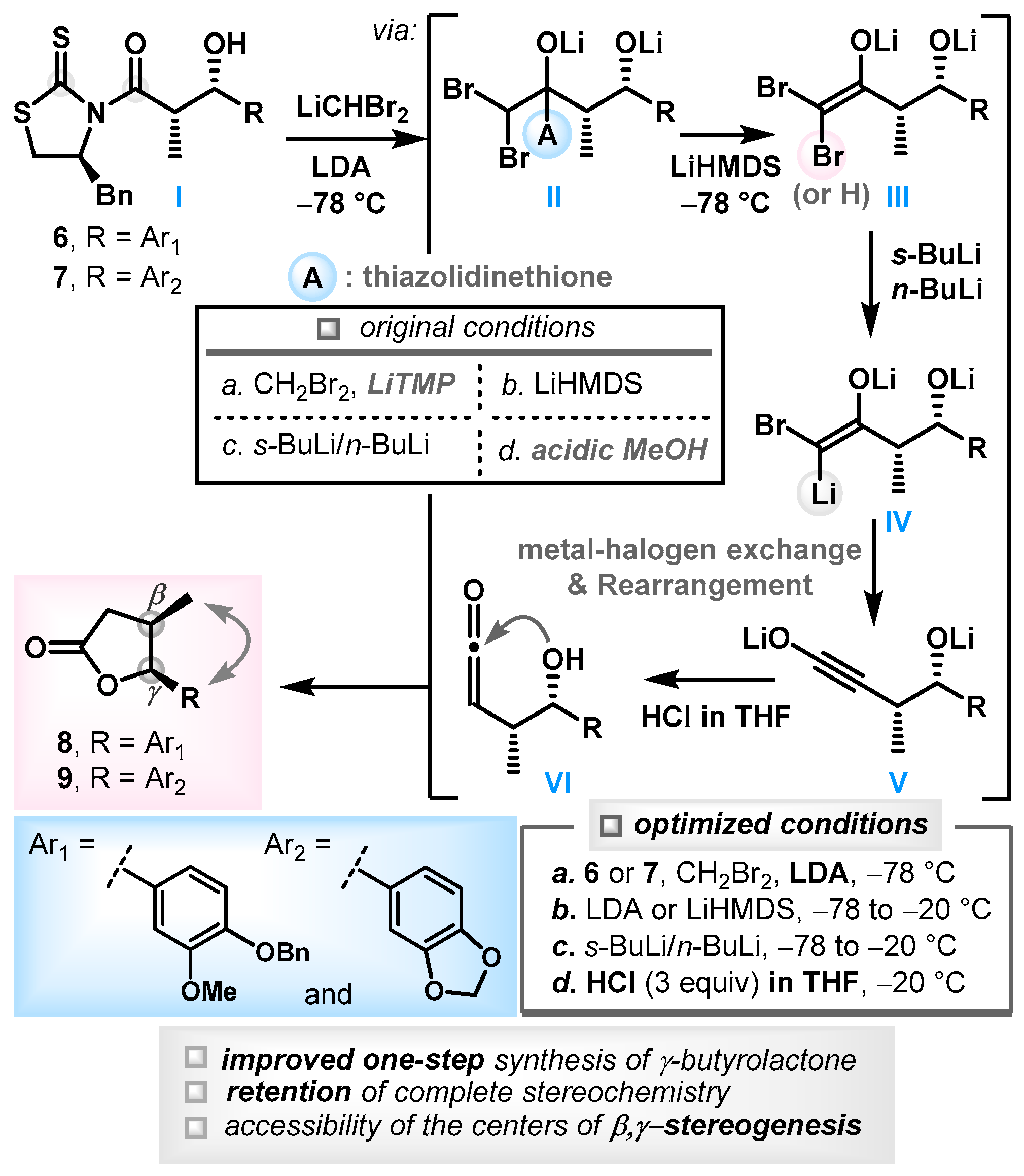
3.2. Synthesis of (-)-Chicanine (1)
(2S,3S)-1-((S)-4-Benzyl-2-thioxothiazolidin-3-yl)-3-(4-(benzyloxy)-3-methoxyphenyl)-3-hydroxy-2-methylpropan-1-one (6): To a cooled (0 °C) solution of (S)-1-(4-benzyl-2-thioxothiazolidin-3-yl)propan-1-one, 10, (670 mg, 2.52 mmol) in CH2Cl2 (13 mL, 0.2 M), titanium(IV) chloride (2.8 mL, 1.0 M in CH2Cl2, 2.8 mmol, 1.1 equiv.) was added. After being stirred for 15 min at the same temperature, i-Pr2NEt (0.53 mL, 3.0 mmol, 1.2 equiv.) was added dropwise, and the reaction mixture was stirred for 40 min at 0 °C. NMP (0.49 mL, 5.1 mmol, 2 equiv.) was added, and the reaction mixture was stirred for an additional 10 min. 4-Benzyloxy-3-methoxybenzaldehyde (1.22 g, 5.04 mmol, 2 equiv.) in CH2Cl2 (10 mL) was added to the enolate. After being stirred for 30 min, the reaction mixture was quenched with saturated aqueous NH4Cl (20 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (20 mL × 3). The combined organic layers were washed with brine (40 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 25% EtOAc/hexane) to provide 6 (1.24 g, 97%) as a yellow foam: [α]D25 +138 (c 1.27, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.31–7.37 (m, 4H); 7.26–7.30 (m, 1H); 7.21–7.25 (m, 2H); 7.16–7.20 (m, 3H); 6.96 (d, J = 1.7 Hz, 1H); 6.77 (d, J = 8.2 Hz, 1H); 6.71 (dd, J = 8.2, 1.8 Hz, 1H); 5.10 (ABX, 2H, J = 12.7 Hz, Δν = 11.9 Hz); 4.82 (p, J = 6.6 Hz, 1H); 4.66 (d, J = 7.8 Hz, 1H); 4.47 (ddd, J = 10.5, 6.5, 3.9 Hz, 1H); 3.88 (s, 3H); 3.10 (dd, J = 13.2, 3.6 Hz, 1H); 2.86 (dd, J = 13.1, 10.8 Hz, 1H); 2.63 (br s, 1H); 2.47 (d, J = 11.3 Hz, 1H); 2.40 (dd, J = 11.3, 6.8 Hz, 1H); 1.39 (d, J = 6.6 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 201.3, 176.8, 149.5, 147.4, 136.7, 136.2, 134.8, 129.2, 128.7, 128.3, 127.8, 127.1, 127.0, 118.5, 113.2, 109.3, 76.8, 70.5, 68.9, 55.8, 46.6, 36.4, 32.1, and 13.0; HRMS (Q–TOF) m/z: 506.1460 ((M–H)+, C28H28NO4S2 requires 506.1460).
