Abstract
By taking advantage of a sequence of oxidative addition/reductive elimination reactions, Pt(II) cyclometalated derivatives are able to promote a rare C(sp2)-C(sp3) bond coupling, resulting in the production of novel methyl-substituted pyridines and bipyridines. Starting from 6-phenyl-2,2′-bipyridine, the step-by-step full sequence of reactions has been followed, leading to the unprecedented 3-methyl-6-phenyl-2,2′-bipyridine, which was isolated and fully characterized. The synthesis involves the following steps: (1) rollover cyclometalation to give the starting complex [Pt(N^C)(DMSO)Me]; (2) the synthesis of a more electron-rich complex [Pt(N^C)(PPh3)Me] by the substitution of DMSO with triphenylphosphine; (3) oxidative addition with methyl iodide to give the Pt(IV) complex [Pt(N^C)(PPh3)(Me)2(I)]; (4) iodide abstraction with silver tetrafluoborate to give an unstable pentacoordinate intermediate, which rapidly evolves through a carbon–carbon reductive coupling, forming a new C(sp3)-C(sp2) bond; (5) finally, the extrusion and characterization of the newly formed 3-methyl-6-phenyl-2,2′-bipyridine. The reaction has been therefore extended to a well-known classical cyclometalating ligand, 2-phenylpyridine, demonstrating that the method is not restricted to rollover derivatives. Following the same step-by-step procedure, 2-phenylpyridine was converted to 2-o-tolyl-pyridine, displaying the potential application of the method to the larger family of classical cyclometalated complexes. The application of this protocol may be useful to convert an array of heterocyclic compounds to their methyl- or alkyl-substituted analogs.
1. Introduction
One of the main goals of organometallic chemistry is the functionalization of C−H bonds mediated by transition metals, particularly when it involves the formation of new carbon–carbon bonds [1]. The C-C coupling is particularly interesting when it implicates the activation of a C-H bonds in organic compounds followed by metalation (the formation of a metal–carbon bond) and successive functionalization (Scheme 1):
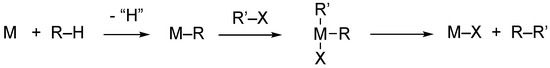
Scheme 1.
Activation, metalation, and functionalization via C-C reductive coupling.
A successive combination of oxidative addition/reductive elimination reactions is often found in catalytic cycles, leading first to the activation of a substrate and second to the formation of new bonds and hence to newly designed compounds. Reductive elimination is usually the key step in C−C bond formation, representing the reaction in which the final product is formed [2].
The cyclometalation reaction is a useful method for C-H bond activation in aromatic heterocycles, leading to particularly stable organometallic complexes known as cyclometalated compounds [3]. These derivatives, characterized by a carbon-heteroatom chelating ligand, have attracted enormous interest for various applications: as catalysts for a large array of reactions [4], in biomedicine as antitumor or antimicrobial agents [5,6], and in the field of innovative materials, such as photoluminescent devices, sensors, switches, etc. [7,8].
Furthermore, after the activation of the C-H bond, the subsequent functionalization of the cyclometalated ligand may facilitate the synthesis of new organic molecules by means of stoichiometric or catalytic processes [9].
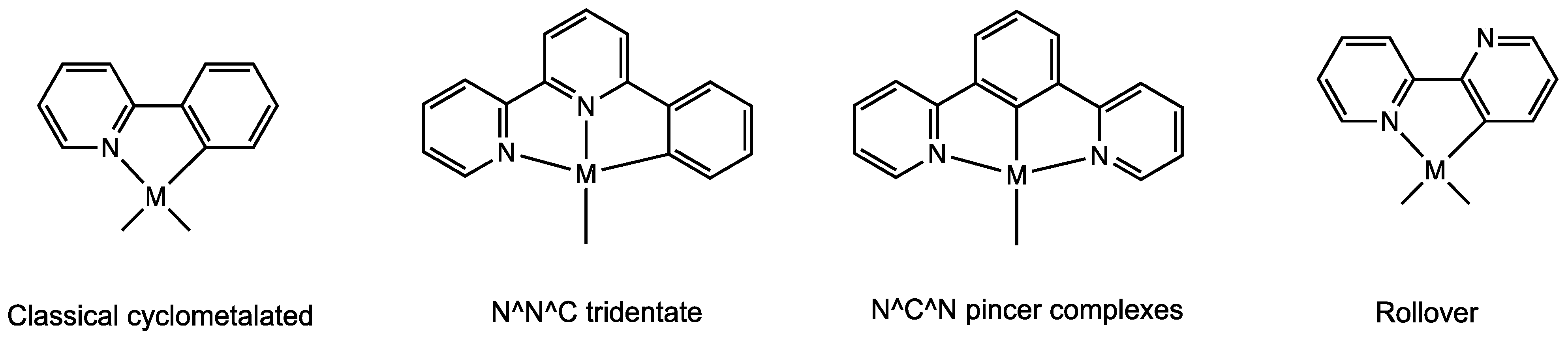
For these reasons, a great variety of subclasses of cyclometalated species have emerged, particularly for nitrogen donors such as pyridines, imines, and amines. In addition to classical C^N donors (e.g., phenylpyridines and phenylamines), the subclasses of tridentate cyclometalates have been thoroughly studied, comprising N^N^C donors [10] (such as 6-phenyl-2,2′-bipyridine) and the so-called “pincer ligands” [11] (e.g., C^N^C, N^C^N donors). One recent and unusual class is that of the rollover complexes, ref. [12] derived from the internal rotation of bidentate heterocyclic ligands, such as 2,2′-bipyridine (Figure 1).
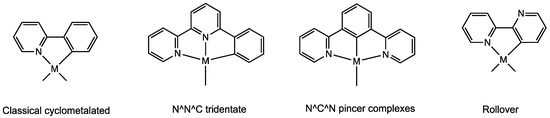
Figure 1.
Some classes of cyclometalated complexes.
Among the various methods, the rollover cyclometalation reaction is a quite new and useful process, suitable for activating heterocyclic donor ligands in remote positions, such as the C3 position in 2,2′-bipyridines (see Figure 1). It is not easy to functionalize these bidentate ligands in certain positions with transition metals owing to their propensity to form stable N^N chelated complexes. In contrast to classical cyclometalated complexes, rollover derivatives contain a free donor atom that confers behaviors not accessible to classical cyclometalated species [13], such as protonation [14], retro-rollover reactions [15,16], successive additional cyclometalation, and polymerization [17].
Rollover complexes were also found to promote C-C bond formation in both stoichiometric (gas phase and in solution) [18,19] and catalytic [20,21,22] reactions. In this regard, we have recently reported that the rollover activation of the C3-H bond in 2,2′-bipyridine Pt(II) rollover complexes might be employed to promote C-C bond formation by means of a sequence of oxidative addition and reductive elimination reactions to give methyl-substituted pyridine rings [23].
Following our previous studies in the field [24], we report some aspects of the behavior of one of the most studied organometallic ligands, 6-phenyl-2,2′-bipyridine (bpyPh), which lead to the activation and functionalization of the remote pyridine C3-H bond through Pt(II) rollover cyclometalation. The reaction has been successfully extended to the corresponding classical ligand, 2-phenylpyridine (pyPh), revealing additional potential applications.
