3-(Benzyloxy)-4-methoxyphenyl)ethanamine (4)
A flask containing anhydrous THF (15 mL) was cooled to 0 °C under N2, after which LiAlH4 (1.52 g, 40.0 mmol, 4 equiv.) was added cautiously. A solution of 2-(benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3; 2.85 g, 10.0 mmol, 1 equiv.) in anhydrous THF (30 mL) was added dropwise at 35 °C. The temperature of the system was controlled at 35 °C under N2 for 4 h. Upon completion, the reaction mixture was cooled to 0 °C and slowly quenched with water (6 mL). After 15 min, 15% (w/w) aq. NaOH (3 mL) was added. The resultant mixture was stirred for 30 min at 20–30 °C, and the mixture was filtered through a Celite pad with anhydrous MgSO4. The filtrate was concentrated, and the residue was purified as follows: methanol (15 mL) was added to the residue and allowed to dissolve completely by stirring. Anhydrous oxalic acid (1.05 g) was also added at 45 °C. Once completely dissolved, ethyl acetate (50 mL) was introduced into the mixture, and a significant amount of white solid was precipitated. The reaction was stirred for 2 h before cooling it down to 20–30 °C, after which we filtered the solution and air dried the obtained filter cake. Next, we adjusted the pH value to approximately 9–10 by adding a solution containing 30% (w/w) aq. NaOH (40 mL) while continuously stirring magnetically for 2 h. The solid was filtered, and then we beat the filter cake with water (50 mL) for 2 h before filtering it once more. Finally, the resulting filter cake was dried by air, which furnished the 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4) (50% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)ethanamine (4): white solid; Rf (MeOH/CH2Cl2 90:10, 0.02% NH3·H2O) = 0.56; purification by flash column chromatography (deactivated silica gel, MeOH/CH2Cl2 95:5) 1H-NMR (400 MHz, CDCl3): δ = 7.39–7.37 (m, 2H), 7.30–7.26 (m, 2H), 7.23–7.20 (m, 1H), 7.96 (d, J = 8.0 Hz, 1H), 6.70–6.66 (m, 2H), 5.06 (s, 2H), 3.76 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 148.30, 148.09, 137.28, 132.27, 128.58 (2C), 127.90, 127.48 (2C), 121.56, 115.14, 112.10, 71.07, 56.10, 43.47, 39.16. HRMS (ESI) calculated for C16H20NO2+ [M + H]+ 258.1489, found 258.1489.
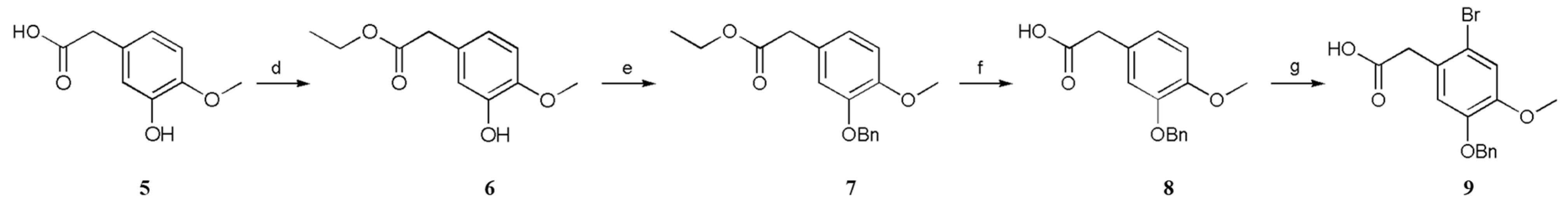
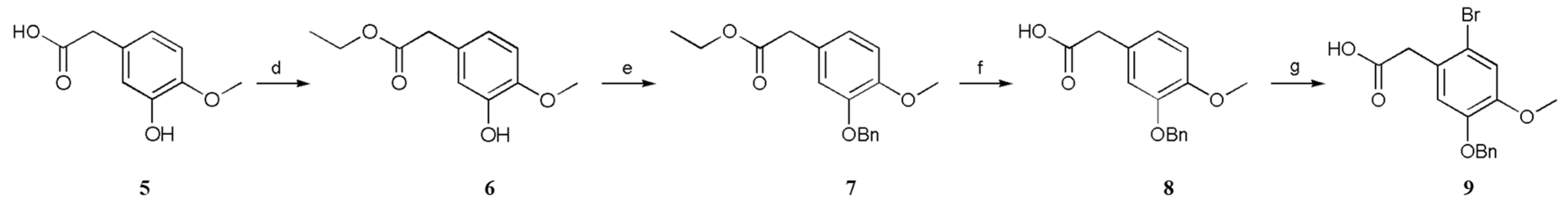
3.2.4. Ethyl 2-(3-Hydroxy-4-methoxyphenyl)acetate (6)
A solution of 2-(3-hydroxy-4-methoxyphenyl)acetic acid (5; 1.82 g, 10.0 mmol, 1 equiv.) and H2SO4 (1.83 g, 1 mL, 18.7 mmol, 1.87 equiv.) in anhydrous EtOH (10 mL) was stirred at 78 °C for 4 h. The mixture was then cooled to 20–30 °C. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (10 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 2 mL), water (2 × 5 mL), and brine (2 × 5 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6) (90% yield).
Ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6): colorless oil; Rf (Hexane/EtOAc 35:65) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 60:40) 1H-NMR (400 MHz, CDCl3): δ = 6.78–6.74 (m, 1H), 6.61–6.57 (m, 2H), 6.49 (br, 1H), 4.01–3.95 (m, 2H), 3.59–3.55 (m, 3H), 3.39–3.35 (m, 2H), 1.10–1.02 (m, 3H); 13C-NMR (100 MHz, CDCl3): δ = 172.10, 146.14, 145.82, 127.17, 120.70, 115.88, 111.06, 60.81, 55.71, 40.58, 13.99. HRMS (ESI) data were calculated for C11H14O4Na+ [M + Na]+ 233.0784, found 233.0788.
3.2.5. Ethyl 2-(3-(Benzyloxy)-4-methoxyphenyl)acetate (7)
A solution of ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6; 2.1 g, 10.0 mmol, 1 equiv.), BnCl (1.52 g, 12.0 mmol, 1.2 equiv.), K2CO3 (4.15 g, 30.0 mmol, 3 equiv.), and KI (4.15 mg, 0.025 mmol, 0.025 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 3 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (20 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure. The resultant crude products were purified by recrystallization (hexane), which furnished ethyl 2-(3-(benzyloxy)-4-methoxyphenyl)acetate (7) (82% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)acetate (7): white solid; Rf (Hexane/EtOAc 35:65) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 60:40) 1H-NMR (400 MHz, CDCl3): δ = 7.45–7.27 (m, 5H), 6.87–6.83 (m, 3H), 5.13 (s, 1H), 4.11 (q, 2H), 3.85 (s, 3H), 3.49 (s, 2H), 1.21 (t, 3H); 13C-NMR (100 MHz, CDCl3): δ = 171.88, 148.93, 148.24, 137.20, 128.62 (2C), 127.93, 127.47 (2C), 126.68, 122.13, 115.18, 111.95, 71.09, 60.88, 56.13, 40.99, 14.28. HRMS (ESI) calculated for C18H21O4+ [M + H]+ 301.1434, found 301.1436.
3.2.6. 2-(3-(Benzyloxy)-4-methoxyphenyl)acetic Acid (8)
A solution of ethyl 2-(3-(benzyloxy)-4-methoxyphenyl)acetate (7; 3.00 g, 10.0 mmol, 1 equiv.) and 20% (w/w) aq. NaOH (40 mL) in anhydrous EtOH (15 mL) was stirred at 78 °C for 3 h. The mixture was then cooled to 20–30 °C. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (10 mL) and water (10 mL). Following this, the 10% (w/w) aq. HCl was added to adjust the pH to 2–3. The precipitate was filtered off. The precipitate was washed with water to neutral, which furnished 2-(3-(benzyloxy)-4-methoxyphenyl)acetic acid (8) (93% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)acetic acid (8): white solid; Rf (Hexane/EtOAc 80:20) = 0.53; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 50:50) 1H-NMR (400 MHz, CDCl3): δ = 7.43–7.27 (m, 5H), 6.84–6.83 (m, 3H), 5.11 (s, 2H), 3.85 (s, 3H), 3.52 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ = 177.96, 149.13, 148.27, 137.04, 128.63 (2C), 127.98, 127.57 (2C), 125.75, 122.29, 115.34, 111.96, 71.15, 56.12, 40.60. HRMS (ESI) calculated for C16H16O4Na+ [M + Na]+ 295.0941, found 295.0944.