(4R,5S)-5-(4-(Benzyloxy)-3-methoxyphenyl)-4-methyldihydrofuran-2(3H)-one (8): To a cooled (−78 °C) solution of i-Pr2NH (1.1 mL, 7.8 mmol, 4.4 equiv.) in THF (4.5 mL, 0.4 M), n-BuLi (4.5 mL, 1.6 M in hexane, 7.1 mmol, 4 equiv.) was added, and the reaction mixture was stirred for 15 min at 0 °C before it was cooled to −78 °C. CH2Br2 (7.8 mL, 1.0 M in THF, 7.8 mmol, 4.4 equiv.) and 6 (904 mg, 1.78 mmol) in THF (7.8 mL) were added dropwise to the above LDA solution. After being stirred for 10 min at −78 °C, the reaction mixture was added to LiHMDS (7.1 mL, 1.0 M in THF, 7.1 mmol, 4 equiv.). After being stirred for 5 min at −78 °C, the reaction mixture was stirred for 90 s at −20 °C before it was cooled to −78 °C. s-BuLi (5.1 mL, 1.4 M in cyclohexane, 7.1 mmol, 4 equiv.) was added dropwise, and the reaction mixture was stirred for 90 s at −20 °C and cooled to −78 °C, followed by the dropwise addition of n-BuLi (4.5 mL, 1.6 M in hexane, 7.1 mmol, 4 equiv.). After being stirred for 5 min, the reaction mixture was stirred for 90 s at −20 °C, warmed to 25 °C in a water bath, and stirred for 30 min before it was added to a solution of HCl (0.44 mL, 5.3 mmol, 3 equiv.) in THF (59 mL, 0.03 M) at −20 °C. After being stirred for 5 min at −20 °C, the reaction mixture was quenched with the addition of saturated aqueous NaHCO3 (30 mL) and diluted with EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (30 mL × 3). The combined organic layers were washed with brine (60 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 33% EtOAc/hexane) to provide 8 (506 mg, 91%) as a colorless oil: [α]D25 −28.0 (c 1.28, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.42–7.45 (m, 2H); 7.34–7.38 (m, 2H); 7.28–7.32 (m, 1H); 6.88 (d, J = 8.3 Hz, 1H); 6.77 (d, J = 2.0 Hz, 1H); 6.68–6.71 (m, 1H); 5.53 (d, J = 5.7 Hz, 1H); 5.15 (s, 2H); 3.88 (s, 3H); 2.79–2.85 (m, 2H); 2.31–2.37 (m, 1H); 0.70 (d, J = 7.0 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 176.8, 149.6, 147.8, 136.9, 129.1, 128.5, 127.8, 127.2, 117.6, 113.8, 109.1, 83.9, 71.0, 56.1, 37.1, 35.1, and 15.1; HRMS (Q–TOF) m/z: 335.1260 ((M + Na)+, C19H20NaO4 requires 335.1259).
(4R,5S)-5-(4-(Benzyloxy)-3-methoxyphenyl)-4-methyl-3-methylenedihydrofuran-2(3H)-one (11): To a cooled (−78 °C) solution of 8 (229 mg, 0.733 mmol) in THF (3.7 mL, 0.2 M), LiHMDS (1.5 mL, 1.0 M in THF, 1.5 mmol, 2 equiv.) was added. After being stirred for 1 h, the reaction mixture was added to Eschenmoser’s salt (407 mg, 2.20 mmol, 3 equiv.) and stirred for 10 min at −78 °C before it was quenched with the addition of saturated aqueous NH4Cl (5 mL) and diluted with Et2O (10 mL). The layers were separated, and the aqueous layer was extracted with Et2O (10 mL × 3). The combined organic layers were washed with brine (20 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was dissolved in CH2Cl2 (9.2 mL, 0.08 M) and saturated NaHCO3 (4.6 mL, 0.16 M). m-CPBA (190 mg, 1.10 mmol, 1.5 equiv.) was added to the above solution. After being stirred for 5 min at 0 °C, the reaction mixture was diluted with saturated aqueous NaHCO3 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (15 mL × 3). The combined organic layers were washed with brine (20 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 13% EtOAc/hexane) to provide 11 (195 mg, 82%) as a colorless oil: [α]D25 +30.0 (c 1.73, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.41−7.44 (m, 2H); 7.34−7.38 -(m, 2H); 7.28−7.32 (m, 1H); 6.86 (d, J = 8.2 Hz, 1H); 6.67 (d, J = 2.0 Hz, 1H); 6.64 (dd, J = 8.3, 1.9 Hz, 1H); 6.32 (d, J = 2.8 Hz, 1H); 5.57 (d, J = 2.6 Hz, 1H); 5.55 (d, J = 8.0 Hz, 1H); 5.14 (s, 2H); 3.86 (s, 3H); 3.35−3.43 (m, 1H); 0.81 (d, J = 7.1 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 170.6, 149.6, 148.1, 140.2, 136.8, 129.3, 128.5, 127.9, 127.3, 121.6, 118.5, 113.7, 109.5, 82.1, 71.0, 56.0, 39.0, and 15.3; HRMS (Q–TOF) m/z: 325.1444 ((M + H)+, C20H21O4 requires 325.1440).
(3S,4R,5S)-5-(4-(Benzyloxy)-3-methoxyphenyl)-3,4-dimethyldihydrofuran-2(3H)-one (12): To a solution of 11 (92.0 mg, 0.284 mmol) in toluene (2.8 mL, 0.1 M), RhCl(PPh3)3 (79 mg, 0.085 mmol, 30 mol%) was added, and the reaction mixture was hydrogenated at 1 atm. After being stirred for 1 h at 25 °C, the reaction mixture was concentrated in vacuo and purified by column chromatography (SiO2, 13% EtOAc/hexane) to provide 12 (83.1 mg, 90%) as a colorless oil: [α]D20 −50.0 (c 0.14, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.43 (d, J = 7.3 Hz, 2H); 7.34–7.38 (m, 2H); 7.27–7.31 (m, 1H); 6.88 (d, J = 8.3 Hz, 1H); 6.82 (d, J = 1.8 Hz, 1H); 6.72 (dd, J = 8.3, 1.5 Hz, 1H); 5.45 (d, J = 5.0 Hz, 1H); 5.14 (s, 2H); 3.88 (s, 3H); 2.94–3.01 (m, 1H); 2.68–2.76 (m, 1H); 1.21 (d, J = 7.2 Hz, 3H); 0.55 (d, J = 7.3 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 179.0, 149.6, 147.6, 136.9, 129.2, 128.5, 127.8, 127.2, 117.4, 113.8, 108.9, 82.1, 71.0, 56.0, 41.0, 40.0, 10.0, and 9.4; HRMS (Q–TOF) m/z: 327.1609 ((M + H)+, C20H23O4 requires 327.1596).