2. Results and Discussion
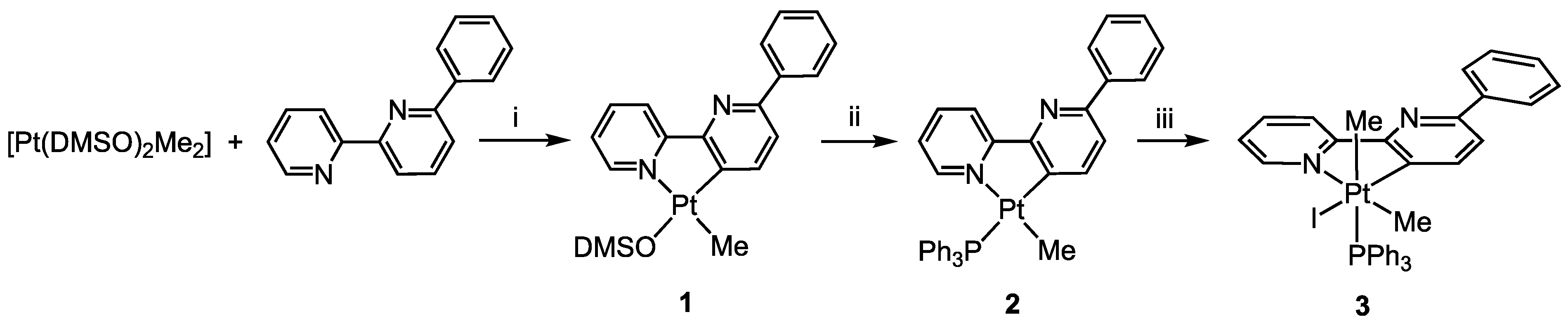
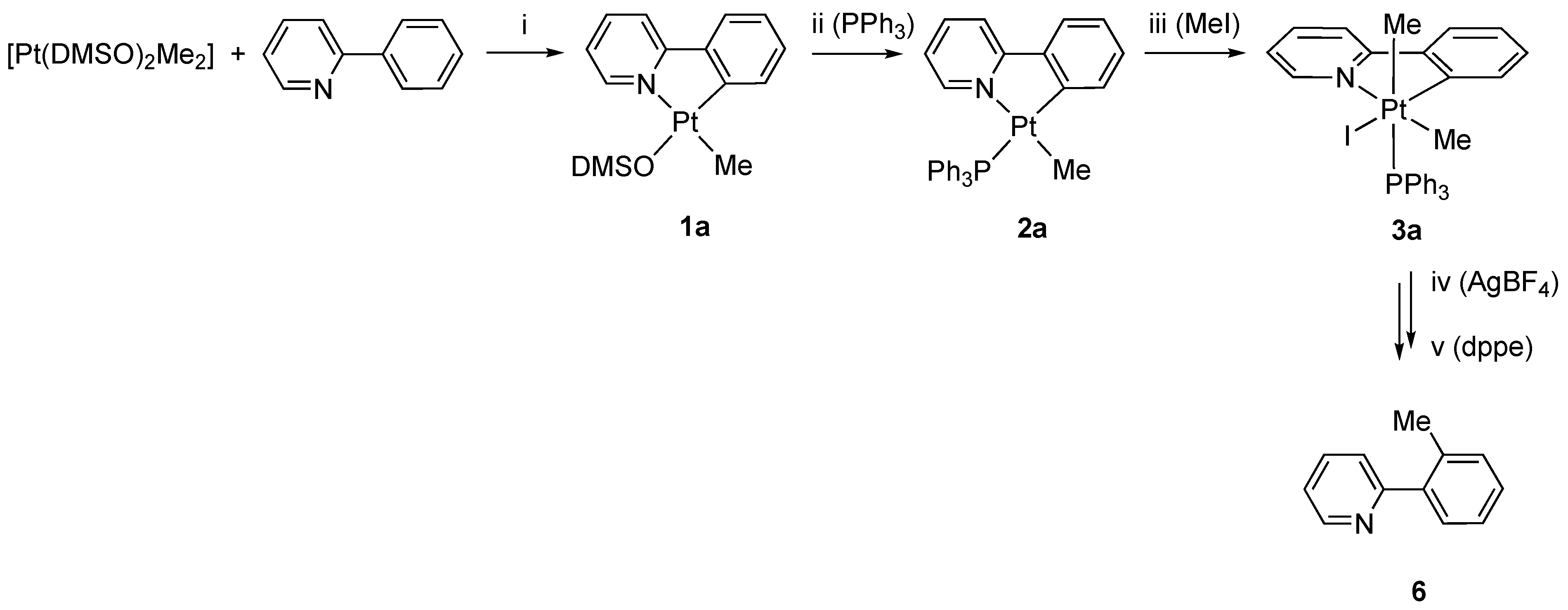
As reported previously, the reaction of the electron-rich Pt(II) precursor [Pt(DMSO)2(Me)2] with 6-phenyl-2,2′-bipyridine, bpyPh, produces the rollover complex [Pt(bpyPh-H)(Me)(DMSO], 1, from which the substitution of the labile DMSO ligand allows the isolation of the corresponding phosphane complex [Pt(bpyPh-H)(Me)(PPh3)], 2 [25]. The reaction can be performed following a “one-pot” procedure by adding PPh3 to the reaction mixture of [Pt(DMSO)2(Me)2] and bpyPh after completion of the rollover process, thus directly isolating 2 in high yield (Scheme 2).
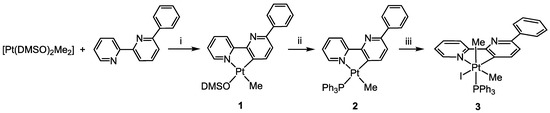
Scheme 2.
Synthesis of complex 3: (i) acetone, 50 °C, 2 h; (ii) acetone, PPh3, r.t., 1 h; (iii) CH2Cl2, MeI, 30 °C, 6 h.
Oxidative addition reactions of alkyl halides to Pt(II) electron-rich complexes have been extensively studied by several authors [26,27,28,29,30]. In particular, cyclometalated Pt(II) complexes are known to result in oxidative addition with methyl iodide, affording the corresponding Pt(IV) derivatives [31]. The reaction usually proceeds through an intermolecular SN2 mechanism at room temperature.
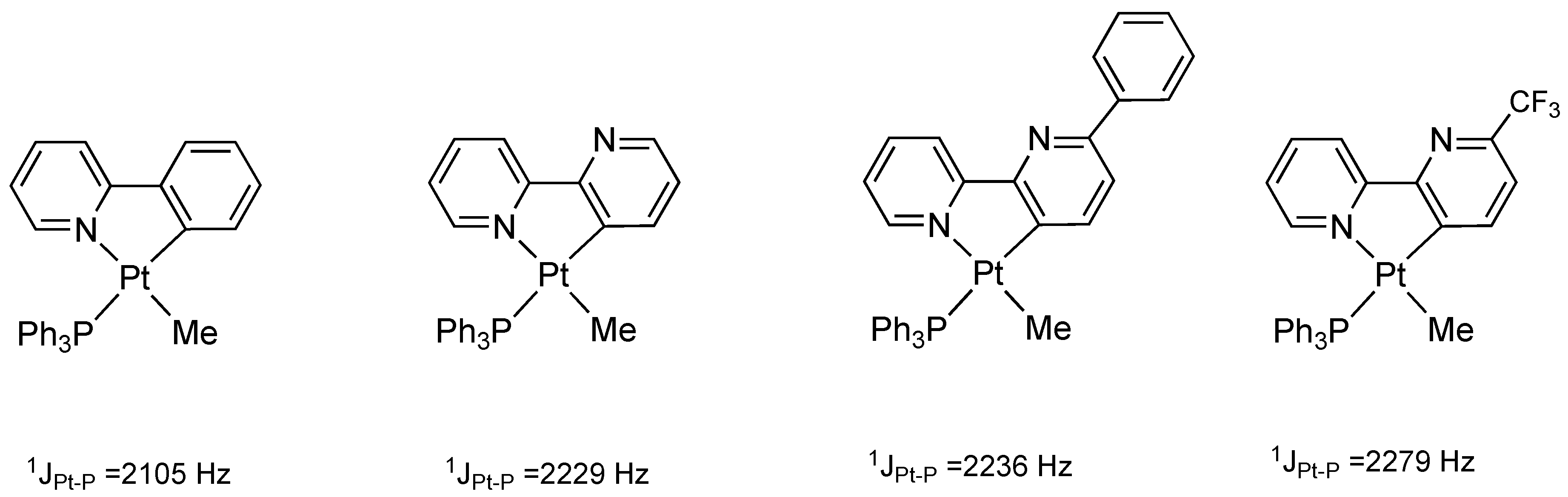
The reaction of complex 2 with MeI does not occur easily: under the same conditions followed by analogous cyclometalated Pt(II) complexes (acetone, room temperature), we were not able to obtain the desired Pt(IV) product. The greater difficulty of the oxidative addition reaction on complex 2 may be explained by the donor properties of the rollover-coordinated 6-phenyl-2,2′-bipyridine. As we recently reported [32], a good indicator of the donor properties of platinum(II) cyclometalated complexes is the direct 195Pt-31P coupling value revealed by 31P NMR spectroscopy. In comparison with related cyclometalated ligands (Figure 2), 6-phenyl-2,2′-bipyridine is a poorer donor than cyclometalated 2-phenylpyridine and rollover-coordinated 2,2′-bipyridine, but better than the worst donor (6-CF3-2,2′-bipyridine). As a consequence, the platinum center in complex 2 is relatively electron-poor compared with similar cyclometalated complexes and the hampered oxidative addition reaction has a plausible justification.
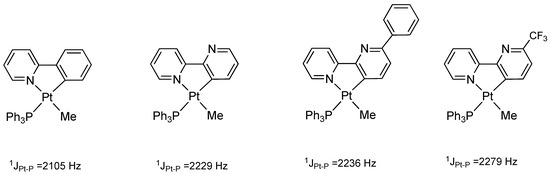
Figure 2.
195Pt-31P NMR coupling constants in [Pt(N^C)(PPh3)Me] complexes as a consequence of N^C ligand donor properties [12,24,25].
It is likely that the presence of the phenyl substituent slows down the reaction, so we raised the temperature and prolonged the reaction time. In this way, we were able to obtain and isolate the Pt(IV) complex [Pt(bpyPh-H)I(Me)2PPh3], 3, in high yield (Scheme 2).
The reaction of 2 with MeI should give a complex with the methyl and iodide ligands entering in a mutually trans position. However, as previously observed, rapid isomerization produced the more stable isomer, with the phosphane ligand in the axial position with respect to the bipyridine metalated plane [33].
