
A Study on the Photoisomerization of (E)-Dehydrozingerone, Its (E)-(E)-C₂ Symmetric Dimer, and Their O-Methylated Derivatives



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
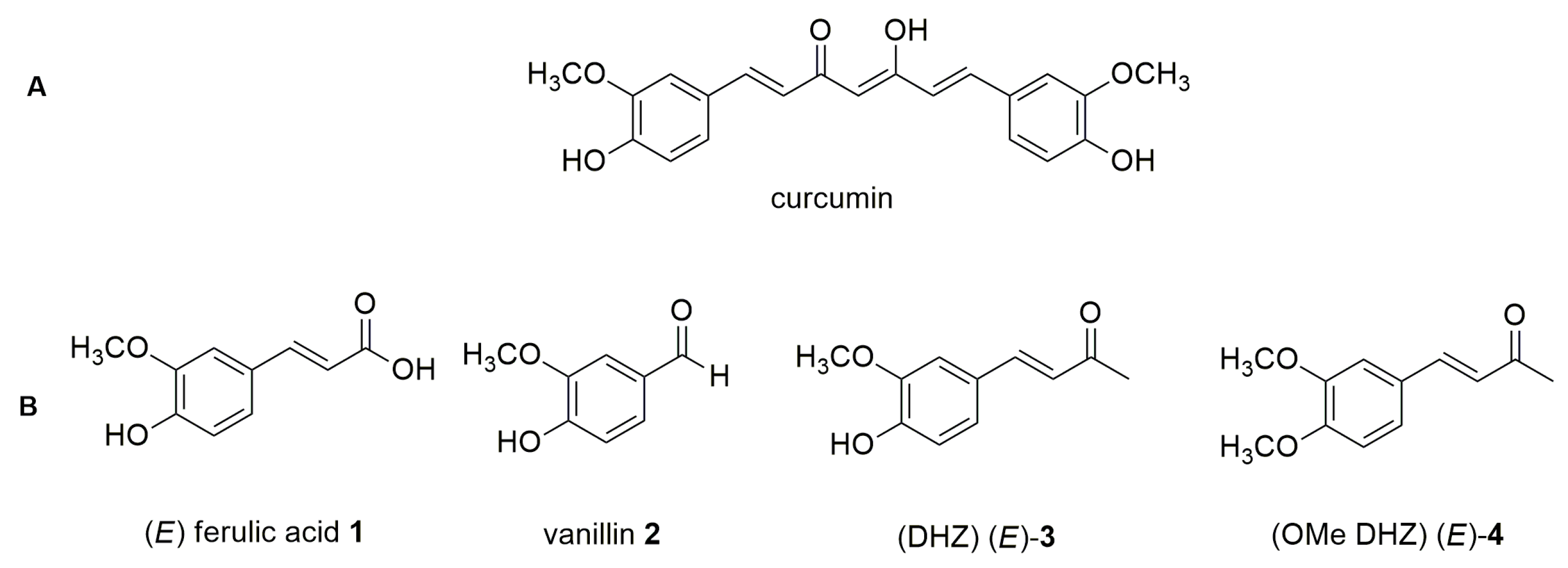