(2S,3R,4S)-2-(4-(Benzyloxy)-3-methoxyphenyl)-3,4-dimethyl-5-(phenylsulfonyl)tetrahydrofuran (13): [DIBALH reduction]: To a cooled (−78 °C) solution of 12 (92.7 mg, 0.284 mol) in toluene (2.8 mL, 0.1 M), DIBALH (0.31 mL, 1.0 M in toluene, 0.31 mmol, 1.1 equiv.) was added. After being stirred for 10 min at −78 °C, the reaction mixture was quenched with MeOH (0.5 mL), followed by the addition of aqueous Rochelle’s salt solution (15 mL), and diluted with Et2O (15 mL). The resulting mixture was stirred for 6 h at 25 °C. The layers were separated, and the aqueous layer was extracted with Et2O (10 mL × 3). The combined organic layers were washed with brine (15 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was filtered through a pad of silica gel (20% EtOAc/hexane) and concentrated in vacuo to provide a crude mixture, which was employed in the next step without further purification. [Sulfonylation]: To a solution of the above crude mixture in CH2Cl2 (3.6 mL, 0.08 M), PhSO2H (80.8 mg, 0.568 mmol, 2 equiv.), CaCl2 (94.6 mg, 0.852 mmol, 3 equiv.), and CSA (0.28 mL, 0.1 M in CH2Cl2, 0.028 mmol, 10 mol%) were added. After being stirred for 2 h at 25 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 (5 mL) and diluted with EtOAc (5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (5 mL × 3). The combined organic layers were washed with brine (10 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 10% EtOAc/hexane) to provide a 1.1:1 anomeric mixture of sulfonate, 13 (97.5 mg, 76%) as a colorless oil: 1H-NMR (500 MHz, CDCl3) δ 7.96–7.99 (m, 2H); 7.91–7.94 (m, 2H); 7.63–7.67 (m, 1H); 7.59–7.63 (m, 1H); 7.54 (t, J = 7.7 Hz, 2H); 7.50 (t, J = 7.7 Hz, 2H); 7.46 (d, J = 7.4 Hz, 2H); 7.42 (d, J = 7.4 Hz, 2H); 7.34–7.40 (m, 5H); 7.28–7.33 (m, 2H); 6.87 (dd, J = 8.2, 1.7 Hz, 1H); 6.84 (d, J = 8.2 Hz, 1H); 6.81 (d, J = 8.3 Hz, 1H); 6.72 (d, J = 1.7 Hz, 1H); 6.64 (dd, J = 8.2, 1.7 Hz, 1H); 5.28 (d, J = 4.5 Hz, 1H); 5.17 (s, 2H); 5.13 (s, 2H); 4.62 (d, J = 8.0 Hz, 1H); 4.60 (d, J = 1.3 Hz, 1H); 4.54 (d, J = 10.5 Hz, 1H); 3.98 (s, 3H); 3.93 (s, 3H); 3.20–3.30 (m, 2H); 2.68–2.77 (m, 1H); 2.42–2.50 (m, 1H); 1.30 (d, J = 7.0 Hz, 3H); 1.20 (d, J = 7.4 Hz, 3H); 0.88 (d, J = 6.9 Hz, 3H); 0.53 (d, J = 7.2 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 150.0, 149.4, 148.4, 147.3, 137.5, 137.4, 137.2, 137.1, 133.8, 132.0, 131.0, 129.4, 129.093, 129.088, 129.0, 128.58, 128.55, 127.9, 127.8, 127.29, 127.27, 120.3, 118.1, 113.6, 113.1, 110.7, 109.8, 100.0, 97.8, 89.9, 85.8, 71.1, 71.0, 56.1, 43.8, 42.3, 38.6, 38.4, 14.6, 14.5, 10.3, and 9.4; HRMS (EI) m/z: 452.1658 ((M)+, C26H28O5S requires 452.1657).
5-((2S,3S,4R,5S)-5-(4-(Benzyloxy)-3-methoxyphenyl)-3,4-dimethyltetrahydrofuran-2-yl)benzo[d][1,3]dioxole (15): To a solution of 3,4-methylenedioxybromobenzene (59.2 mg, 0.294 mmol, 3.2 equiv.) and Mg turnings (6.7 mg, 0.276 mmol, 3 equiv.) in THF (0.9 mL, 0.1 M), I2 (46 μL, 0.1 M in THF, 4.6 μmol, 5 mol%) was added, and the resulting mixture was stirred for 30 min at 80 °C (the oil bath temperature) to afford 14 and then treated with ZnBr2 (1.5 mL, 0.2 M in THF, 0.29 mmol, 3.2 equiv.) via a cannula at 25 °C, and the resulting mixture was stirred for 30 min at 25 °C before it was added to 13 (41.6 mg, 0.092 mmol) in THF (0.9 mL, 0.1 M). After being stirred for 1 h at 25 °C, the reaction mixture was quenched with saturated aqueous NH4Cl (5 mL) and diluted with EtOAc (5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (5 mL × 3). The combined organic layers were washed with brine (10 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 10% EtOAc/hexane) to provide 15 (33.8 mg, 85%) as a colorless oil: [α]D25 −86.6 (c 0.15, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.44 (d, J = 7.4 Hz, 2 H); 7.34–7.38 (m, 2H); 7.27–7.31 (m, 1H); 6.93 (dd, J = 4.3, 1.6 Hz, 2H); 6.85 (d, J = 8.2 Hz, 1H); 6.82 (dd, J = 8.0, 1.5 Hz, 1H); 6.77 (dd, J = 8.0, 2.6 Hz, 2H); 5.94 (ABq, 2H, J = 1.4 Hz, Δν = 3.1 Hz); 5.42 (d, J = 4.4 Hz, 1H); 5.14 (s, 2H); 4.62 (d, J = 9.3 Hz, 1H); 3.89 (s, 3H); 2.36–2.46 (m, 2H); 1.00 (d, J = 6.5 Hz, 3H); 0.61 (d, J = 7.0 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 149.4, 147.8, 146.89, 146.85, 137.3, 137.2, 133.8, 128.5, 127.7, 127.3, 119.5, 118.0, 113.8, 109.8, 107.9, 106.4, 100.9, 85.7, 84.7, 71.1, 56.0, 47.6, 43.4, 11.8, and 9.4; HRMS (Q–TOF) m/z: 433.2020 ((M + H)+, C27H29O5 requires 433.2015).