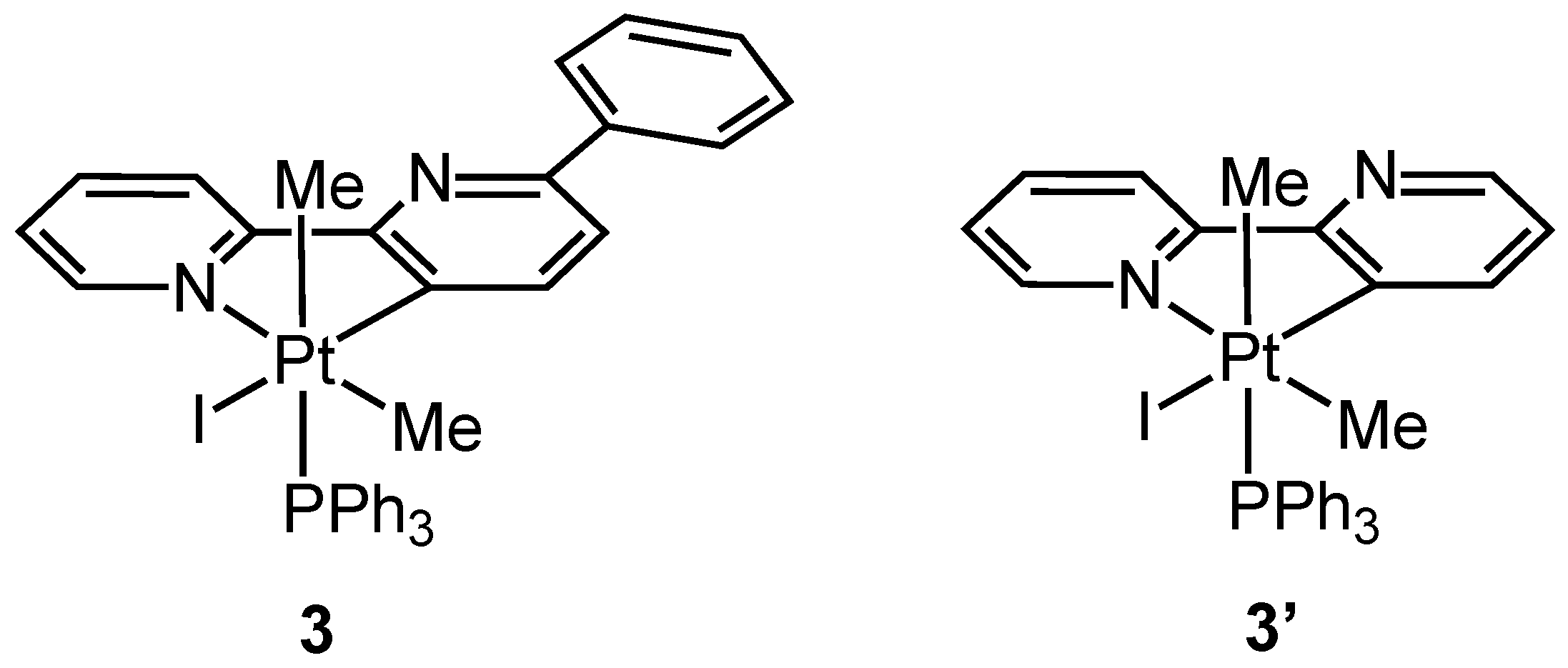
Complex 3 was isolated and characterized analytically and spectroscopically, in particular, by means of NMR spectroscopy. The 31P NMR spectrum of 3 shows a singlet with satellites at −10.95 ppm, with a Pt-P coupling constant value, 1JPt-P = 962.5 Hz, in agreement with a Pt(IV) species, but smaller than that found in the corresponding simple 2,2′-bipyridine rollover complex 3′, [Pt(bpy-H)(Me)2I(PPh3)] (1JPt-P = 991 Hz) (Figure 3).
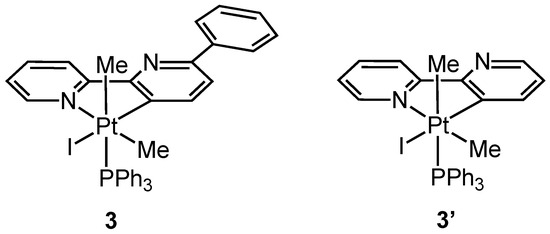
Figure 3.
Pt(IV) derivatives 3 and 3′.
The 1H NMR spectrum shows two different coordinated methyls as doublets with satellites due to coupling to 195Pt and 31P nuclei. Comparison with NMR data for 3′ shows that the shielded methyl (δ 1.24 ppm, 2JPt-H = 60.0 Hz) is coordinated in the “axial” position (with respect to the bipyridine plane), whereas the deshielded methyl (δ 1.70 ppm, 2JPt-H = 70.5 Hz) is in the equatorial position. The H6′ signal is strongly deshielded (δ 9.62 ppm, JPt-H ca 11 Hz), as expected, due to proximity to the iodide ligand [34,35].
Treatment of Pt(IV) Complexes with Silver Salts
Compound 3 is stable in the presence of air and moisture, both in the solid state and in solution.
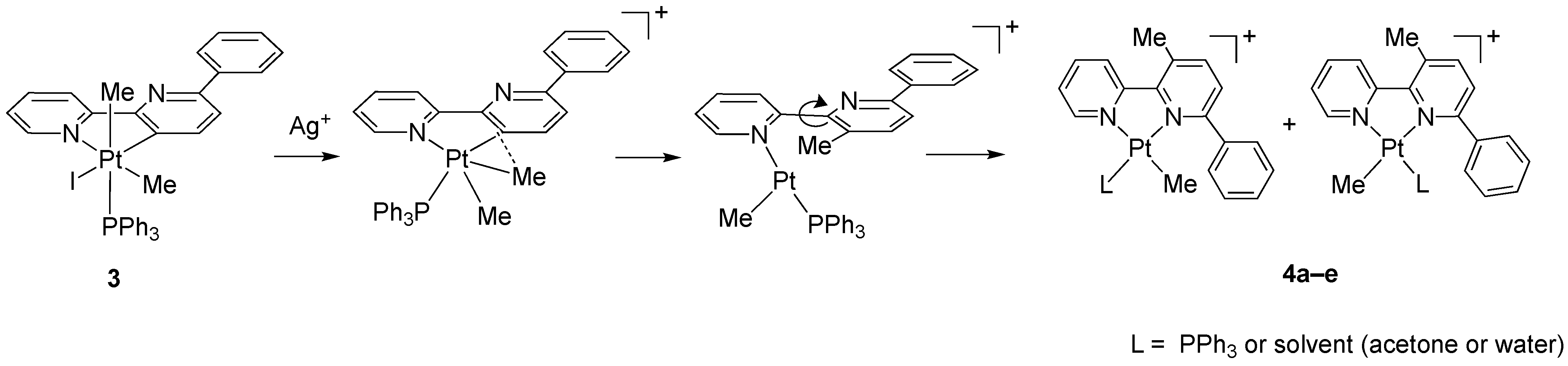
The reaction of 3 with AgBF4 in acetone resulted in the abstraction of the coordinated iodide and the precipitation of solid AgI, which was removed by filtration. The resulting coordinatively unsaturated, five-coordinate species is unstable and rearranges following a reductive elimination process, through C(sp3)-C(sp2) coupling, and a subsequent “retro-rollover” process, involving the rotation of the substituted pyridine, allows the coordination of the second nitrogen atom to prepare N^N chelated adducts (Scheme 3).
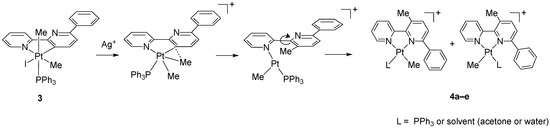
Scheme 3.
Reaction of 3 with AgBF4 (acetone, room temperature, 3 h).
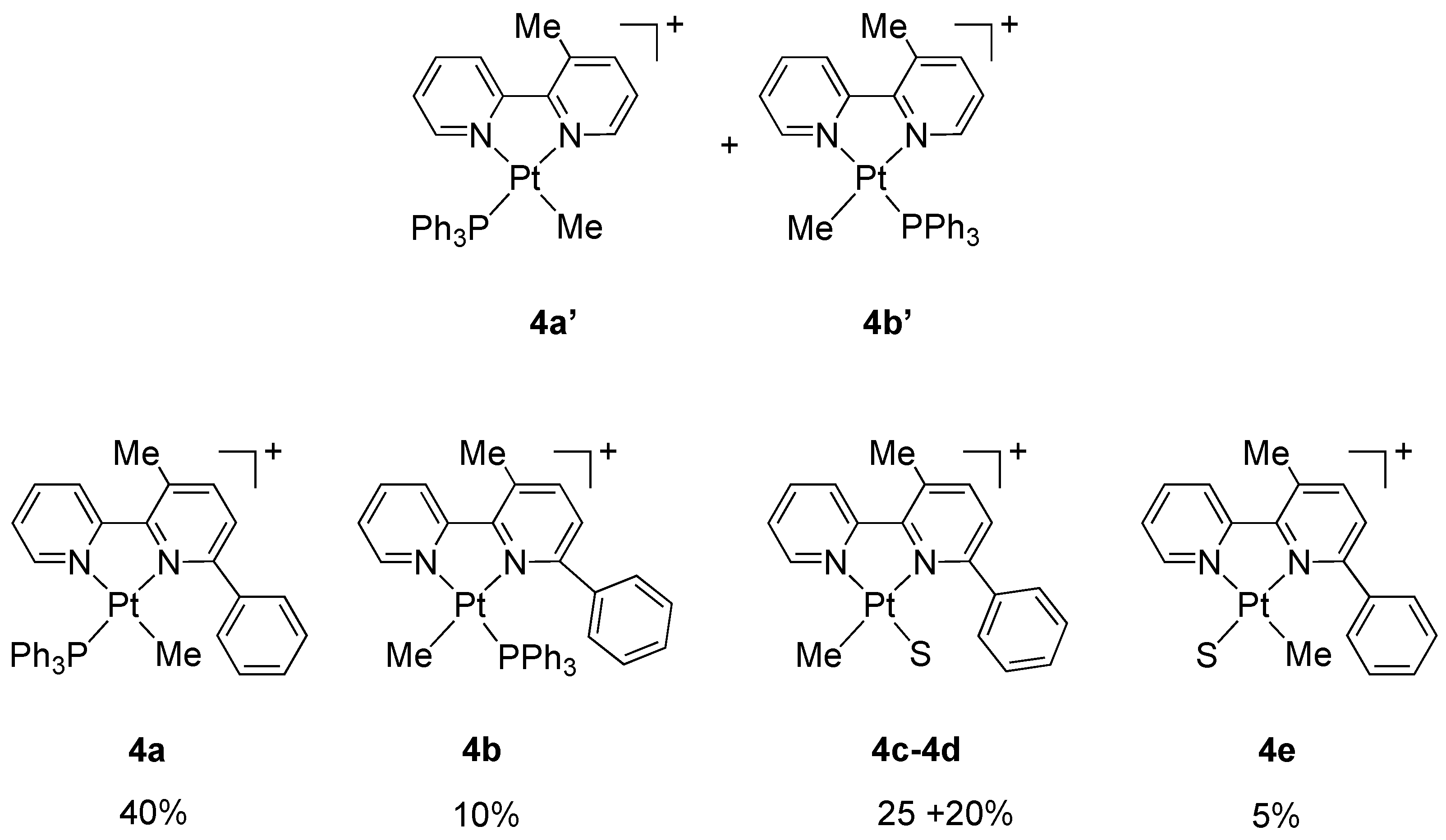
On the basis of our previous experience, we expected to obtain the two geometric isomers of the adduct [Pt(N^N)(PPh3)Me]+, as in the case of unsubstituted 2,2′-bipyridine, where the two species 4a′ and 4b′ were isolated in a 1:1 molar ratio (Figure 4). However, in the case of 6-phenyl-2,2′-bipyridine, likely due to the presence of the bulky phenyl substituent, the situation is more complex. The 1H NMR spectrum of the isolated product showed a mixture of at least five species in an approximate 8:5:4:2:1 molar ratio (Supplementary Material S3). All species contain a coordinated 3-methyl-6-phenyl-2,2′-bipyridine resulting from the desired C(sp3)−C(sp2) coupling, as shown by singlets around 3.0 ppm in a 1:1 molar ratio with the corresponding Pt-CH3 signals.