1. Introduction
2. Results and Discussion
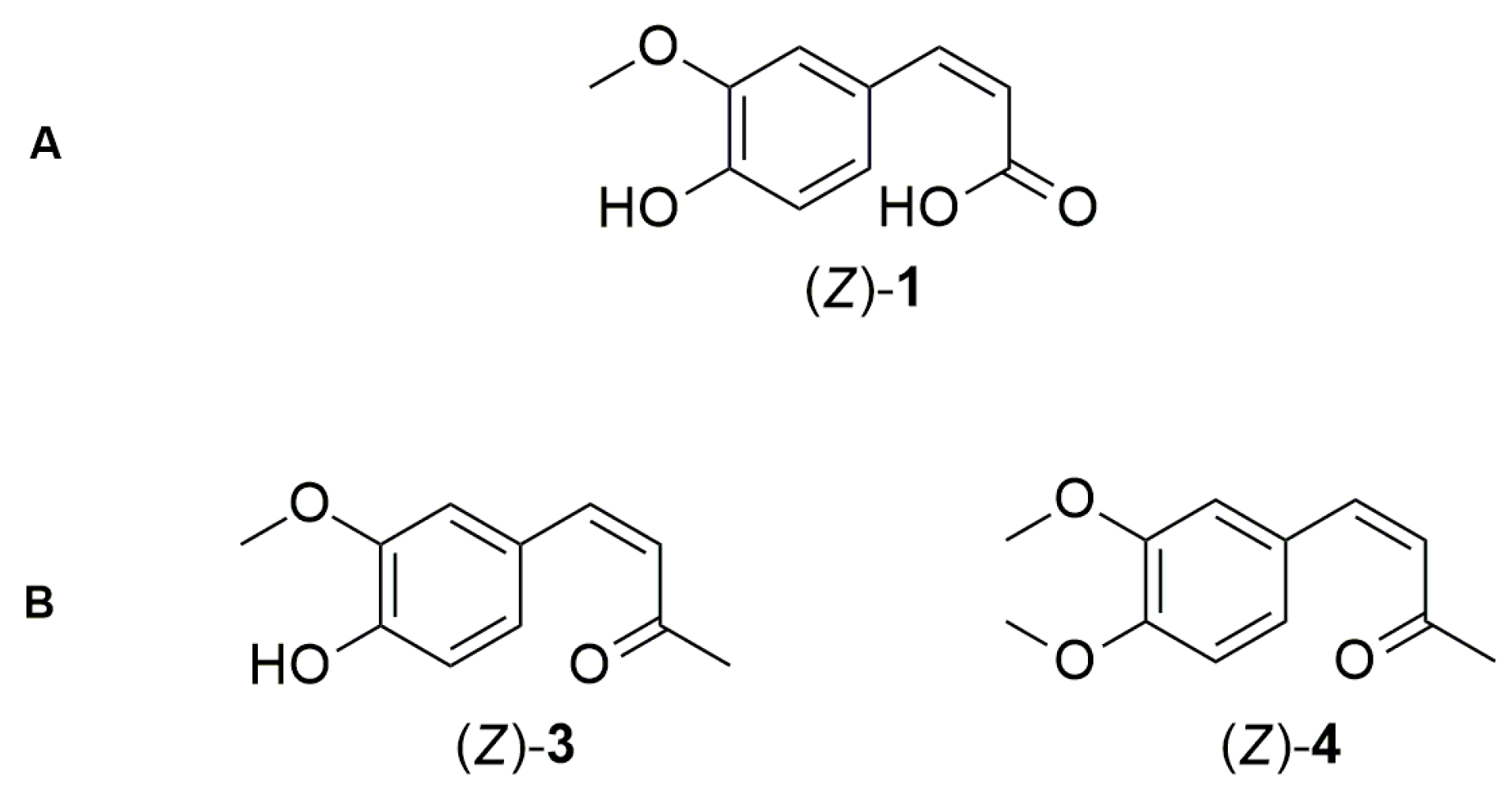