(-)-Chicanine (
1): To a solution of
15 (24.8 mg, 0.057 mol) in MeOH (1.4 mL, 0.04 M), Pd/C (10%, 124 mg) was added, and the reaction mixture was hydrogenated at 1 atm. After being stirred for 2 h at 25 °C, the reaction mixture was filtered through a pad of celite and concentrated in vacuo. The residue was purified by column chromatography (SiO
2, 25% EtOAc/hexane) to provide natural (-)-chicanine (
1, 18.1 mg, 93%) as a colorless oil for which the spectral data were identical to those of the known synthetic
1 [11,39]: [α]
D25 –139 (
c 0.12, CHCl
3) vs. [α]
D25 −134.3 (
c 1.01, CHCl
3) [
39];
1H-NMR (500 MHz, CDCl
3) δ 6.92 (dd,
J = 5.8, 1.5 Hz, 2H); 6.88 (d,
J = 8.1 Hz, 1H); 6.82 (dd,
J = 8.0, 1.5 Hz, 1H); 6.78 (d,
J = 7.9 Hz, 1H); 6.77 (dd,
J = 8.2, 1.6 Hz, 1H); 5.94 (ABq, 2H,
J = 1.3 Hz, Δν = 3.2 Hz); 5.55 (s, 1H); 5.43 (d,
J = 4.3 Hz, 1H); 4.62 (d,
J = 9.3 Hz, 1H); 3.88 (s, 3H); 2.36–2.46 (m, 2H); 0.99 (d,
J = 6.4 Hz, 3H); 0.61 (d,
J = 7.0 Hz, 3H);
13C-NMR (125 MHz, CDCl
3) δ 147.8, 146.9, 146.2, 144.3, 137.2, 132.5, 119.5, 118.8, 113.9, 108.7, 108.0, 106.4, 100.9, 85.7, 84.8, 55.9, 47.6, 43.4, 11.8, and 9.4; HRMS (Q–TOF)
m/
z: 343.1559 ((M + H)
+, C
20H
23O
5 requires 343.1545).
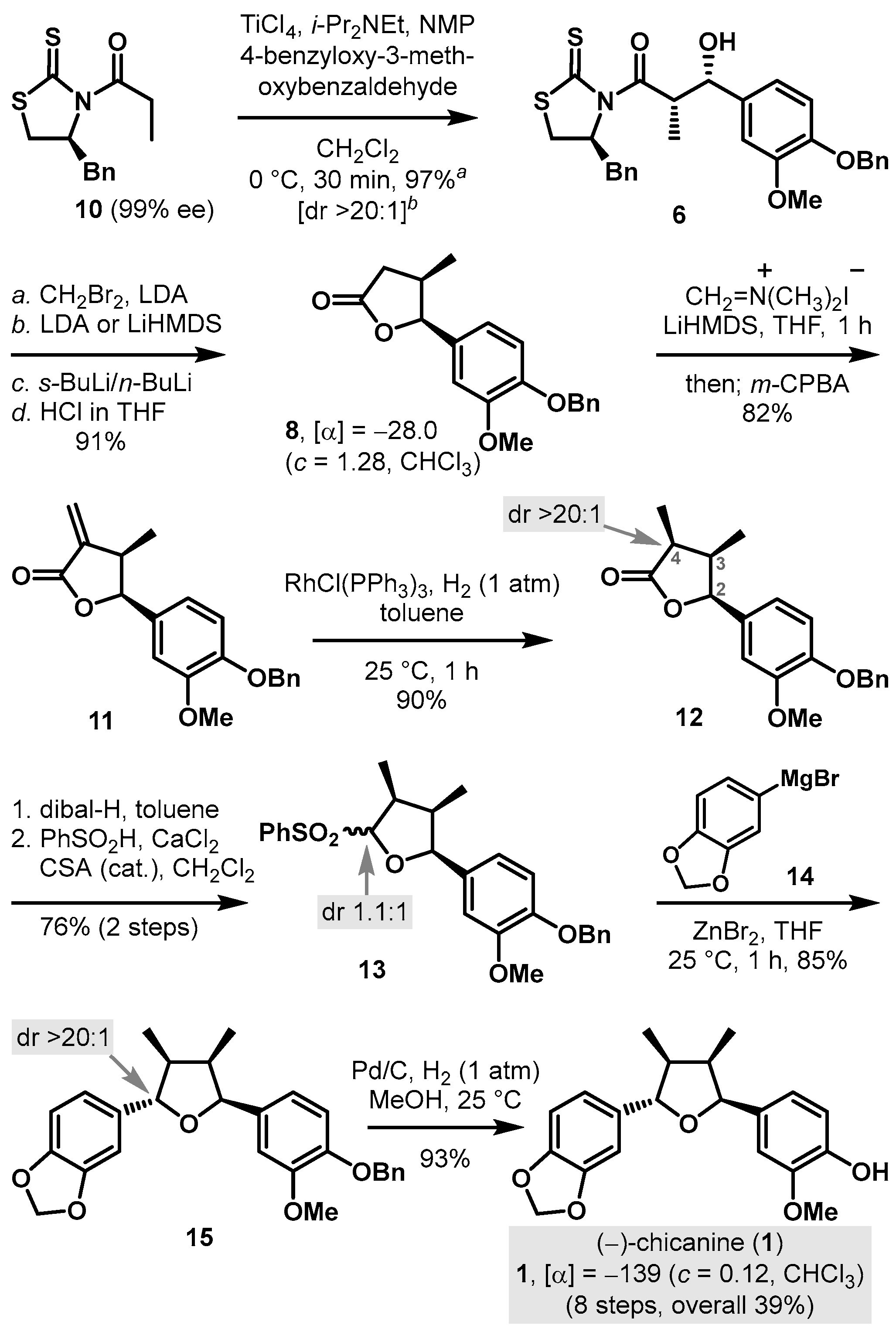
3.3. Synthesis of (+)-Fragransin A2 (2) and (+)-Galbelgin (3)
(3R,4R,5S)-5-(4-(Benzyloxy)-3-methoxyphenyl)-3,4-dimethyldihydrofuran-2(3H)-one (16): To a cooled (−78 °C) solution of 8 (187 mg, 0.599 mmol) in THF (6.0 mL, 0.1 M), LiHMDS (1.2 mL, 1.0 M in THF, 1.2 mmol, 2 equiv.) was added. After being stirred for 1 h, the reaction mixture was added to MeI (56.0 μL, 0.899 mmol, 1.5 equiv.) and stirred for 10 min at −78 °C before it was quenched with the addition of saturated aqueous NH4Cl (10 mL) and diluted with EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 25% EtOAc/hexane) to provide 16 (187 mg, 96%) as a colorless oil: [α]D25 +28.5 (c 1.72, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.42 (d, J = 7.3 Hz, 2H); 7.33−7.37 (m, 2H); 7.27−7.31 (m, 1H); 6.87 (d, J = 8.2 Hz, 1H); 6.66 (d, J = 1.9 Hz, 1H); 6.64 (dd, J = 8.2, 1.8 Hz, 1H); 5.48 (d, J = 7.6 Hz, 1H); 5.13 (s, 2H); 3.86 (s, 3H); 2.41−2.49 (m, 1H); 2.30−2.37 (m, 1H); 1.27 (d, J = 7.1 Hz, 3H); 0.75 (d, J = 6.9 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 179.7, 149.5, 147.9, 136.8, 129.1, 128.4, 127.8, 127.2, 118.0, 113.6, 109.4, 82.3, 70.9, 56.0, 42.3, 39.9, 14.5, and 13.6; HRMS (Q–TOF) m/z: 327.1612 ((M + H)+, C20H23O4 requires 327.1596).