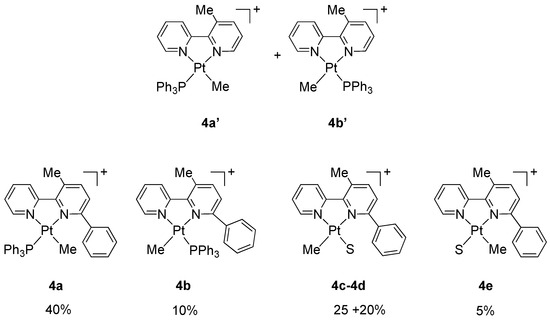
Figure 4.
Adducts 4a′ and 4b′ obtained from 2,2′-bipyridine [23] and suggested formulations for species 4a–e obtained from 6-phenyl-2,2′-bipyridine (this work). S = acetone or water, counter anion BF4−.
A tentative analysis of the complex mixture may explain the signals as follows: two species, present in a relative 4:1 molar ratio (ca 40% and 10% of the total) may be described as the geometric isomers of the adduct [Pt(N^N)(PPh3)Me]+ (N^N = 3-methyl-6-phenyl-2,2′-bipyridine, 4a and 4b). This interpretation is based on the presence of two coordinated methyls appearing as doublets with satellites, due to coupling with 195Pt and 31P nuclei: one of the signals, corresponding to the major species, is highly shielded (4a, ca 40%, δ −0.48 ppm, JP-H = 3.8 Hz, JPt-H = 69.5 Hz) as it is in proximity of the shielding cone of the phenyl substituent of the bipyridine, whereas the other signal (4b, ca 10%, δ 0.86 ppm, JP-H = 3.6 Hz, JPt-H = ca 70 Hz) appears in the usual region at around 0.9–1.1 ppm. Two singlets, at 2.99 and 3.07 ppm, with the same integral values and 4:1 molar ratio, may be assigned to the methyls on the C3 position in the central pyridine ring. In the 31P NMR spectrum, these species gave singlets with satellites at 17.57 ppm (4a, JPt-P = 4581 Hz) and 12.49 ppm (4b, JPt-P = 4397 Hz), with the same 4:1 molar ratio. Other smaller signals in the 31P NMR spectrum, e.g., a singlet at 31.72 ppm, were not interpreted.
In the 1H NMR spectrum, two other compounds, 4c and 4d, in an approximate 5:4 molar ratio, (ca. 25% and 20% of the total, respectively), may tentatively be described as solvato species [Pt(N^N)(S)Me]+ (S = acetone or water) generated from detachment of the PPh3 ligand; these species show two singlets with satellites, indicating the absence of coordinated PPh3 (4c, 25%: δ 1.04 ppm, JPt-H = 71.5 Hz; 4d, 20%: δ 0.96 ppm, JPt-H = 67.5 Hz). Two singlets at 2.63 and 2.88 ppm (4c and 4d respectively, Me on C3) indicates that also these species are derived from the C-C coupling. Furthermore, two signals at 3.80 and 2.19 ppm (the latter overlapping with free acetone) may support the assignment of 4c and 4d as solvato species. Finally, a small singlet with satellites, at −0.17 ppm (ca. 5%, JPt-H = 66.5 Hz), may be a solvato species (4e) with a methyl coordinated close to the phenyl substituent.
It is worth mentioning that C(sp2)-C(sp3) formation by reductive elimination is relatively rare, both catalytically and stoichiometrically [36,37,38,39,40,41,42,43]. In our previous study [23] of 2,2′-bipyridine, 2-pyridylquinoline, and 6-methyl-2,2′-bipyridine, we tried to determine which of the two methyl ligands coordinated to platinum(IV) was involved in the coupling. However, the oxidative addition reaction with deuterated CD3I produced a 50:50 distribution of CH3 and CD3 coordinated in the axial and equatorial positions, explainable by a rapid isomerization of the pentacoordinated Pt(IV) complex [Pt(N^C)(CD3)(CH3)(PPh3)]+ before the coordination of iodide.
Here, our main interest consisted in the isolation of the new 3-methyl-6-phenyl-2,2′-bipyridine, so we did not devote further efforts to the characterization of species 4a–e, and proceeded with the decomplexation of the ligand from the metal.
The free-substituted 2,2′-bipyridine 5 was isolated by reaction of the 4a–e species with dppe (1,2-bis-diphenylphosphinoethane). The chelating diphosphane easily displaces the bipyridine ligand so that the organic species can be isolated in good yield (ca. 80%) and analyzed.
To the best of our knowledge, compound 5 has not previously appeared in the literature and its synthesis is unprecedented. For this reason, we performed a thorough characterization in solution by means of mono- and two-dimensional 1H and 13C NMR spectroscopy (1H, 13C, H-H COSY, H-H NOESY, H-C HMQC, H-C HMBC).
The 1H and 13C NMR spectra clearly show the methyl in position 3 (1H, 2.58 ppm, 13C, 19.98 ppm). The aromatic region shows all the expected hydrogens, with 4, 2, and 5-proton spin systems for the aromatic rings, for which a COSY H-H spectrum allowed the assignment of all 1H NMR signals.
In addition, 1H-13C HSQC and HMBC spectra allowed the assignment of 13C signals. A complete assignment is reported in Figure 5.