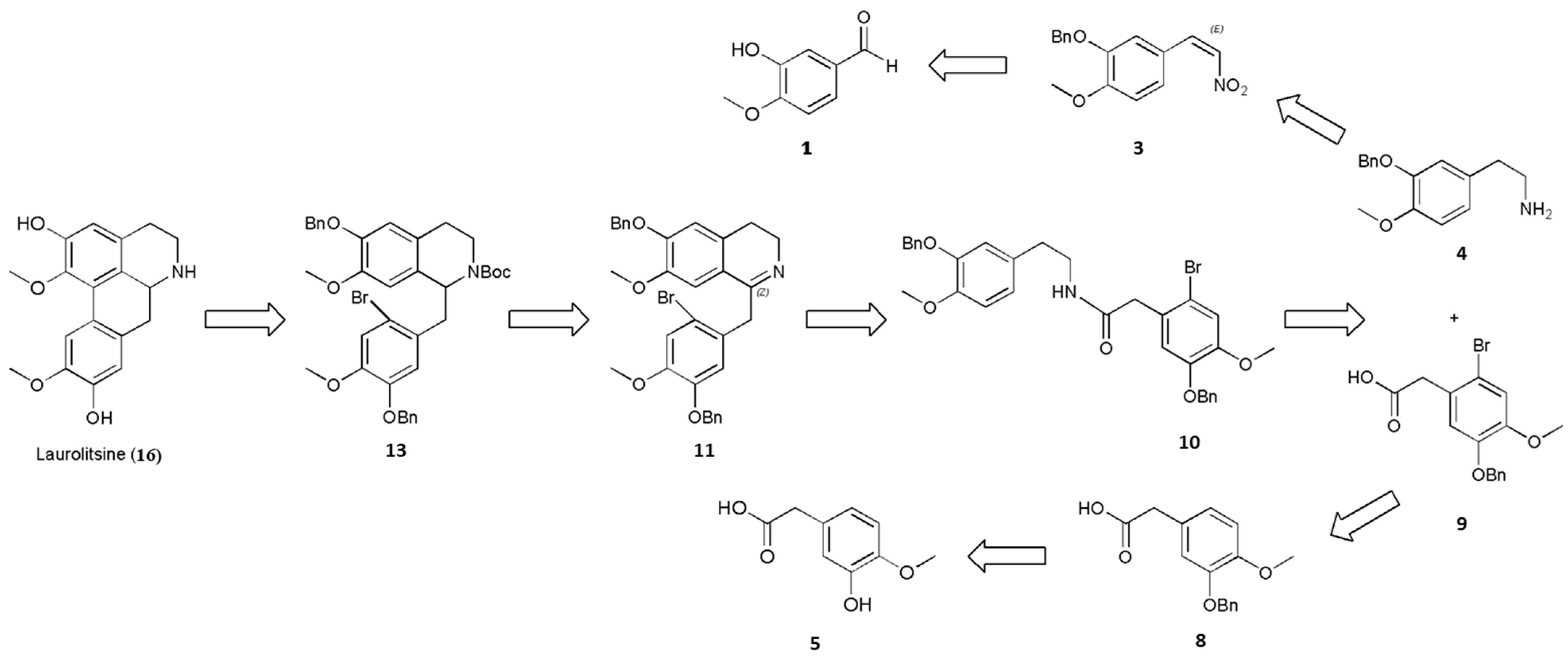
3.2.7. 2-(5-(Benzyloxy)-2-bromo-4-methoxyphenyl)acetic Acid (9)
Bromine (11.2 mmol, 1.12 equiv.) was cautiously added to a solution of 2-(3-(benzyloxy)-4-methoxyphenyl)acetic acid (8; 2.72 g, 10.0 mmol, 1 equiv.) and anhydrous sodium acetate (1.36 g, 34.0 mmol, 3.4 equiv.) in acetic acid (15 mL), and the mixture was stirred for 1 h at 20–30 °C. The precipitate was filtered off and washed with water to neutral pH, which furnished 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9) (61% yield).
2-(5-(Benzoxy)-2-bromo-4-methoxyphenyl)acetic acid (9): white solid; Rf (Hexane/EtOAc 65:35) = 0.46; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 85:15) 1H-NMR (400 MHz, CDCl3): δ = 8.02 (br, 1H), 7.41–7.29 (m, 5H), 7.05 (s, 1H), 6.82 (s, 1H), 5.09 (s, 2H), 3.84 (s, 3H), 3.70 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ = 176.87, 149.80, 147.63, 136.56, 128.71 (2C), 128.18, 127.56 (2C), 125.25, 116.74, 116.04, 115.80, 71.39, 56.32, 40.85. HRMS (ESI) calculated for C16H15BrO4Na+ [M + Na]+ 373.0046, found 373.0050.