(2S,3R,4R)-2-(4-(Benzyloxy)-3-methoxyphenyl)-5-methoxy-3,4-dimethyltetrahydrofuran (17): To a cooled (−78 °C) solution of 16 (18.3 mg, 0.073 mmol) in toluene (1.8 mL, 0.04 M), DIBALH (80 μL, 1.0 M in toluene, 0.080 mmol, 1.1 equiv.) was added. After being stirred for 10 min at −78 °C, the reaction mixture was diluted with MeOH (3.7 mL, 0.02 M) before it was added to HC(OMe)3 (80 μL, 0.73 mmol, 10 equiv.) and p-TsOH (6.3 mg, 0.037 mmol, 50 mol%). After being stirred for 12 h at 25 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 (10 mL) and diluted with EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine (15 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 10% EtOAc/hexane) to provide a 1.3:1 anomeric mixture of cyclic methyl acetal, 17, (18.4 mg, 95%) as a colorless oil: For the major diastereomer: 1H-NMR (500 MHz, CDCl3) δ 7.42–7.45 (m, 2H); 7.33–7.38 (m, 2H); 7.30 (d, J = 7.1 Hz, 1H); 6.84 (d, J = 8.2 Hz, 1H); 6.74 (d, J = 1.7 Hz, 1H); 6.67 (dd, J = 8.2, 1.7 Hz, 1H); 5.12–5.17 (m, 2H); 5.04 (d, J = 4.7 Hz, 1H); 3.88, (s, 3H); 3.43 (s, 3H); 2.24–2.33 (m, 1H); 1.81–1.91 (m, 1H); 1.03 (d, J = 6.8 Hz, 3H); 0.56 (d, J = 7.0 Hz, 3H); for the minor diastereomer: 1H-NMR (500 MHz, CDCl3) δ 7.42–7.45 (m, 2H); 7.33–7.38 (m, 2H); 7.28 (d, J = 6.9 Hz, 1H); 7.06 (d, J = 1.7 Hz, 1H); 6.83 (d, J = 8.2 Hz, 1H); 6.76 (dd, J = 8.3, 1.7 Hz, 1H); 5.12–5.17 (m, 2H); 4.75 (d, J = 5.1 Hz, 1H); 3.90, (s, 3H); 3.56 (s, 3H); 2.10–2.18 (m, 1H); 1.81–1.91 (m, 1H); 1.09 (d, J = 6.9 Hz, 3H); 0.64 (d, J = 7.0 Hz, 3H); for 17: 13C-NMR (125 MHz, CDCl3) δ 149.23, 149.18, 147.1, 146.9, 137.3, 137.2, 133.9, 133.8, 128.4, 127.67, 127.65, 127.23, 127.20, 118.9, 118.8, 113.6, 113.3, 112.5, 110.7, 110.4, 106.1, 84.7, 83.1, 71.0, 56.5, 55.9, 55.7, 54.8, 44.7, 44.2, 44.1, 41.3, 14.8, 14.5, 14.2, and 11.5; HRMS (Q–TOF) m/z: 365.1730 ((M + Na)+, C21H26NaO4 requires 365.1729).
4-((2R,3R,4R,5R)-5-(4-(Benzyloxy)-3-methoxyphenyl)-3,4-dimethyltetrahydrofuran-2-yl)-2-methoxyphenol (18A): To a cooled (−78 °C) solution of 17 (42.8 mg, 0.125 mmol) and 2-methoxyphenol, 18, (78.0 mg, 0.625 mmol, 5 equiv.) in CH2Cl2 (1.3 mL, 0.1 M), BF3∙OEt2 (93 μL, 0.75 mmol, 6 equiv.) was added. After being stirred for 30 min at −20 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 (5 mL) and diluted with CH2Cl2 (5 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (5 mL × 3). The combined organic layers were washed with brine (10 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 17% EtOAc/hexane) to provide 18A (44.0 mg, 81%) as a white oil: [α]D25 +52.1 (c 0.23, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.43 (d, J = 7.4 Hz, 2H); 7.33–7.37 (m, 2H); 7.27–7.31 (m, 1H); 6.98 (s, 1H); 6.94 (d, J = 1.6 Hz, 1H); 6.89 (d, J = 8.1 Hz, 1H); 6.86 (d, J = 1.7 Hz, 1H); 6.85 (s, 2H); 5.58 (br s, 1H); 5.15 (s, 2H); 4.63 (dd, J = 9.1, 1.9 Hz, 2H); 3.92 (s, 3H); 3.91 (s, 3H); 1.72–1.83 (m, 2H); 1.04 (d, J = 5.1 Hz, 6H); 13C-NMR (125 MHz, CDCl3) δ 149.7, 147.6, 146.6, 145.1, 137.2, 135.6, 134.3, 128.5, 127.7, 127.3, 119.3, 118.6, 114.0, 113.8, 109.8, 108.5, 88.4, 88.2, 71.1, 56.0, 55.9, 51.0, 50.9, 13.9, and 13.8; HRMS (EI) m/z: 434.2093 ((M)+, C27H30O5 requires 434.2093).