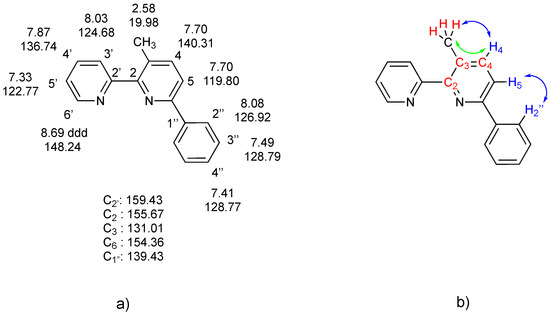
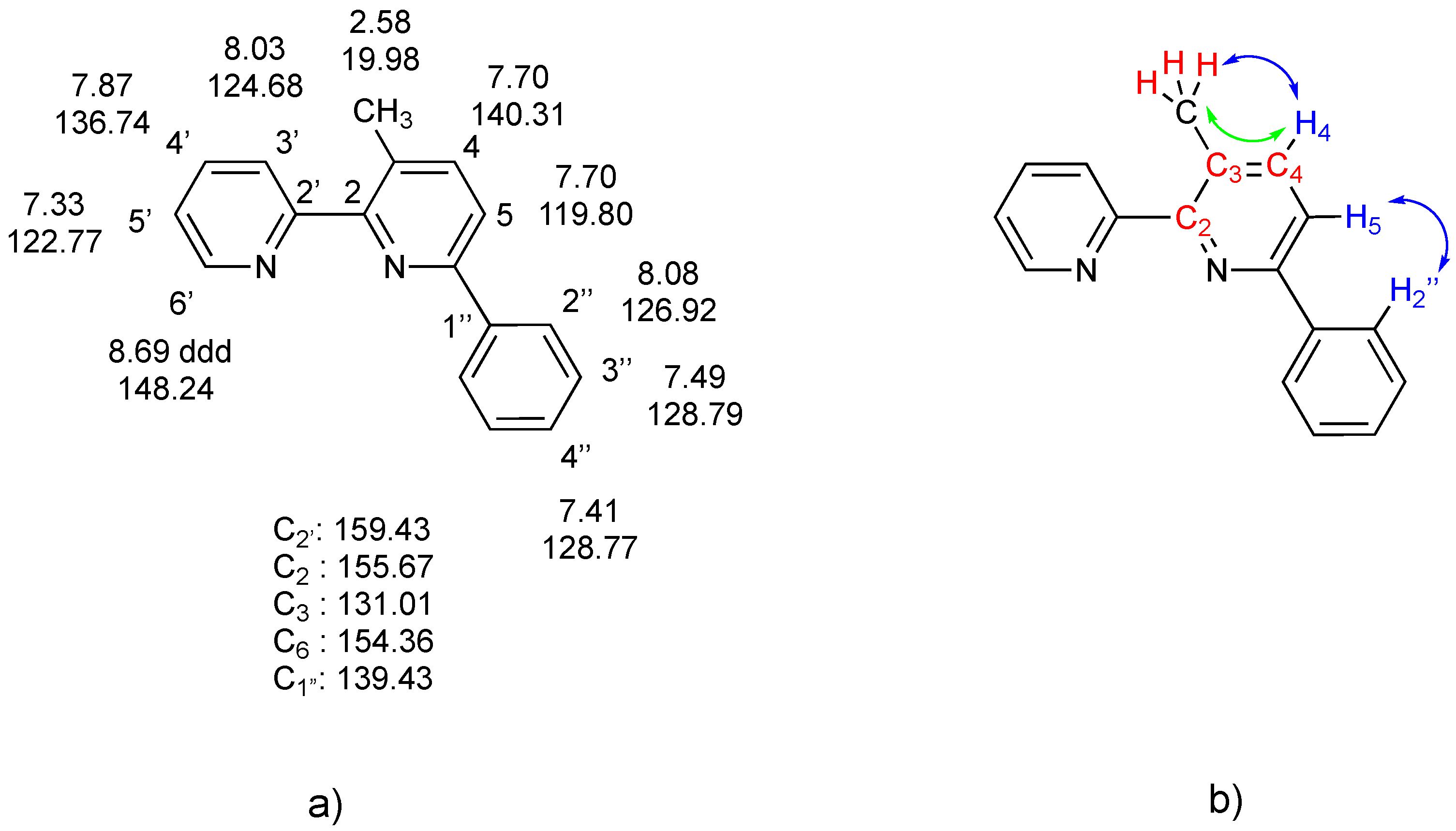
Figure 5.
(a) (left): 3-Methyl-6-phenyl-2,2-bipyridine, 5, along with numbering scheme and complete 1H and 13C NMR assignment (CDCl3 solution) deduced from 2D NMR spectra; (b) (right) interactions revealed by 2D NMR spectra: (i) in red, proximity of CH3 protons with C2, C3, and C4, shown by cross peaks in the HMBC spectrum; (ii) in blue, NOE contacts between CH3 and H2” with the H4/H5 spin system (NOESY spectrum); (iii) in green, long-range interactions of CH3 protons and H5, revealed by the COSY spectrum.
In particular, the 2D 1H-13C HMBC NMR spectrum provided fundamental information, revealing intense cross peaks for 3J(C-H) coupling, as well as weak peaks for 2J(C-H) and 4J(C-H) couplings. As an example, the 1H methyl singlet at 2.58 ppm shows cross peaks with signals at 155.52 (C2), 130.9 (C3), and 140.16 (C4) ppm, supporting the given formulation. The analysis of the other peaks allowed complete assignment. The bonding of the methyl group on the C3 position is confirmed by the 2D COSY spectrum, which shows a weak long-range interaction between the methyl protons at 2.58 ppm and the H4 signal at 7.70 ppm (part of an AB system with the H5 hydrogen).
The 1H NMR spectrum of 5 in CD2Cl2 slightly separated the H4 and H5 signals in the AB system (δ 7.73, 7.75 ppm, JAB = 8.1 Hz). A NOESY spectrum showed NOE contacts between the AB system and the methyl at 2.60 ppm, as well as with the H2” protons; a detailed analysis, hampered by the tightness of the AB system, allowed assignment of the H4 and H5 signals (δ 7.73 and 7.75 ppm, respectively).
The GC-MS spectrum (EI) of 5 shows peaks at m/z 245.2 (100%, [M-H]+) and 246.2 ([M]+, calculated for C17H14N2 246.12).
Functionalization in the C3 position of 2,2′-bipyridines is not so common: the methods reported in literature comprise Negishi [44] and Suzuki–Miyaura coupling [45], in addition to co-cyclotrimerization reactions [46]. In our previous investigation, we reported that only rollover complexes were able to follow the sequence of reactions also reported in this paper. The intermediate adducts [Pt(N^N)(PPh3)Me]+, analogous to 4a–e, were stabilized by chelation, a behavior not available for classical ligands such as 2-phenylpyridine. However, the retro-rollover process should follow the C-C coupling, so we analyzed in more detail the reaction starting from the classical analog [Pt(N^C)Me(PPh3)] complex derived from 2-phenylpyridine, 6 (Scheme 4).
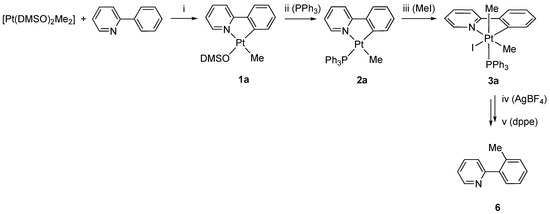
Scheme 4.
Synthesis of 6: (i) toluene, 110 °C, 3 h; (ii) toluene, PPh3, r.t., 1 h; (iii) CH2Cl2, MeI, 30 °C, 6 h; (iv) CH2Cl2/acetone 5:1, AgBF4, 3 h, r.t.; (v) CH2Cl2/acetone 5:1, dppe, 15′.
Following the same reaction sequence used for 1–4, we obtained the Pt(IV) species 3a, which was isolated in the solid state and analyzed by means of NMR spectroscopy.
In this case, we did not try to isolate adduct species analogous to 4a–e, as we were mainly interested in the isolation of the final organic compound. Owing to the different solubility of 3a with respect to 3, the reaction with silver tetrafluoborate was performed in a CH2Cl2/acetone 5/1 molar ratio mixture. Analogous to the previous case, the addition of AgBF4 followed by displacement with dppe, 1,2-bis(diphenylphosphine)ethane, led to the isolation of 2-ortho-tolylpyridine, 6, derived from the C(sp3)-C(sp2) coupling. Compound 6 was purified by acid–base extraction and characterized by NMR spectroscopy. The spectroscopic data, in agreement with the previously reported data for 6, ref. [47] revealed, inter alia, the presence of the methyl group at 2.41 and 20.26 ppm, in the 1H and 13C spectra, respectively.
Furthermore, the GC-MS spectrum (EI) shows peaks at m/z 168.1 (100%, [M-H]+) and 169.1 ([M]+, calculated for C12H11N 169.09).
The obtained result is significant because, to the best of our knowledge, this is a new strategy for C-C coupling in a classical cyclometalated complex, following this sequence of reactions. Moreover, this result suggests the possibility of extending the functionalization to a wide range of cyclometalated ligands.
3. Experimental Section
All the solvents were purified and dried according to standard procedures [48]. Cis-[Pt(DMSO)2(Me)2] was synthesized according to reference [49,50]. Elemental analyses were performed with a PerkinElmer elemental analyzer 240B.1H, 13C{1H}, and 31P{1H} NMR spectra were recorded using a Bruker Avance III 400 spectrometer operating at 400.132, 100.623 and 161.967 MHz, respectively. Chemical shifts are given in ppm relative to internal TMS for 1H and 13C{1H} and external 85% H3PO4 for 31P{1H}; J values are given in Hz. 1H-1H COSY, 1H-1H NOESY, 1H-13C HSQC, and 1H-13C HMBC experiments were performed by means of standard pulse sequences.
GC-MS analyses were carried out using an Agilent GC 7820A equipped with an Agilent MSD 5977E (Column: Zebron ZB-5 60 m × 0.25 mm, i.d. × 0.25 µm).
Preparations
Synthesis of 2, [Pt(bpyPh-H)Me(PPh3)]
Complex 2 was obtained as previously reported [20], following a one-pot procedure, adding PPh3 after completion of the reaction between cis-[Pt(DMSO)2(Me)2] and 6-phenyl-2,2′-bipyridine to give complex 1 as an intermediate. Yield 95%. 1H NMR: (400.1 MHz, CDCl3): δ (ppm) 8.55 (d, 1H, H6′), 8.33 (dd with sat, 3JPt-H = 47.1 Hz), 8.20–7.38 (m, aromatics), 6.71 (m, 1H), 0.79 (d with sat, 3H, 2JPt-H = 83.2 Hz, CH3). 31P NMR: (136.0 MHz, CDCl3): δ (ppm) 32.49 (s with sat, 1JPt-P = 2236 Hz, PPh3).