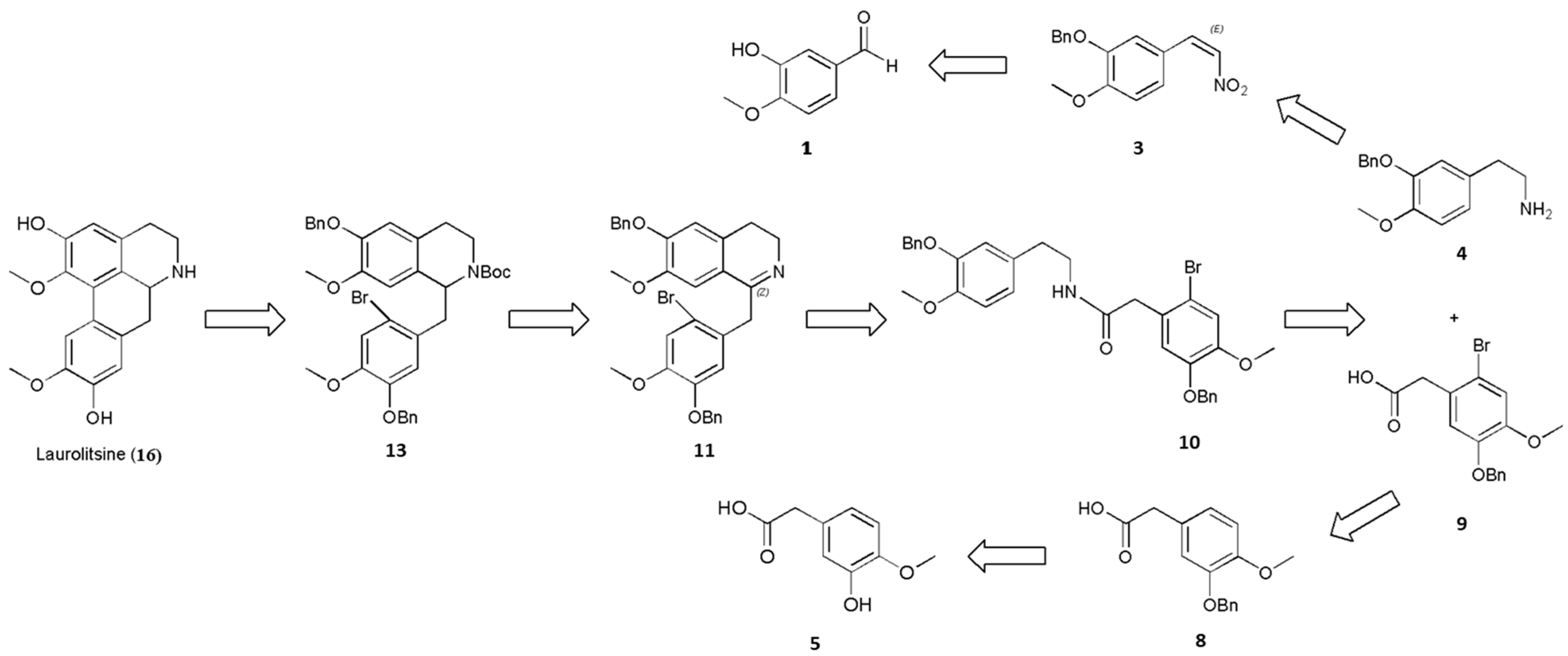
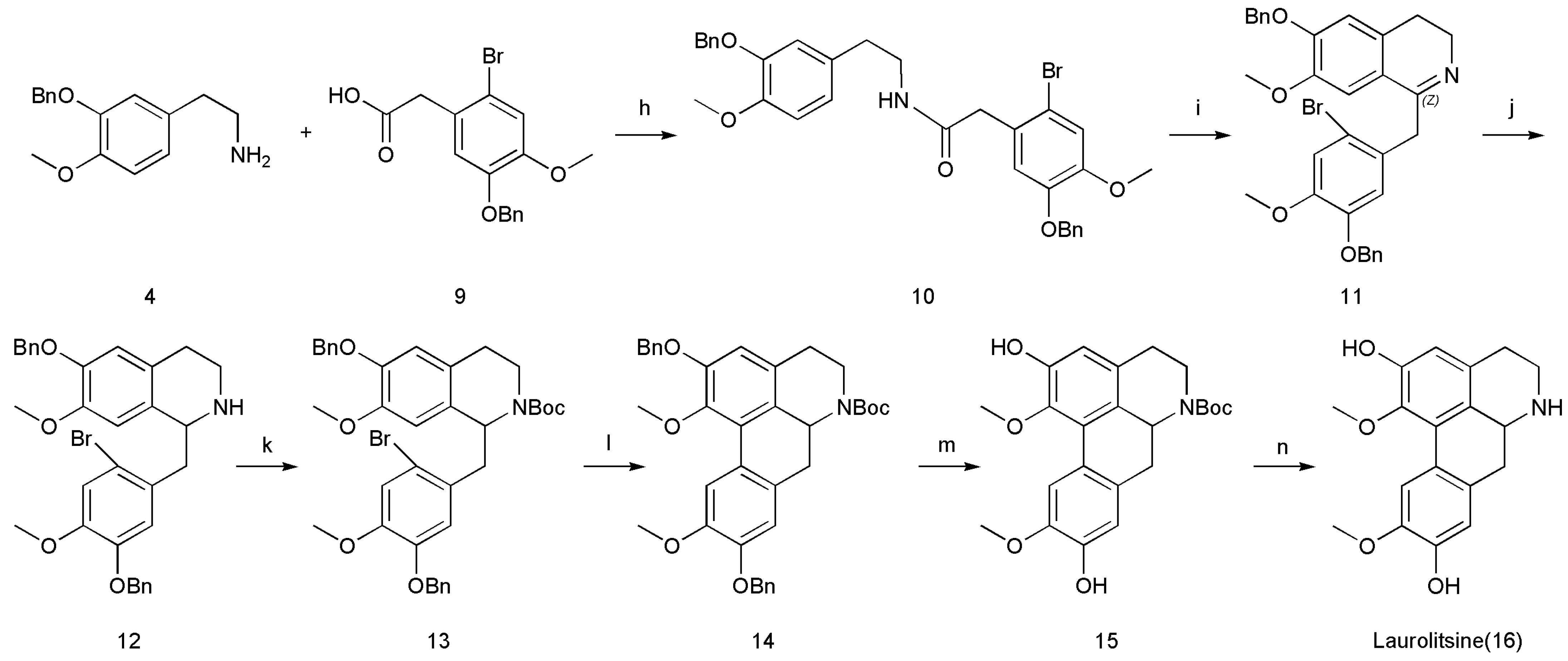
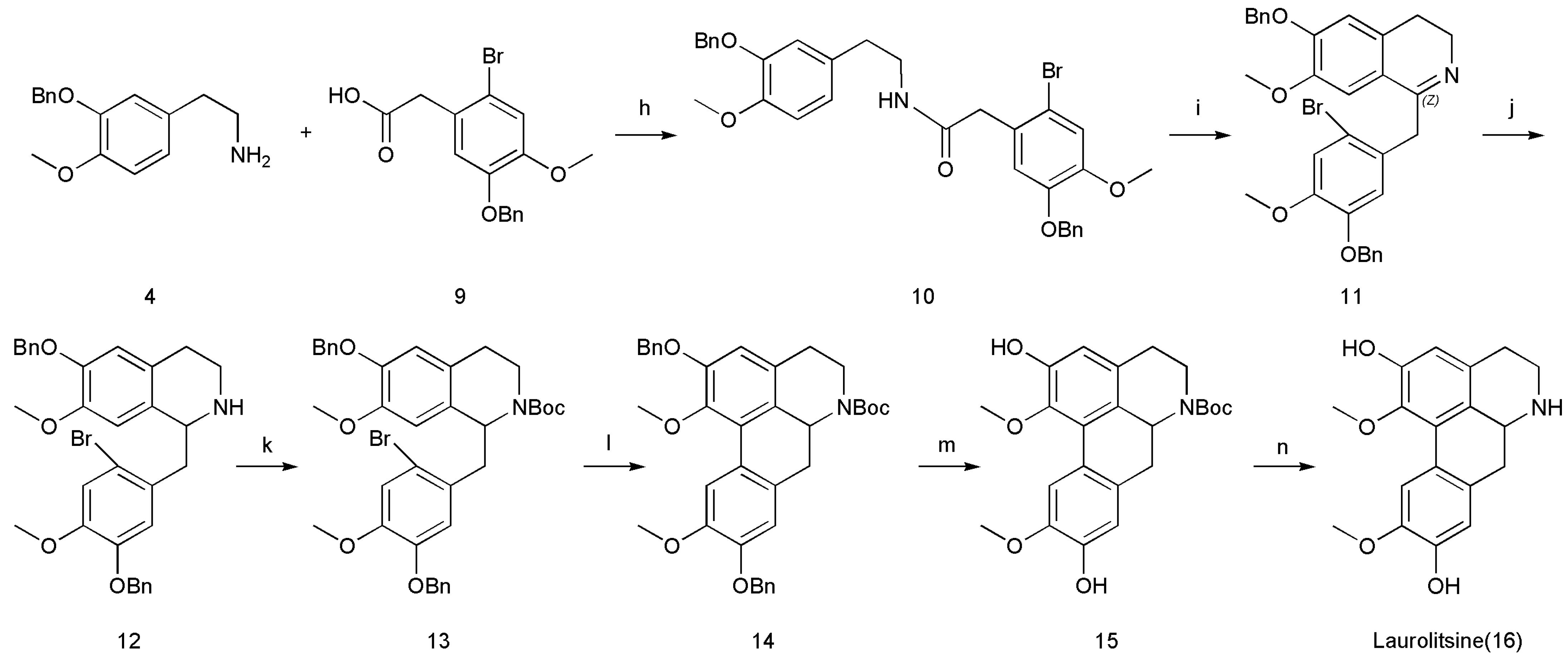
3.2.8. N-(3-(Benzoxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10)
Next, 1-hydroxybenzotriazole (2.03 g, 15.0 mmol, 1.5 equiv.) was added to a solution of 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4; 2.57 g, 10.0 mmol, 1 equiv.) and 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9; 3.69 g, 10.5 mmol, 1.05 equiv.) in dry DMF (50 mL). The reaction mixture was cooled to 0 °C, after which 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (2.3 g, 12.0 mmol, 1.2 equiv.) was added. The reaction mixture was warmed slowly to 20–30 °C and then stirred for 4 h before being quenched by the addition of NaHCO3 (25 mL, sat., aq.) and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated to afford N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10) (74% yield).