(+)-Fragransin A
2 (
2): To a solution of
18A (11 mg, 0.025 mmol) in MeOH (1.3 mL, 0.02 M), Pd/C (10%, 55 mg) was added, and the reaction mixture was hydrogenated at 1 atm. After being stirred for 30 min at 25 °C, the reaction mixture was filtered through a pad of celite and concentrated in vacuo. The residue was purified by column chromatography (SiO
2, 50% EtOAc/hexane) to provide natural (+)-fragransin A
2 (
2, 8.2 mg, 95%) as a colorless oil for which the spectral data were identical to those of the known synthetic
2 [15,18]: [α]
D25 +75.0 (
c 0.12, CHCl
3) vs. [α]
D25 +86.9 (
c 0.88, CHCl
3) [
15];
1H-NMR (500 MHz, CDCl
3) δ 6.95 (d,
J = 1.6 Hz, 2H); 6.90 (d,
J = 8.0 Hz, 2H); 6.87 (dd,
J = 8.1, 1.7 Hz, 2H); 5.57 (s, 2H); 4.63 (d,
J = 9.2 Hz, 2H); 3.92 (s, 6H); 1.72–1.80 (m, 2H); 1.04 (d,
J = 6.0 Hz, 6H);
13C-NMR (125 MHz, CDCl
3) δ 146.6, 145.1, 134.3, 119.4, 113.9, 108.4, 88.3, 55.9, 51.0, and 13.8; HRMS (Q–TOF)
m/
z: 367.1523 ((M + Na)
+, C
20H
24NaO
5 requires 367.1521).
(+)-Galbelgin (
3): To a cooled (0 °C) solution of
2 (8.0 mg, 0.023 mmol) in DMF/THF (2:1, a total of 0.6 mL, 0.038 M), NaH (3.7 mg, 60% dispersion in mineral oil, 0.092 mmol, 4 equiv.) was added. After being stirred for 10 min at 0 °C, the reaction mixture was added to MeI (58 μL, 1.0 M in THF, 0.058 μmol, 2.5 equiv.) and stirred for 30 min at 25 °C before it was quenched with the addition of saturated aqueous NH
4Cl (5 mL) and diluted with EtOAc (5 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (5 mL × 3). The combined organic layers were washed with brine (5 mL × 1), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO
2, 25% EtOAc/hexane) to provide natural (+)-galbelgin (
3, 8.5 mg, 99%) as a colorless oil for which the spectral data were identical to those of the known synthetic
3 [10,18,19,20]: [α]
D25 +83.3 (
c 0.06, CHCl
3) vs. [α]
D +80.0 (
c 0.5, CHCl
3) [
20];
1H-NMR (500 MHz, CDCl
3) δ 6.96 (d,
J = 1.9 Hz, 2H); 6.92 (dd,
J = 8.2, 1.9 Hz, 2H); 6.84 (d,
J = 8.2 Hz, 2H); 4.66 (d,
J = 9.2 Hz, 2H); 3.91 (s, 6H); 3.88 (s, 6H); 1.75–1.84 (m, 2H); 1.05 (d,
J = 6.0 Hz, 6H);
13C-NMR (125 MHz, CDCl
3) δ 149.0, 148.5, 134.9, 118.6, 110.8, 109.1, 88.3, 55.89, 55.87, 51.0, and 13.8; HRMS (Q–TOF)
m/
z: 373.2016 ((M + H)
+, C
22H
29O
5 requires 373.2015).
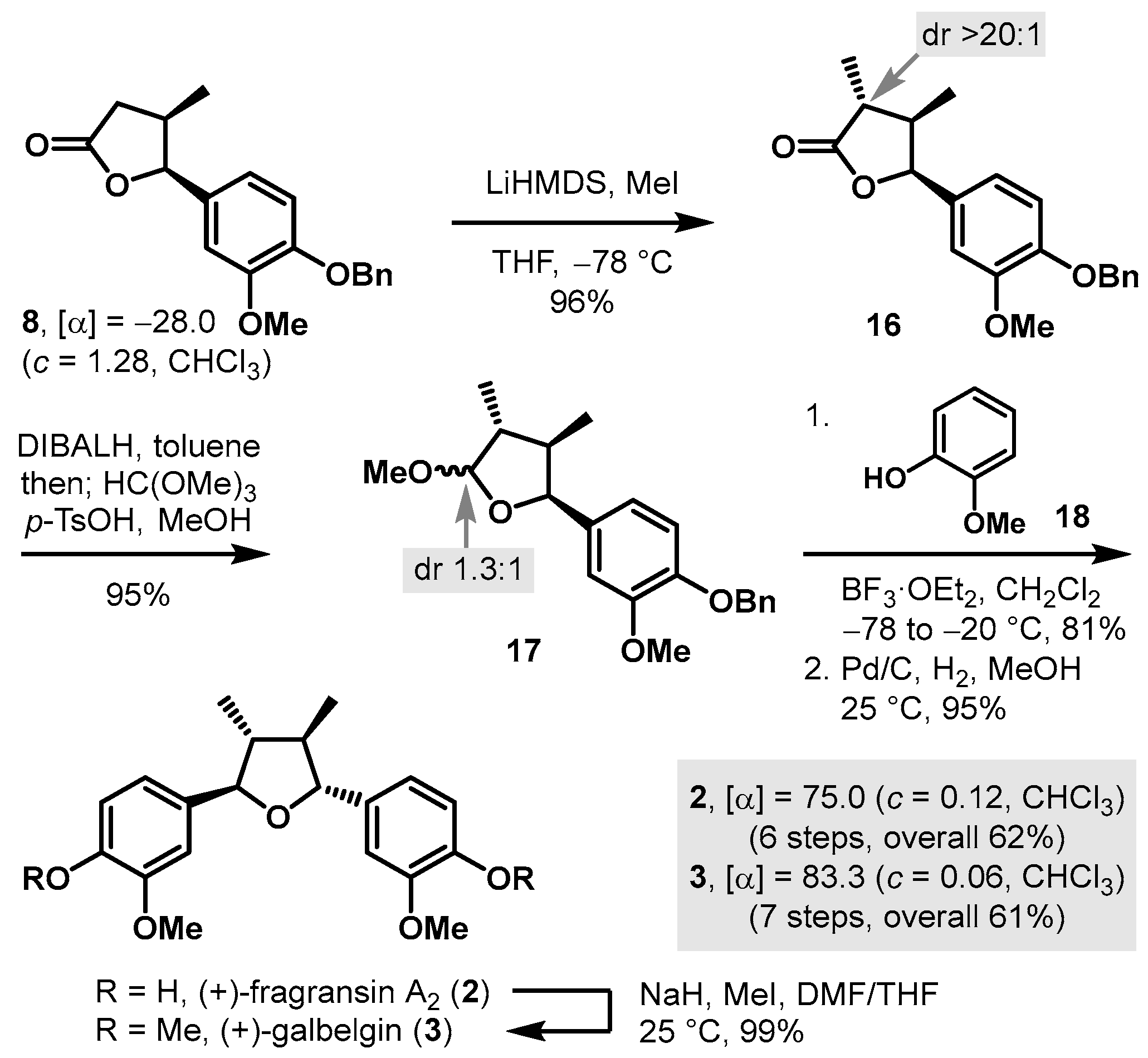
3.4. Synthesis of (+)-Talaumidin (4) and (+)-Galbacin (5)
(2S,3S)-3-(Benzo[d][1,3]dioxol-5-yl)-1-((S)-4-benzyl-2-thioxothiazolidin-3-yl)-3-hydroxy-2-methylpropan-1-one (7). To a cooled (0 °C) solution of (S)-1-(4-benzyl-2-thioxothiazolidin-3-yl)propan-1-one, 10, (590 mg, 2.22 mmol) in CH2Cl2 (11 mL, 0.2 M), titanium(IV) chloride (2.4 mL, 1.0 M in CH2Cl2, 2.4 mmol, 1.1 equiv.) was added. After being stirred for 15 min at the same temperature, i-Pr2NEt (0.47 mL, 2.7 mmol, 1.2 equiv.) was added dropwise, and the reaction mixture was stirred for 40 min at 0 °C. NMP (0.43 mL, 4.4 mmol, 2 equiv.) was added, and the reaction mixture was stirred for an additional 10 min. Piperonal (667 mg, 4.44 mmol, 2 equiv.) in CH2Cl2 (5 mL) was added to the enolate. After being stirred for 30 min, the reaction mixture was quenched with saturated aqueous NH4Cl (10 mL) and diluted with CH2Cl2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with brine (20 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 25% EtOAc/hexane) to provide 7 (775 mg, 84%) as a yellow oil: [α]D25 +156 (c 1.16, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.31–7.35 (m, 2H); 7.26–7.29 (m, 1H); 7.22–7.26 (m, 2H); 6.87 (d, J = 1.6 Hz, 1H); 6.78 (dd, J = 8.0, 1.5 Hz, 1H); 6.73 (d, J = 8.0 Hz, 1H); 5.93 (ABq, 2H, J = 1.4 Hz, Δν = 1.8 Hz); 4.89 (ddd, J = 10.6, 6.8, 4.0 Hz, 1H); 4.81 (d, J = 6.2 Hz, 1H); 4.73 (p, J = 6.6 Hz, 1H); 3.17 (dd, J = 13.2, 3.8 Hz, 1H); 2.93–2.98 (m, 3H); 2.76 (d, J = 11.4 Hz, 1H); 1.30 (d, J = 6.7 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 201.1, 177.0, 147.5, 146.9, 136.2, 135.6, 129.3, 128.8, 127.1, 119.5, 107.9, 106.7, 100.9, 75.4, 68.8, 46.3, 36.5, 32.2, and 12.1; HRMS (Q–TOF) m/z: 416.0997 ((M + H)+, C21H22NO4S2 requires 416.0990).