Synthesis of 3, [Pt(bpyPh-H)I(Me)2PPh3]
To a solution of [Pt(bpyPh-H)Me(PPh3)] (300.0 mg; 0.42 mmol) in 15 mL of dichloromethane, methyl iodide (159.0 μL; 2.52 mmol) was added under stirring. The solution was heated to 30 °C for 6 h, under an argon atmosphere. After completion, the solution was concentrated to a small volume and treated with hexane. The precipitate formed was filtered off and dried to give the analytical sample (331.0 mg; Yield: 93%). Elemental analysis: calculated for C36H32IN2PPt. (%): C, 51.13; H, 3.81; N, 3.31; found: C, 51.26; H, 3.92; N, 3.19. 1H NMR: (400.1 MHz, CDCl3): δ (ppm) 9.65 (dd with sat, 1H, 2JH-H = 5.6 Hz JPt-H = 11.5 Hz, H6′); 8.42 (d, 1H), 8.07 (m, 2H), 7.80 (td, 1H), 7.57–7.05 (m, 21H + CDCl3), 7.06 (dd, 1H), 1.64 (d with sat, 3H, 2JP-H = 7.7 Hz,2JPt-H = 70.45 Hz, CH3). 1.17 (d with sat, 3H, 2JP-H = 7.4 Hz, 2JPt-H = 60.0 Hz, CH3).31P NMR: (136.0 MHz, CDCl3): δ (ppm) −10.9 (s with sat, 1JPt-P = 962.5 Hz, PPh3).
Reaction of 3 with AgBF4
To a solution of 3 (255.3 mg; 0.30 mmol) in acetone (20 mL) AgBF4 (61.4 mg; 0.30 mmol) was added under stirring at room temperature in an argon atmosphere. After three hours, the formed AgI was removed by filtration over celite. The filtered solution was concentrated to a small volume and treated with a hexane–diethyl ether 1:5 mixture to give a precipitate that was filtered off and dried to give the analytical sample.
The 1H NMR spectrum of the obtained product shows the presence of at least five species, 4a (40%), 4b (10%), 4c (25%), 4d (20%), 4e (5%), tentatively identified as [Pt(N^N)L(Me)]+ cationic adducts (L = PPh3, water, or acetone) where the N^N ligand is 3-methyl-6-phenyl-2,2′-bipyridine (5).
Selected 1H and 31P NMR data (CDCl3, δ (ppm))
4a (40%), [Pt(Pt(k2-N,N-3-methyl-6-phenyl-2,2′-bipyridine)(PPh3)(Me)]+ (isomer with Pt-Me cis to Ph-bipy): 1H NMR: δ −0.47 (d with sat, 3H, JP-H = 3.8 Hz, JPt-H = 69.5 Hz, Pt-Me); 2.99 (s, 3H, Me-bipy) 31P NMR: δ 17.57 (s with sat, JPt-P = 4581 Hz).
4b (10%), [Pt(Pt(k2-N,N-3-methyl-6-phenyl-2,2′-bipyridine)(PPh3)(Me)]+ (isomer with Pt-Me trans to Ph-bipy): δ 0.86 (d with sat, 3H, JP-H = 3.6 Hz, JPt-H = ca 70 Hz, Pt-Me); 3.07 (s, 3H, Me-bipy). 31P NMR: δ 12.49 (s with sat, JPt-P = 4397 Hz).
4c (25%), [Pt(Pt(k2-N,N-3-methyl-6-phenyl-2,2′-bipyridine)(S)(Me)]+ (S = water or acetone, isomer with Pt-Me trans to Ph-bipy). 1H NMR: δ 1.04 (s with sat, 3H, JPt-H = 71.5 Hz, Pt-Me); 2.63 (s, 3H, Me-bipy).
4d (20%), [Pt(Pt(k2-N,N-3-methyl-6-phenyl-2,2′-bipyridine)(S)(Me)]+ (S = water or acetone, isomer with Pt-Me trans to Ph-bipy). 1H NMR: δ 0.96 (s with sat, 3H, JPt-H = 67.5 Hz, Pt-Me); 2.88 (s, 3H, Me-bipy).
4e (5%), [Pt(Pt(k2-N,N-3-methyl-6-phenyl-2,2′-bipyridine)(S)(Me)]+ (S = water or acetone, isomer with Pt-Me trans to Ph-bipy), 1H NMR: δ -0.15 (s with sat, JPt-H = 66.5 Hz, Pt-Me); 2.85 (s, 3H, Me-bipy).
Synthesis of 5, 3-methyl-6-phenyl-2,2′-bipyridine
To a solution of 3 (255.3 mg; 0.30 mmol) in acetone (20 mL), AgBF4 (61.4 mg; 0.30 mmol) was added under stirring at room temperature in an argon atmosphere. After three hours, the formed AgI was removed by filtration over celite. To the filtered solution, a 10% excess of dppe (132.1 mg; 0.303 mmol) was added and the solution was left under stirring for 15′. Then the mixture was evaporated to dryness. The organic product was solubilized with diethyl ether, filtered and purified by an acid–base extraction in order to eliminate traces of dppe, anhydrified with Na2SO4, filtered and evaporated to dryness to give the analytical sample with a 75% yield. Complete 1H and 13C NMR assignment was enabled by analysis of 1D and 2D NMR spectra (1H, 13C, H-H COSY, H-H NOESY, H-C HSQC, H-C HMBC).
1H NMR: (400.1 MHz, CDCl3): δ (ppm) 8.69 (ddd, 1H, JH-H = 4.8, 1.7, 0.9 Hz, H6′), 8.08 (m, 1H, H2″), 8.03 (dt, 1H, JH-H = 8.0, 0.9 Hz, H3′), 7.87 (td, 1H, JH-H = 7.8, 1.8 Hz H4′), 7.70 (AB system, 2H, H5+H4, JH5-H4 = 8.1 Hz), 7.49 (m, 2H, H3″), 7.41 (m, 1H, H4″), 7.33 (ddd, 1H, JH-H = 7.7, 4.8, 1.2 Hz, H5′), 2.58 (s, 3H, CH3). 1H NMR (400.1 MHz, CD2Cl2): selected signals δ (ppm) 8.71 (ddd, 1H, H6′), 7.73(H4, 1H, AB system, JAB = 8.1 Hz); 7.75 (H5, 1H, AB system, JAB = 8.1 Hz); 2.60 (s, 3H, CH3).
13C NMR: (100.1 MHz, CDCl3) δ (ppm) 159.43 (C2); 155.67 (C2′); 154.36 (C6); 148.24 (C6′); 140.31 (C4); 139.43 (C1″); 136.74 (C4′); 131.01 (C3); 128.79 (C3″); 128.77 (C4″); 126.92 (C2″); 124.68 (C3′); 122.77 (C5′); 119.80 (C5); 19.98 (CH3).
GC-MS (EI): C17H14N2, found m/z 245.2 [M-H]+ (calculated for M+ 246.1).
Synthesis of 2a, [Pt(pyPh-H)Me(PPh3)]
To a solution of cis-[Pt(DMSO)2(Me)2] (210.0 mg; 0.55 mmol) in 15 mL of anhydrous toluene, 0.10 mL of 2-phenylpyridine (0.70 mmol) was added under constant stirring. The solution was heated, under an argon atmosphere, to a temperature of 110 °C for 3 h. After cooling to room temperature, a 10% excess of triphenylphosphine was added and allowed to react for one hour. The obtained solution was concentrated to a small volume and treated with diethyl ether. The precipitate formed was filtered off and washed with hexane to produce the analytical sample (0.174 g; yield: 50%). 1H and 31P NMR data match those previously reported [51].
Selected 1H NMR data: (400.1 MHz, CDCl3): δ (ppm) 077 (d with sat, 3H, 3JP-H = 7.9 Hz, 2JPt-H = 83.5 Hz, Pt-Me). 8.00 (m with sat, 1H, JPt-H = 51 Hz, H3′), 31P NMR (136.0 MHz, CDCl3): δ (ppm) 33.10 (s with sat, JPt-P = 2105 Hz).
Synthesis of 3a, [Pt(pyPh-H)I(Me)2(PPh3)]
To a solution of [Pt(pyPh-H)Me(PPh3)] (100.0 mg; 0.16 mmol) in 15 mL of dichloromethane, an excess of CH3I (59.6 μL; 0.96 mmol) was added, under constant stirring in an argon atmosphere. The solution was heated to 30 °C for 6 h. After completion, the solution was concentrated to small volume and treated with hexane. The precipitated formed was filtered off and dried to give the analytical sample (0.083 g; yield: 68%). 1H and 31P NMR data match those previously reported. Ref. [33] Selected NMR data (CDCl3): δ(ppm) 1.25 (d, 3H, 2JPt-H = 60.8 Hz, 3JP-H = 7.5 Hz, Me trans to P), 1.65 (d, 3H, 2JPtH = 71.1 Hz, 3JP-H = 7.7 Hz, Me trans to N), 9.72 (ddd, 1H, JH-H) 5.5 Hz, 3JPt-H = 10.5 Hz, H6′, adjacent to N). 31P NMR (136.0 MHz, CDCl3): −9.34 (s, 1JPt-P = 967 Hz).