N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10): white solid; Rf (Hexane/EtOAc 65:35) = 0.62; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 85:15) 1H-NMR (400 MHz, CDCl3): δ = 7.43–7.28 (m, 10H), 6.99 (s, 1H), 6.78 (s, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 6.58 (dd, J = 8.1, 2.0 Hz, 1H), 5.33 (t, J = 5.8 Hz, 1H), 5.08 (s, 2H), 5.06 (s, 2H), 3.82 (s, 3H), 3.81 (s, 3H), 3.49 (s, 2H), 3.37 (q, J = 7.0 Hz, 2H), 2.60 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 169.83, 149.73, 148.46, 148.30, 147.81, 137.15, 136.42, 131.05, 128.70 (2C), 128.62 (2C), 128.19, 127.96, 127.54 (2C), 127.47 (2C), 126.49, 121.39, 116.43, 116.11, 115.48, 114.69, 111.97, 71.17, 71.10, 56.29, 56.10, 43.64, 40.72, 34.94, 29.78. HRMS (ESI) data were calculated for C32H33BrNO5+ [M + H]+ 590.1537, found 590.1538.
3.2.9. 1-(5-(Benzoxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11)
A solution of N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10; 5.91 g, 10.0 mmol, 1 equiv.) and POCl3 (1.84 g, 12.0 mmol, 1.2 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 4 h. The mixture was then cooled to 20–30 °C and quenched slowly with water (6 mL). The solvent was removed under reduced pressure. The resultant crude products were purified by column chromatography on silica gel (CH2Cl2), which furnished 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11) (71% yield).
1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11): White solid; Rf (CH2Cl2/MeOH 95:5) = 0.52; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, CDCl3): δ = 7.42–7.18 (m, 10H), 7.03 (s, 1H), 6.90 (s, 1H), 6.79 (s, 1H), 6.62 (s, 1H), 5.15 (s, 2H), 5.00 (s, 2H), 4.04 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 3.59 (t, J = 7.5 Hz, 2H), 2.43 (t, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 165.65, 150.04, 148.98, 147.99, 147.70, 136.71, 136.68, 131.43, 129.43, 128.76 (2C), 128.54 (2C), 128.13, 127.88, 127.28 (2C), 127.16 (2C), 121.61, 115.78, 114.87, 114.69, 112.31, 109.75, 70.97, 70.85, 56.38, 56.27, 47.21, 42.05, 25.68. HRMS (ESI) calculated for C32H31BrNO4+ [M + H]+ 572.1431, found 572.1432.
3.2.10. 1-(5-(Benzoxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12)
NaBH4 (0.57 g, 15.0 mmol, 1.5 equiv.) was added slowly to a stirred ice-cooled solution of 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11; 5.72 g, 10.0 mmol, 1.0 equiv.) in EtOH (30 mL) at 0 °C. The mixture was stirred at 0 °C for 1 h and then at 20–30 °C for 2 h. The mixture was cooled to 0 °C and diluted with water (10 mL), after which the precipitate was filtered. The precipitate was washed with water to neutral, which furnished 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12) (72% yield).
1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12): white solid; Rf (CH2Cl2/MeOH 90:10) = 0.55; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, DMSO-d6): δ = 9.16 (br, 1H), 7.41–7.32 (m, 10H), 7.28 (s, 1H), 7.18 (s, 1H), 6.88 (s, 1H), 6.36 (s, 1H), 5.07 (d, J = 11.6 Hz, 1H), 5.03 (s, 2H), 4.98 (d, J = 11.6 Hz, 1H), 4.53 (t, J = 7.4 Hz, 2H), 3.75 (s, 3H), 3.53 (s, 3H), 3.41–3.33 (m, 2H), 3.20–3.15 (m, 2H), 3.02–2.95 (m, 1H), 2.87–2.79 (m, 1H); 13C-NMR (100 MHz, DMSO-d6): δ = 149.59, 147.90, 147.80, 137.47, 137.13, 130.19, 129.00 (2C), 128.96 (2C), 128.58 (2C), 128.43, 128.31 (2C), 127.74, 125.02, 117.68, 116.44, 115.43, 113.93, 110.55, 70.74, 70.34, 56.57, 55.88, 54.35, 39.19, 29.61, 29.12, 27.08, 25.37. HRMS (ESI) data were calculated for C32H33BrNO4+ [M + H]+ 574.1588, found 574.1587.