(4R,5S)-5-(Benzo[d][1,3]dioxol-5-yl)-4-methyldihydrofuran-2(3H)-one (9): To a cooled (−78 °C) solution of i-Pr2NH (0.58 mL, 4.1 mmol, 4.4 equiv.) in THF (2.3 mL, 0.4 M), n-BuLi (2.3 mL, 1.6 M in hexane, 3.8 mmol, 4 equiv.) was added, and the reaction mixture was stirred for 15 min at 0 °C before it was cooled to −78 °C. CH2Br2 (4.1 mL, 1.0 M in THF, 4.1 mmol, 4.4 equiv.) and 7 (390 mg, 0.939 mmol) in THF (4.0 mL) were added dropwise to the above LDA solution. After being stirred for 10 min at −78 °C, the reaction mixture was treated with LiHMDS (1.9 mL, 1.0 M in THF, 1.9 mmol, 2 equiv.). After being stirred for 5 min at −78 °C, the reaction mixture was stirred for 90 s at −20 °C before it was cooled to −78 °C. s-BuLi (1.3 mL, 1.4 M in cyclohexane, 1.9 mmol, 2 equiv.) was added dropwise, and the reaction mixture was stirred for 90 s at −20 °C and cooled to −78 °C, followed by the dropwise addition of n-BuLi (2.3 mL, 1.6 M in hexane, 3.8 mmol, 4 equiv.). After being stirred for 5 min, the reaction mixture was stirred for 90 s at –20 °C, warmed to 25 °C in a water bath, and stirred for 30 min before it was added to a solution of HCl (86 μL, 2.8 mmol, 3 equiv.) in THF (31 mL, 0.03 M) at −20 °C. After being stirred for 5 min at −20 °C, the reaction mixture was quenched with the addition of saturated aqueous NaHCO3 (10 mL), and diluted with EtOAc (10 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (10 mL × 3). The combined organic layers were washed with brine (15 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 20% EtOAc/hexane) to provide 9 (194 mg, 94%) as a colorless oil: [α]D25 −12.5 (c 0.40, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 6.76 (d, J = 8.0 Hz, 1H); 6.67 (d, J = 1.4 Hz, 1H); 6.65 (dd, J = 8.0, 1.6 Hz, 1H); 5.92 (s, 2H); 5.46 (d, J = 5.9 Hz, 1H); 2.74–2.82 (m, 2H); 2.25–2.32 (m, 1H); 0.67 (d, J = 7.0 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 176.5, 147.7, 147.1, 129.8, 118.7, 108.0, 105.9, 101.0, 83.8, 36.8, 34.8, and 14.9; HRMS (Q–TOF) m/z: 221.0821 ((M + H)+, C12H13O4 requires 221.0814).
(3R,4R,5S)-5-(Benzo[d][1,3]dioxol-5-yl)-3,4-dimethyldihydrofuran-2(3H)-one (9A): To a cooled (−78 °C) solution of 9 (210 mg, 0.954 mmol) in THF (9.5 mL, 0.1 M), LiHMDS (1.9 mL, 1.0 M in THF, 1.9 mmol, 2 equiv.) was added. After being stirred for 1 h, the reaction mixture was added to MeI (71.2 μL, 1.15 mmol, 1.2 equiv.), and the reaction mixture was stirred for 10 min at −78 °C before it was quenched with the addition of saturated aqueous NH4Cl (15 mL) and diluted with EtOAc (15 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (15 mL × 3). The combined organic layers were washed with brine (30 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 25% EtOAc/hexane) to provide 9A (206 mg, 92%) as a colorless oil: [α]D25 +30.1 (c 1.72, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 6.73 (d, J = 7.8 Hz, 1H); 6.55−6.58 (m, 2H); 5.90−5.92 (m, 2H); 5.40 (d, J = 7.8 Hz, 1H); 2.41 (dp, J = 10.3, 7.0 Hz, 1H); 2.28 (dq, J = 10.3, 7.0 Hz, 1H); 1.21 (d, J = 7.1 Hz, 3H); 0.71 (d, J = 7.0 Hz, 3H); 13C-NMR (125 MHz, CDCl3) δ 179.5, 147.7, 147.2, 129.8, 119.1, 107.9, 106.1, 101.1, 82.2, 42.1, 39.6, 14.3, and 13.4; HRMS (Q–TOF) m/z: 235.0993 ((M + H)+, C13H15O4 requires 235.0970).