Synthesis of 6, 2(o-tolyl)pyridine
To a solution of [Pt(pyPh-H)I(Me)2(PPh3)] (60.0 mg; 0.080 mmol) in a 5:1 mixture of dichloromethane acetone, AgBF4 (17.2 mg, 0.088 mmol) was added under constant stirring. The solution was stirred at room temperature, under an argon atmosphere, for three hours. At the end, the resulting suspension was filtered to remove the formed AgI and a 10% excess of 10% dppe (35.06 mg; 0.088 mmol) is added. The solution was stirred at room temperature for 15′, then evaporated to dryness, solubilized with diethyl ether, and filtered. The filtered solution was evaporated to dryness and purified by means of an acid–base extraction, followed by anhydrification with Na2SO4, filtration, and evaporation to dryness, to produce the analytical sample (0.015 g; yield: 94%). Selected NMR data. 1H NMR: (400.1 MHz, CDCl3): δ (ppm) 8.72 (dd, 1H, H6′), 7.81 (td, 1H, H4′), 2.41 (s, 3H, CH3).13C NMR: (100.1 MHz, CDCl3) δ (ppm) 159.83 (Cq); 148.91; 139.98 (Cq); 136.78, 135.92, 130.93, 129.80, 128.63, 126.07, 124.47, 121.93, 20.42 (CH3). NMR data match those previously reported (ref. [47]).
GC-MS (EI): C12H11N, found m/z 168.1 [M-H]+ (calculated for M+ 169.1). Traces (<1%) of triphenylphosphine oxide (m/z 278.1).
4. Conclusions
The coupling reaction reported here constitutes a new synthetic method for the preparation of methyl-substituted heterocycles: in principle, a large array of heterocyclic donors, able to support classical or rollover cyclometalation may follow the sequence of reactions reported here, taking advantage of a rollover/retro-rollover reaction pathway.
The reductive elimination step involves a rather rare C(sp3)-C(sp2) coupling in place of the more common C(sp3)-C(sp3) one. The reaction has now been demonstrated not to be restricted to rollover complexes but may have a more general application.
The next steps in this research will be the extension of the reaction to other cyclometalated heterocyclic donors and to other organic halides in order to add different groups in the C-C coupling. It will also be useful for revealing the factors governing the process, such as the electronic and steric influence of the ligands coordinated to the platinum center.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules29030707/s1. NMR spectra of compounds 2, 3, 4, 5, 2a, 3a, 4a and 6: S1–S13; GC-MS spectra of 5 and 6: S14 and S15.
Author Contributions
Conceptualization, A.Z.; Methodology, A.Z. and S.S.; Formal analysis, A.Z., S.S., G.S. and L.M.; Investigation, A.Z., L.M. and G.S.; Resources, A.Z. and S.S.; Data curation, A.Z. and L.M.; Writing—original draft, A.Z.; Writing—review & editing, A.Z., S.S., L.M. and G.S.; Supervision, A.Z.; Project administration, A.Z.; Funding acquisition, A.Z. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Università degli Studi di Sassari, “Fondo di Ateneo per la Ricerca 2020”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available from the corresponding author on request.
Acknowledgments
A.Z. and S.S. gratefully acknowledge the Università degli Studi di Sassari for financial support (“Fondo di Ateneo per la Ricerca 2020”). Maurizio Solinas and Daniela Giunta of Institute of Biomolecular Chemistry of C.N.R. (Research site of Sassari) are gratefully acknowledged for the GC-MS spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Liu, B.; Romine, A.M.; Rubel, C.Z.; Engle, K.M.; Shi, B.F. Transition-Metal-Catalyzed, Coordination-Assisted Functionalization of Nonactivated C(sp3)-H Bonds. Chem. Rev. 2021, 121, 14957–15074. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 7th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2019; ISBN 978-1-119-46588-1. [Google Scholar]
- Albrecht, M. Cyclometalation using d-block transition metals: Fundamental aspects and recent trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, C.S.; Illam, P.M.; Donthireddy, S.N.R.; Rit, A. Recent Advances in the Syntheses and Catalytic Applications of Homonuclear Ru-, Rh-, and Ir-Complexes of CNHC^C Cyclometalated Ligands. Chem. Eur. J. 2021, 27, 16581–16600. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, S.; Kuhn, F.E.; Casini, A. Cyclometalated complexes of platinum and gold with biological properties: State-of-the-art and future perspectives. Curr. Med. Chem. 2018, 25, 437–461. [Google Scholar] [CrossRef]
- Chakraborty, P.; Oosterhuis, D.; Bonsignore, R.; Casini, A.; Olinga, P.; Scheffers, D.J. An Organogold Compound as Potential Antimicrobial Agent against Drug-Resistant Bacteria: Initial Mechanistic Insights. ChemMedChem 2021, 16, 3060–3070. [Google Scholar] [CrossRef]
- Yoshida, M.; Kato, M. Cation-controlled luminescence behavior of anionic cyclometalated platinum(II) complexes. Coord. Chem. Rev. 2020, 408, 213194. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Lin, S.; Lou, J.; Zhang, Q. Recent development and application of cyclometalated iridium (iii) complexes as chemical and biological probes. Dalton Trans. 2021, 50, 6410–6417. [Google Scholar] [CrossRef]
- Lyons, T.W.; Sanford, M.S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 2010, 110, 1147–1169. [Google Scholar] [CrossRef]
- Dikova, Y.M.; Yufit, D.S.; Williams, J.A.G. Platinum (IV) Complexes with Tridentate, NNC-Coordinating Ligands: Synthesis, Structures, and Luminescence. Inorg. Chem. 2023, 62, 1306–1322. [Google Scholar] [CrossRef]
- Albrecht, M.; van Koten, G. Platinum Group Organometallics Based on ‘’Pincer’’ Complexes: Sensors, Switches, and Catalysts. Angew. Chem. Int. Ed. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Zucca, A.; Pilo, M.I. Rollover cyclometalation as a valuable tool for regioselective C-H bond activation and functionalization. Molecules 2021, 26, 328. [Google Scholar] [CrossRef] [PubMed]
- Crosby, S.H.; Clarkson, G.J.; Rourke, J.P. Reactions of a Platinum(II) Agostic Complex: Decyclometalation, Dicyclometalation, and Solvent-Switchable Formation of a Rollover Complex. Organometallics 2011, 30, 3603–3609. [Google Scholar] [CrossRef]
- Zucca, A.; Cordeschi, D.; Stoccoro, S.; Cinellu, M.A.; Minghetti, G.; Chelucci, G.; Manassero, M. Platinum(II)-cyclometalated “roll-over” complexes with a chiral pinene-derived 2,2′-bipyridine. Organometallics 2011, 30, 3064–3074. [Google Scholar] [CrossRef]
- Maidich, L.; Zuri, G.; Stoccoro, S.; Cinellu, M.A.; Masia, M.; Zucca, A. Mesoionic complexes of platinum(II) derived from “rollover” cyclometalation: A delicate balance between Pt-C(sp3) and Pt-C(sp2) bond cleavage as a result of different reaction conditions. Organometallics 2013, 32, 438–448. [Google Scholar] [CrossRef]
- Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. “Roll-over” Cyclometalation of 2,2′-Bipyridine Platinum(II) Complexes in the Gas Phase: A Combined Experimental and Computational Study. Chem. Eur. J. 2008, 14, 11050–11060. [Google Scholar] [CrossRef] [PubMed]
- Skapski, A.C.; Sutcliffe, V.F.; Young, G.B. Roll-over’ 3-Metallation of Co-ordinated 2,2′-Bipyridyl in the Thermal Rearrangement of Diaryl(bipyridyl)platinum(II) Complexes: Molecular- Structure of (m-bidyl)[PtPh(Butpy)]2. J. Chem. Soc. Chem. Commun. 1985, 9, 609–611. [Google Scholar] [CrossRef]
- Butschke, B.; Schwarz, H. On the activation of chloromethanes by cyclometalated [Pt(bipy—H)]+ in the gas phase: A mechanistic study. Int. J. Mass Spectrom. 2011, 306, 108–113. [Google Scholar] [CrossRef]
- Butschke, B.; Schlangen, M.; Schröder, D.; Schwarz, H. Platinum (II)-mediated dehydrosulfurization and oxidative carbon—Carbon coupling in the gas-phase decomposition of thioethers. Int. J. Mass Spectrom. 2009, 283, 3–8. [Google Scholar] [CrossRef]