3.2.11. Tert-Butyl 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13)
Triethylamine (1.32 g, 13.0 mmol, 1.3 equiv.) and Boc2O (2.62 g, 12.0 mmol, 1.2 equiv.) were added to a stirred solution of 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12; 5.75 g, 10.0 mmol, 1 equiv.) in CH2Cl2 (15 mL) at 20–30 °C, and the resulting mixture was stirred for 2 h and washed sequentially with water (2 × 10 mL) and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13) (80% yield).
Tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13): white solid; Rf (CH2Cl2/MeOH 97:3) = 0.52; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, CDCl3): δ = 7.44–7.27 (m, 10H), 7.06 (s, 1H), 6.72 (s, 1H), 6.63 (s, 1H), 6.55 (s, 1H), 5.24 (dd, J = 10.1, 3.8 Hz, 1H), 5.11–5.05 (m, 4H), 4.27–4.21 (m, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.18–3.13 (m, 2H), 2.85–2.79 (m, 2H), 2.57–2.52 (m, 1H), 1.20 (s, 9H); 13C-NMR (400 MHz, CDCl3): δ = 154.17, 149.84, 146.58, 143.65, 130.39, 130.28, 127.43, 123.82, 123.01, 115.68, 114.98, 112.55, 79.48, 67.53, 59.61, 56.34, 56.10, 30.02, 28.63 (3C), 28.22, 25.64. HRMS (ESI) calculated for C37H41BrNO6+ [M + H]+ 674.2112, found 674.2114.
3.2.12. Tert-Butyl 1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (14)
Pd(OAc)2 (4.49 g, 2.0 mmol, 0.2 equiv.), the ligand di-tert-butyl(methyl)phosphonium tetrafluoroborate (9.89 g, 4.0 mmol, 0.4 equiv.), and Cs2CO3 (9.77 g, 3.0 mmol, 3 equiv.) were added to a solution of tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13; 6.75 g, 10.0 mmol, 1 equiv.) in 1,4-dioxane (40 mL) by purging with nitrogen for 8 h at 101 °C. After cooling to 20–30 °C, the solvent was removed under reduced pressure. The precipitate was extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was dried (anhydrous Na2SO4), and the precipitate was loaded onto a deactivated silica gel column (200–300 mesh) and eluted with hexane/EtOAc (90:10, v/v) to afford 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-,1,1-dimethylethyl ester (14) (53%).
6H-Dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-Tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-, 1,1-dimethylethyl ester (14): white solid; Rf (Hexanes/EtOAc 80:20) = 0.64; purification by flash column chromatography (deactivated silica gel, Hexanes/EtOAc 94:6) 1H-NMR (400 MHz, CDCl3): δ = 8.17 (s, 1H), 7.47–7.27 (m, 10H), 6.77 (s, 1H), 6.67 (s, 1H), 5.19–5.08 (m, 4H), 4.63–4.60 (m, 1H), 4.38–4.35 (m, 1H), 3.91 (s, 3H), 3.70 (s, 3H), 2.92–2.68 (m, 5H), 2.59 (d, J = 14.7 Hz, 1H), 1.43 (s, 9H); 13C-NMR (400 MHz, CDCl3): δ = 154.28, 149.23, 148.14, 147.81, 147.19, 137.18, 136.96, 130.35, 129.53, 128.75 (2C), 128.66 (2C), 128.05, 127.96, 127.37 (2C), 127.33 (2C), 126.71, 117.20, 116.02, 115.71, 114.15, 110.71, 79.47, 71.68, 71.13, 56.55, 56.27, 54.33, 42.12, 36.48, 28.53, 28.17 (3C). HRMS (ESI) calculated for C37H39NO6Na+ [M + Na]+ 616.2670, found 616.2667.
3.2.13. Tert-Butyl 6H-Dibenzo[de,g]quinoline-6-carboxylic acid,4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-,1,1-dimethylethyl Ester (15)
Palladium 10% on carbon (wetted with ca. 55% water) (Pd/C) (1.18 g) and HOAc (0.5 mL) were added to a solution of 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-, and 1,1-dimethylethyl ester (14; 5.94 g, 10.0 mmol, 1 equiv.) in THF (40 mL) by purging with hydrogen for 8 h at 20–30 °C. The precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (40 mL) and washed sequentially with water (2 × 20 mL) and brine (2 × 20 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished 6H-dibenzo[de,g]quinoline-6-carboxylic acid and 4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-, 1,1-dimethylethyl ester (15) (82% yield).