5-((2S,3R,4R)-5-Methoxy-3,4-dimethyltetrahydrofuran-2-yl)benzo[d][1,3]dioxole (19): To a cooled (−78 °C) solution of 9A (180 mg, 0.770 mmol) in toluene (19 mL, 0.04 M), DIBALH (0.85 mL, 1.0 M in toluene, 0.85 mmol, 1.1 equiv.) was added. After being stirred for 10 min at −78 °C, the reaction mixture was diluted with MeOH (38 mL, 0.02 M) before it was added to HC(OMe)3 (0.84 mL, 7.7 mmol, 10 equiv.) and p-TsOH (66.3 mg, 0.385 mmol, 50 mol%). After being stirred for 12 h at 25 °C, the reaction mixture was quenched with saturated aqueous NaHCO3 (30 mL) and diluted with EtOAc (30 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (30 mL × 3). The combined organic layers were washed with brine (45 mL × 1), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, 10% EtOAc/hexane) to provide a 1.5:1 anomeric mixture of cyclic methyl acetal, 19, (190 mg, 99%) as a colorless oil: For the major diastereomer: 1H-NMR (500 MHz, CDCl3) δ 6.75 (s, 1H); 6.68 (d, J = 1.6 Hz, 1H); 6.64 (dd, J = 8.0, 1.2 Hz, 1H); 5.92 (s, 2H); 5.13 (d, J = 8.9 Hz, 1H); 5.01 (d, J = 4.7 Hz, 1H); 3.41 (s, 3H); 2.22–2.31 (m, 1H); 1.78–1.88 (m, 1H); 1.01 (d, J = 6.8 Hz, 3H); 0.56 (d, J = 7.0 Hz, 3H); for the minor diastereomer: 1H-NMR (500 MHz, CDCl3) δ 6.91 (s, 1H); 6.75 (s, 1H); 6.74 (s, 1H); 5.93 (s, 2H); 5.10 (d, J = 7.6 Hz, 1H); 4.71 (d, J = 5.1 Hz, 1H); 3.53 (s, 3H); 2.11 (dp, J = 9.0, 7.1 Hz, 1H); 1.78–1.88 (m, 1H); 1.07 (d, J = 6.9 Hz, 3H); 0.63 (d, J = 7.0 Hz, 3H); for 19: 13C-NMR (125 MHz, CDCl3) δ 147.4, 147.3, 146.5, 146.4, 134.73, 134.66, 119.9, 112.4, 107.7, 107.2, 106.2, 100.81, 100.76, 84.5, 83.2, 56.6, 54.8, 44.7, 44.1, 43.9, 41.3, 14.9, 14.4, 14.3, and 11.5.
(+)-Talaumidin (
4): To a cooled (−78 °C) solution of
19 (56.5 mg, 0.226 mmol) and 2-methoxyphenol,
20, (140 mg, 1.13 mmol, 5 equiv.) in CH
2Cl
2 (2.3 mL, 0.1 M), BF
3∙OEt
2 (0.17 mL, 1.36 mmol, 6 equiv.) was added. After being stirred for 30 min at −20 °C, the reaction mixture was quenched with saturated aqueous NaHCO
3 (10 mL) and diluted with CH
2Cl
2 (10 mL). The layers were separated, and the aqueous layer was extracted with CH
2Cl
2 (10 mL × 3). The combined organic layers were washed with brine (15 mL × 1), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO
2, 17% EtOAc/hexane) to provide natural (+)-talaumidin (
4, 67.9 mg, 88%) as a colorless oil for which the spectral data were identical to those of the known synthetic
4 [18,39,41]: [α]
D25 +75.9 (
c 0.16, CHCl
3) vs. [α]
D20 +88.3 (
c 2.1, CHCl
3) [
39];
1H-NMR (500 MHz, CDCl
3) δ 6.93 (dd,
J = 4.5, 1.6 Hz, 2H); 6.89 (d,
J = 8.1 Hz, 1H); 6.82–6.87 (m, 2H); 6.78 (d,
J = 7.9 Hz, 1H); 5.94–5.95 (m, 2H); 5.61 (s, 1H); 4.62 (d,
J = 9.2 Hz, 2H); 3.91 (s, 3H); 1.71–1.82 (m, 2H); 1.04 (d,
J = 6.2 Hz, 3H); 1.02 (d,
J = 6.2 Hz, 3H);
13C-NMR (125 MHz, CDCl
3) δ 147.8, 147.0, 146.6, 145.1, 136.6, 134.1, 119.7, 119.4, 114.0, 108.6, 108.0, 106.6, 100.9, 88.4, 88.2, 56.0, 51.2, 50.9, 13.84, and 13.81; HRMS (EI)
m/
z: 342.1465 ((M)
+, C
20H
22O
5 requires 342.1467).
(+)-Galbacin (
5): To a cooled (−78 °C) solution of
19A (39.6 mg, 0.158 mmol) and 1,2-methylenedioxybenzene,
21, (97 mg, 0.79 mmol, 5 equiv.) in CH
2Cl
2 (2 mL, 0.08 M), BF
3∙OEt
2 (0.12 mL, 0.95 mmol, 6 equiv.) was added. After being stirred for 30 min at −20 °C, the reaction mixture was quenched with saturated aqueous NaHCO
3 (5 mL) and diluted with CH
2Cl
2 (5 mL). The layers were separated, and the aqueous layer was extracted with CH
2Cl
2 (5 mL × 3). The combined organic layers were washed with brine (10 mL × 1), dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO
2, 50% CH
2Cl
2/hexane) to provide natural (+)-galbacin (
5, 47.3 mg, 88%) as a colorless oil for which the spectral data were identical to those of the known synthetic
5 [15,20,42]: [α]
D25 +121 (
c 0.22, CHCl
3) vs. [α]
D28 +110 (
c 1.0, CHCl
3) [
20];
1H-NMR (500 MHz, CDCl
3) δ 6.92 (d,
J = 1.5 Hz, 2H); 6.83 (dd,
J = 8.0, 1.5 Hz, 2H); 6.77 (d,
J = 7.9 Hz, 2H); 5.94 (s, 4H); 4.60 (d,
J = 9.2 Hz, 2H); 1.70–1.80 (m, 2H); 1.02 (d,
J = 6.0 Hz, 6H);
13C-NMR (125 MHz, CDCl
3) δ 147.7, 146.9, 136.3, 119.7, 107.9, 106.5, 100.9, 88.2, 51.0, and 13.7; HRMS (EI)
m/
z: 340.1310 ((M)
+, C
20H
20O
5 requires 340.1311).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}