- Kwak, J.; Ohk, Y.; Jung, Y.; Chang, S. Rollover Cyclometalation Pathway in Rhodium Catalysis: Dramatic NHC Effects in the C–H Bond Functionalization. J. Am. Chem. Soc. 2012, 134, 17778–17788. [Google Scholar] [CrossRef]
- Shibata, T.; Takayasu, S.; Yuzawa, S.; Otani, T. Rh(III)-Catalyzed C–H Bond Activation along with “Rollover” for the Synthesis of 4-Azafluorenes. Org. Lett. 2012, 14, 5106–5109. [Google Scholar] [CrossRef]
- Taghizadeh Ghoochany, L.; Kerner, C.; Farsadpour, S.; Menges, F.; Sun, Y.; Niedner-Schatteburg, G.; Thiel, W.R. C–H Activation at a Ruthenium(II) Complex—The Key Step for a Base-Free Catalytic Transfer Hydrogenation? Eur. J. Inorg. Chem. 2013, 24, 4305–4317. [Google Scholar] [CrossRef]
- Zucca, A.; Maidich, L.; Canu, L.; Petretto, G.L.; Stoccoro, S.; Cinellu, M.A.; Clarkson, G.J.; Rourke, J.P. Rollover-assisted C(sp2)-C(sp3) bond formation. Chemistry. A Eur. J. 2014, 20, 5501–5510. [Google Scholar] [CrossRef]
- Maidich, L.; Dettori, G.; Stoccoro, S.; Cinellu, M.A.; Rourke, J.P.; Zucca, A. Electronic and Steric Effects in Rollover C–H Bond Activation. Organometallics 2015, 34, 817–828. [Google Scholar] [CrossRef]
- Minghetti, G.; Stoccoro, S.; Cinellu, M.A.; Soro, B.; Zucca, A. Activation of a C-H Bond in a Pyridine Ring. Reaction of 6-Substituted 2,2′-Bipyridines with Methyl and Phenyl Platinum(II) Derivatives: N,C(3)-“Rollover” Cyclometalation. Organometallics 2003, 22, 4770–4777. [Google Scholar] [CrossRef]
- Anderson, C.M.; Crespo, M.; Jennings, M.C.; Lough, A.J.; Ferguson, G.; Puddephatt, R.J. Competition between Intramolecular Oxidative Addition and Ortho Metalation in Organoplatinum(II) Compounds: Activation of Aryl-Halogen Bonds. Organometallics 1991, 10, 2672–2679. [Google Scholar] [CrossRef]
- Nabavizadeh, S.M.; Habibzadeh, S.; Rashidi, M.; Puddephatt, R.J. Oxidative Addition of Ethyl Iodide to a Dimethylplatinum(II) Complex: Unusually Large Kinetic Isotope Effects and Their Transition-State Implications. Organometallics 2010, 29, 6359–6368. [Google Scholar] [CrossRef]
- Habibzadeh, S.; Rashidi, M.; Masoud Nabavizadeh, S.; Mahmoodi, L.; Hosseini, F.N.; Puddephatt, R.J. Steric and Solvent Effects on the Secondary Kinetic α-Deuterium Isotope Effects in the Reaction of Methyl Iodide with Organoplatinum(II) Complexes: Application of a Second-Order Technique in Measuring the Rates of Rapid Processes. Organometallics 2010, 29, 82–88. [Google Scholar] [CrossRef]
- Rendina, L.M.; Puddephatt, R.J. Oxidative Addition Reactions of Organoplatinum(II) Complexes with Nitrogen-Donor Ligands. Chem. Rev. 1997, 97, 1735–1754. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.J.; Nasrabadi, H.; Nabavizadeh, S.M.; Rashidi, M.; Puddephatt, R.J. Reactivity and Mechanism in the Oxidative Addition of Allylic Halides to a Dimethylplatinum(II) Complex. Organometallics 2012, 31, 2357–2366. [Google Scholar] [CrossRef]
- Nabavizadeh, S.M.; Amini, H.; Jame, F.; Khosraviolya, S.; Shahsavari, H.R.; Hosseini, F.N.; Rashidi, M. Oxidative Addition of MeI to Some Cyclometalated Organoplatinum(II) Complexes: Kinetics and Mechanism. J. Organomet. Chem. 2012, 698, 53–61. [Google Scholar] [CrossRef]
- Maidich, L.; Pilo, M.I.; Rourke, J.P.; Clarkson, G.J.; Canu, P.; Stoccoro, S.; Zucca, A. Classical vs. non-classical cyclometalated Pt(II) complexes. Molecules 2022, 27, 7249. [Google Scholar] [CrossRef]
- Maidich, L.; Zucca, A.; Clarkson, G.J.; Rourke, J.P. Oxidative Addition of MeI to a Rollover Complex of Platinum(II): Isolation of the Kinetic Product. Organometallics 2013, 32, 3371–3375. [Google Scholar] [CrossRef]
- Byers, P.K.; Canty, A.J. Synthetic routes to methylpalladium(II) and dimethylpalladium(Ii) chemistry and the synthesis of new nitrogen donor ligand systems. Organometallics 1990, 9, 210–220. [Google Scholar] [CrossRef]
- Kaupp, M.; Malkina, O.L.; Malkin, V.G.; Pyykkö, P. How do spin-orbit-induced heavy-atom effects on nmr chemical shifts function? Validation of a simple analogy to spin-spin coupling by density functional theory (DFT) calculations on some iodo compounds. Chem. Eur. J. 1998, 4, 118–126. [Google Scholar] [CrossRef]
- Shaw, P.A.; Rourke, J.P. Selective C–C Coupling at a Pt(IV) Centre: 100% Preference for sp2–sp3 over sp3–sp3. Dalton. Trans. 2017, 46, 4768–4776. [Google Scholar] [CrossRef]
- Madison, B.L.; Thyme, S.B.; Keene, S.; Williams, B.S. Mechanistic Study of Competitive sp3-sp3 and sp2-sp3 Carbon−Carbon Reductive Elimination from a Platinum (IV) Center and the Isolation of a C−C Agostic Complex. J. Am. Chem. Soc. 2007, 129, 9538–9539. [Google Scholar] [CrossRef] [PubMed]
- Crespo, M.; Anderson, C.M.; Kfoury, N.; Font-Bardia, M.; Calvet, T. Reductive Elimination from Cyclometalated Platinum(IV) Complexes To Form Csp2–Csp3 Bonds and Subsequent Competition between Csp2–H and Csp3–H Bond Activation. Organometallics 2012, 31, 4401–4404. [Google Scholar] [CrossRef]
- Anderson, C.M.; Crespo, M.; Kfoury, N.; Weinstein, M.A.; Tanski, J.M. Regioselective C–H Activation Preceded by Csp2–Csp3 Reductive Elimination from Cyclometalated Platinum(IV) Complexes. Organometallics 2013, 32, 4199–4207. [Google Scholar] [CrossRef]
- Wang, T.; Alfonso, B.J.; Love, J.A. Platinum(II)-Catalyzed Cross-Coupling of Polyfluoroaryl Imines. Org. Lett. 2007, 9, 5629–5631. [Google Scholar] [CrossRef]
- Wang, T.; Love, J. Insight into the mechanism of platinum-catalyzed cross-coupling of polyfluoroaryl imines. Organometallics 2008, 27, 3290–3296. [Google Scholar] [CrossRef]
- Buckley, H.L.; Sun, A.D.; Love, J.A. User-Friendly Precatalyst for the Methylation of Polyfluoroaryl Imines. Organometallics 2009, 28, 6622–6624. [Google Scholar] [CrossRef]
- Wang, T.; Keyes, L.; Patrick, B.O.; Love, J. Exploration of the Mechanism of Platinum(II)-Catalyzed C–F Activation: Characterization and Reactivity of Platinum(IV) Fluoroaryl Complexes Relevant to Catalysis. Organometallics 2012, 31, 1397–1407. [Google Scholar] [CrossRef]
- Fang, Y.-Q.; Hanan, G.S. Rapid and Efficient Synthesis of Functionalized Bipyridines. Synlett 2003, 6, 852–854. [Google Scholar] [CrossRef]
- Guetz, C.; Luetzen, A. Synthesis of 2, 2′-Bipyridines via Suzuki-Miyaura Cross-Coupling. Synthesis 2010, 1, 85–90. [Google Scholar]
- Sankio Chemical Co Ltd. Process for the Production of 2-Pyridylpyridine Derivatives. Current Patent Assignee: FUJIFILM HOLDINGS CORP. Patent US6504032 B1, 7 January 2003. [Google Scholar]
- Hogg, A.; Wheatley, M.; Domingo-Legarda, P.; Carral-Menoyo, A.; Cottam, N.; Larrosa, I. Ruthenium-Catalyzed Monoselective C−H Methylation and d3-Methylation of Arenes. J. Am. Chem. Soc. Au 2022, 2, 2529–2538. [Google Scholar] [CrossRef]
- Longman Scientific and Technical. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Scientific & Technical, Longman Group: Harlow, UK, 1989. [Google Scholar]
- Eaborn, C.; Kundu, K.; Pidcock, A.J. Synthesis of platinum(II) alkyl and aryl complexes from K2[PtCl4] and tetraorganotin compounds in dimethyl sulphoxide. Chem. Soc. Dalton Trans. 1981, 4, 933–938. [Google Scholar] [CrossRef]
- Baquero, E.A.; Rodríguez-Zúñiga, A.; Flores, J.C.; Temprado, M.; de Jesús, E. Revisiting the synthesis of trans-Pt(dmso)2ClMe and cis-Pt(dmso)2Me2: Experimental and DFT studies. J. Organomet. Chem. 2019, 896, 108–112. [Google Scholar] [CrossRef]
- Jamali, S.; Nabavizadeh, S.M.; Rashidi, M. Binuclear Cyclometalated Organoplatinum Complexes Containing 1,1′-Bis(diphenylphosphino)ferrocene as Spacer Ligand: Kinetics and Mechanism of MeI Oxidative Addition. Inorg. Chem. 2008, 47, 5441–5452. [Google Scholar] [CrossRef] [PubMed]
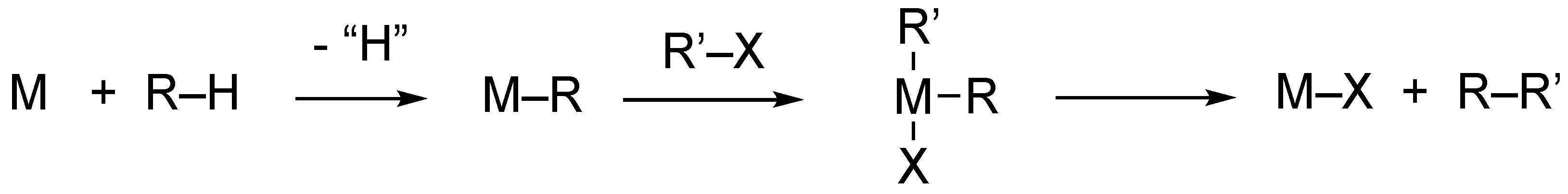
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).