6H-Dibenzo[de,g]quinoline-6-carboxylic acid,4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-,1,1-dimethylethyl ester (15): white solid; Rf (Hexanes/EtOAc 66:34) = 0.46; purification by flash column chromatography (deactivated silica gel, Hexanes/EtOAc 80:20) 1H-NMR (400 MHz, DMSO-d6): δ = 9.18 (s, 1H), 9.16 (s, 1H), 7.89 (s, 1H), 6.65 (s, 1H), 6.54 (s, 1H), 4.38–4.33 (m, 1H), 4.15 (d, J = 12.7 Hz, 1H), 3.75 (s, 3H), 3.51 (s, 3H), 2.66–2.49 (m, 5H), 1.37 (s, 9H); 13C-NMR (100 MHz, DMSO-d6): δ = 154.17, 149.84, 146.58, 143.65, 130.39, 130.28, 127.43, 123.82, 123.01, 115.68, 114.98, 112.55, 79.48, 67.53, 59.61, 56.34, 56.10, 30.02, 28.63 (3C), 28.22, 25.64. HRMS (ESI) calculated for C23H27NO6Na+ [M + H]+ 436.1731, found 436.1729.
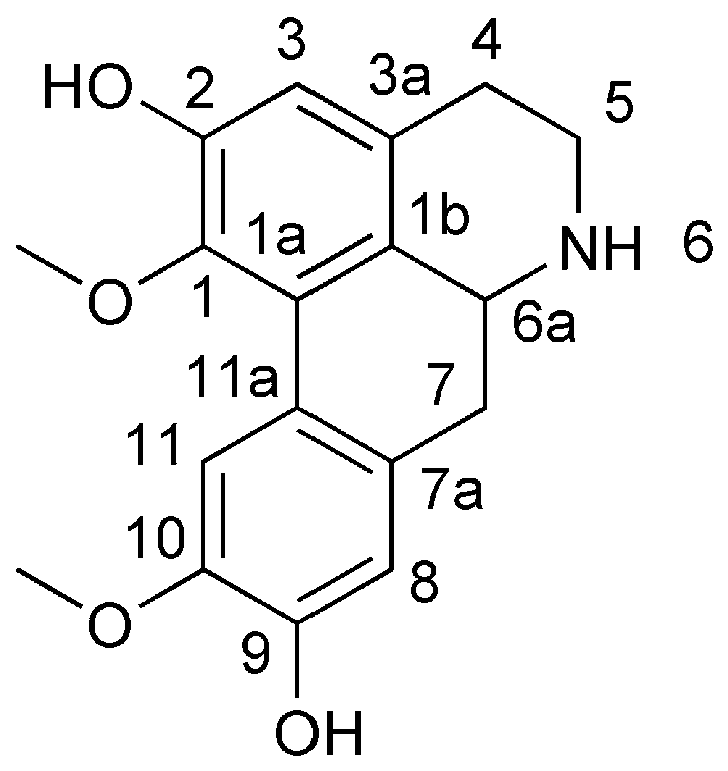
3.2.14. 5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16)
ZnBr2 (9.01 g, 40.0 mmol, 4 equiv.) was added to a solution of 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-, 1,1-dimethylethyl ester (15; 4.13 g, 10.0 mmol, 1 equiv.) in dry CH2Cl2 (50 mL) under a nitrogen atmosphere, and the mixture was stirred at 20–30 °C for 48 h. The mixture was then quenched with a solution of saturated NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 250 mL). The combined organic layer was dried (anhydrous Na2SO4) and concentrated under reduced pressure to afford the resultant crude products. Then, the resultant crude products were purified by column chromatography on silica gel (CH2Cl2/MeOH 90:10), which furnished 5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16) (49% yield).
5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16): brown solid; Rf (CH2Cl2/MeOH 80:20) = 0.45; purification by flash column chromatography (deactivated silica gel, CH2Cl2/MeOH 90:10) 1H-NMR (400 MHz, DMSO-d6): δ = 10.21 (br, 1H), 9.48 (s, 1H), 9.37 (s, 1H), 7.85 (s, 1H), 6.73 (s, 1H), 6.62 (s, 1H), 4.09–4.06 (m, 1H), 3.73 (s, 3H), 3.55 (s, 3H), 3.46–3.44 (m, 1H), 3.13–3.02 (m, 2H), 2.89–2.84 (m, 1H), 2.81–2.73 (m, 2H); 13C-NMR (100 MHz, DMSO-d6): δ = 151.23, 147.01, 146.88, 143.85, 127.11, 127.07, 126.78, 122.73, 120.16, 115.75, 114.94, 112.54, 59.87, 56.16, 52.42, 40.61, 32.82, 25.14. HRMS (ESI) calculated for C18H20NO4+ [M + H]+ 314.1387, found 314.1384.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}