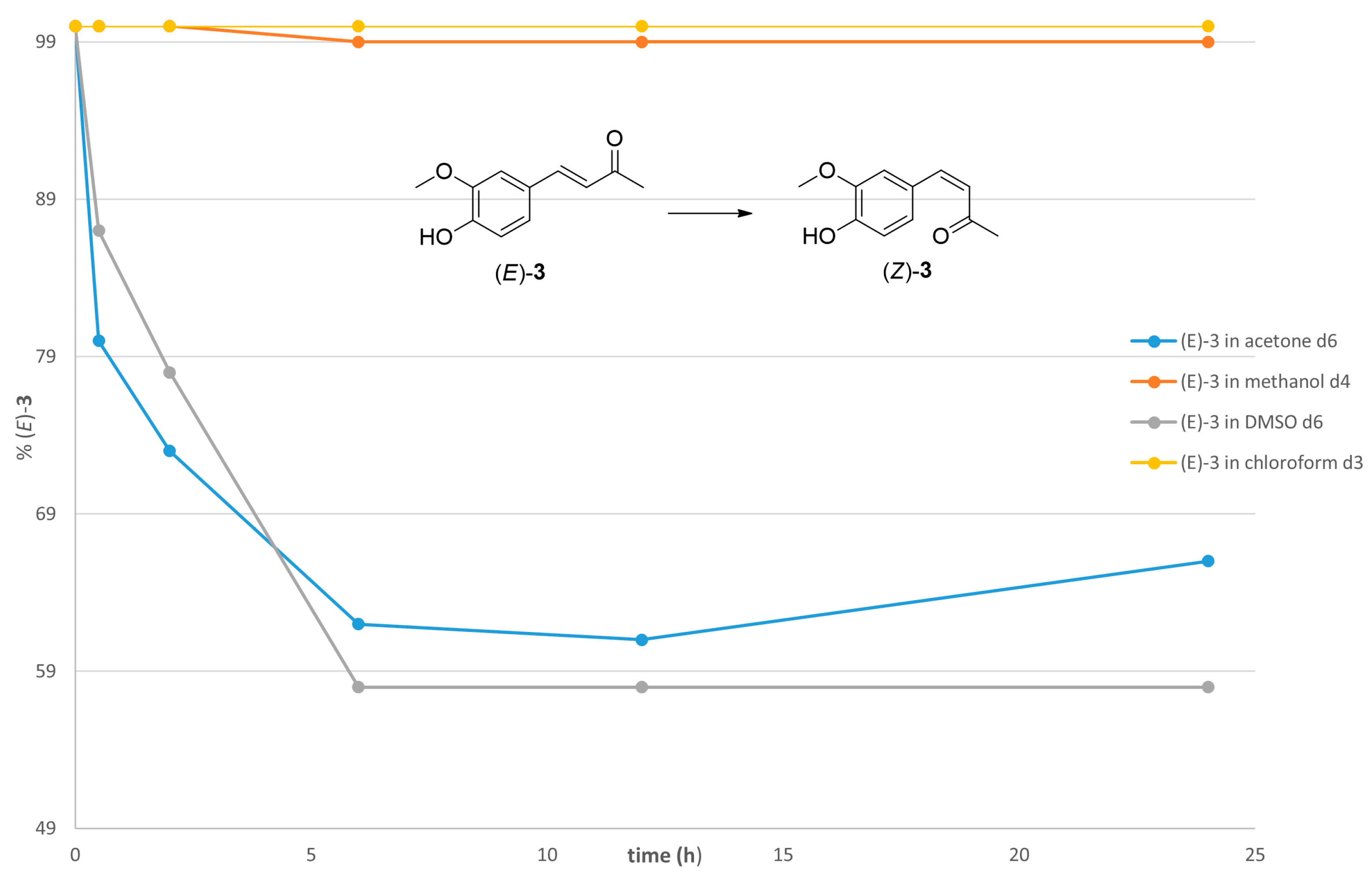
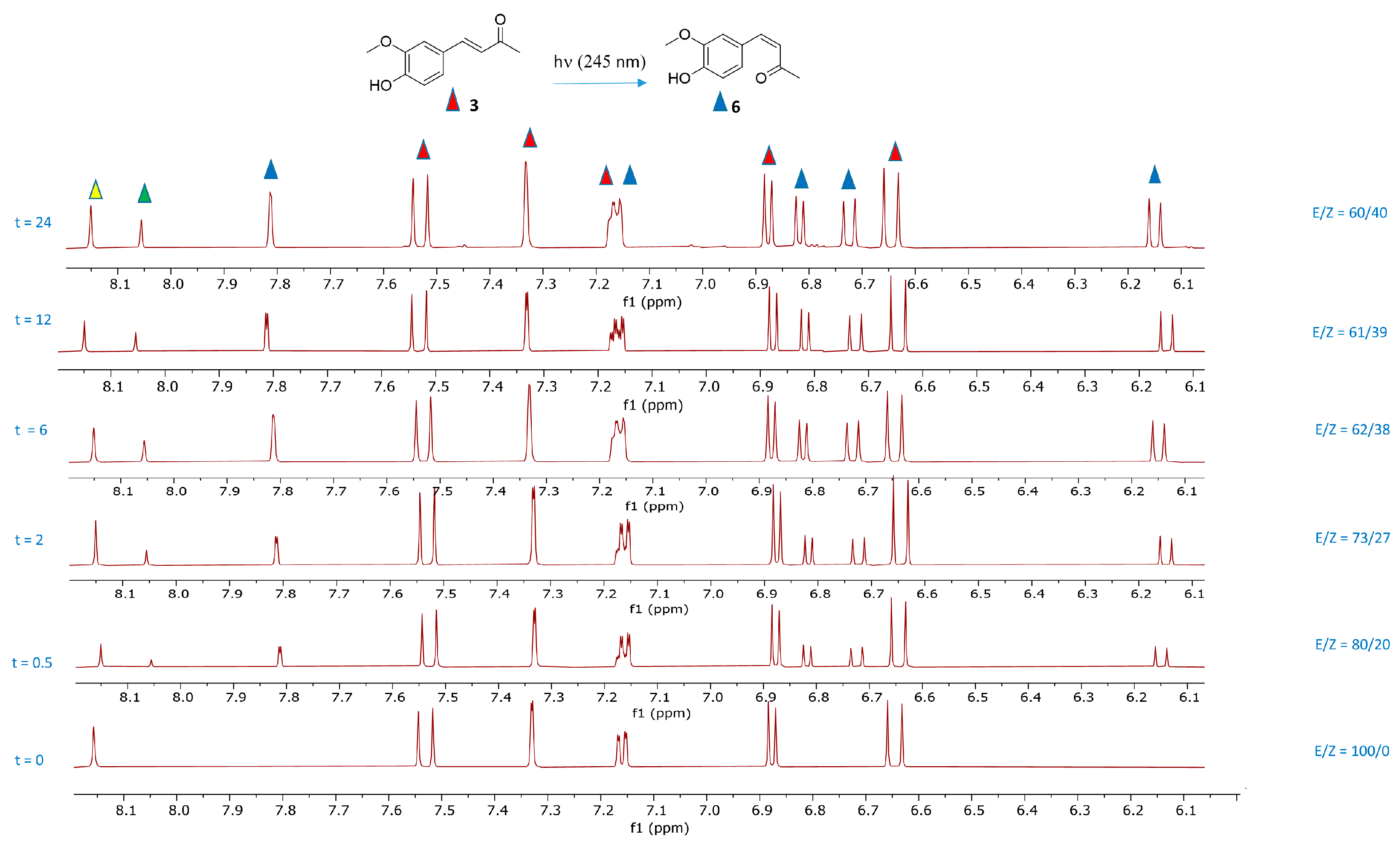
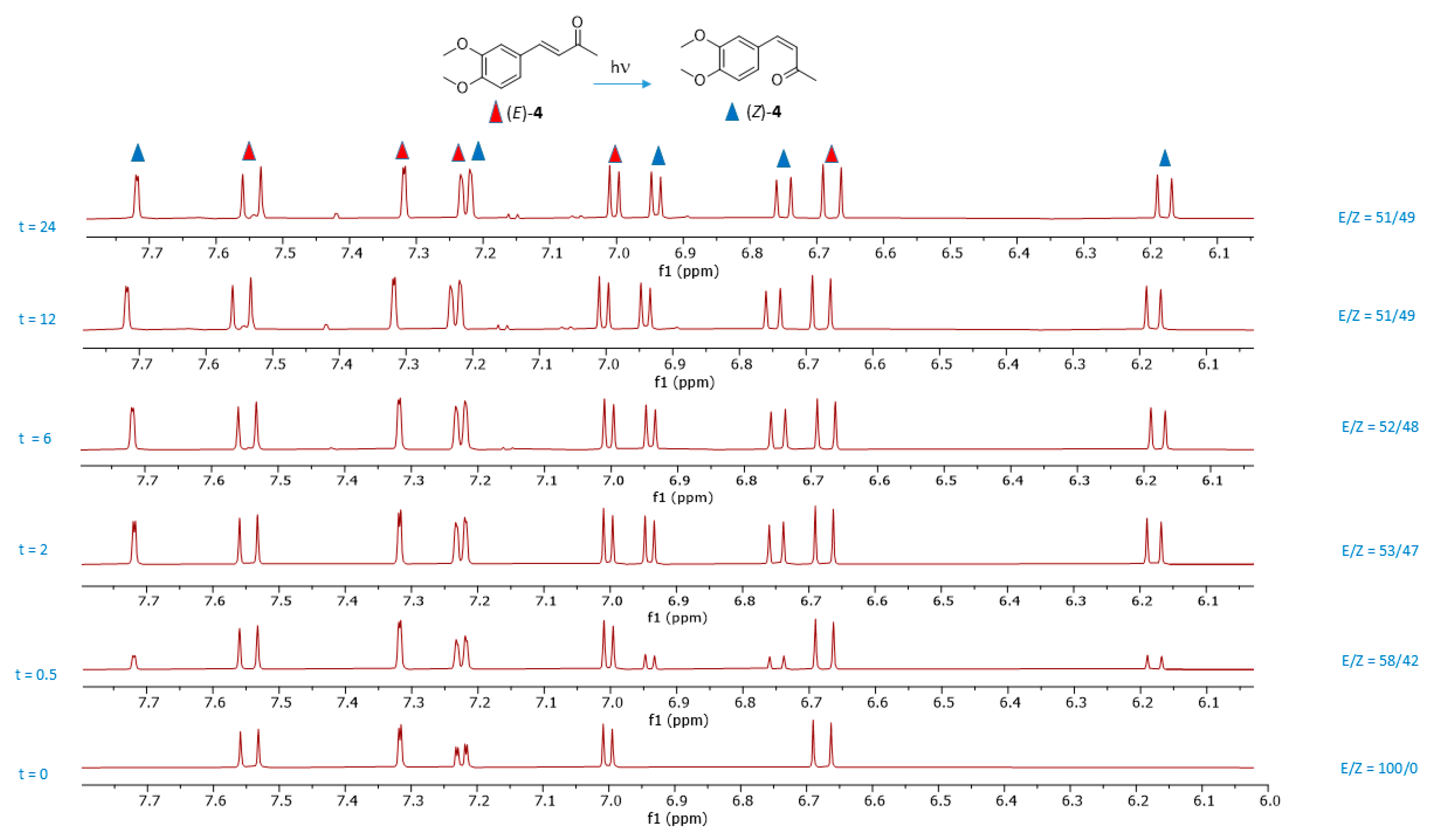
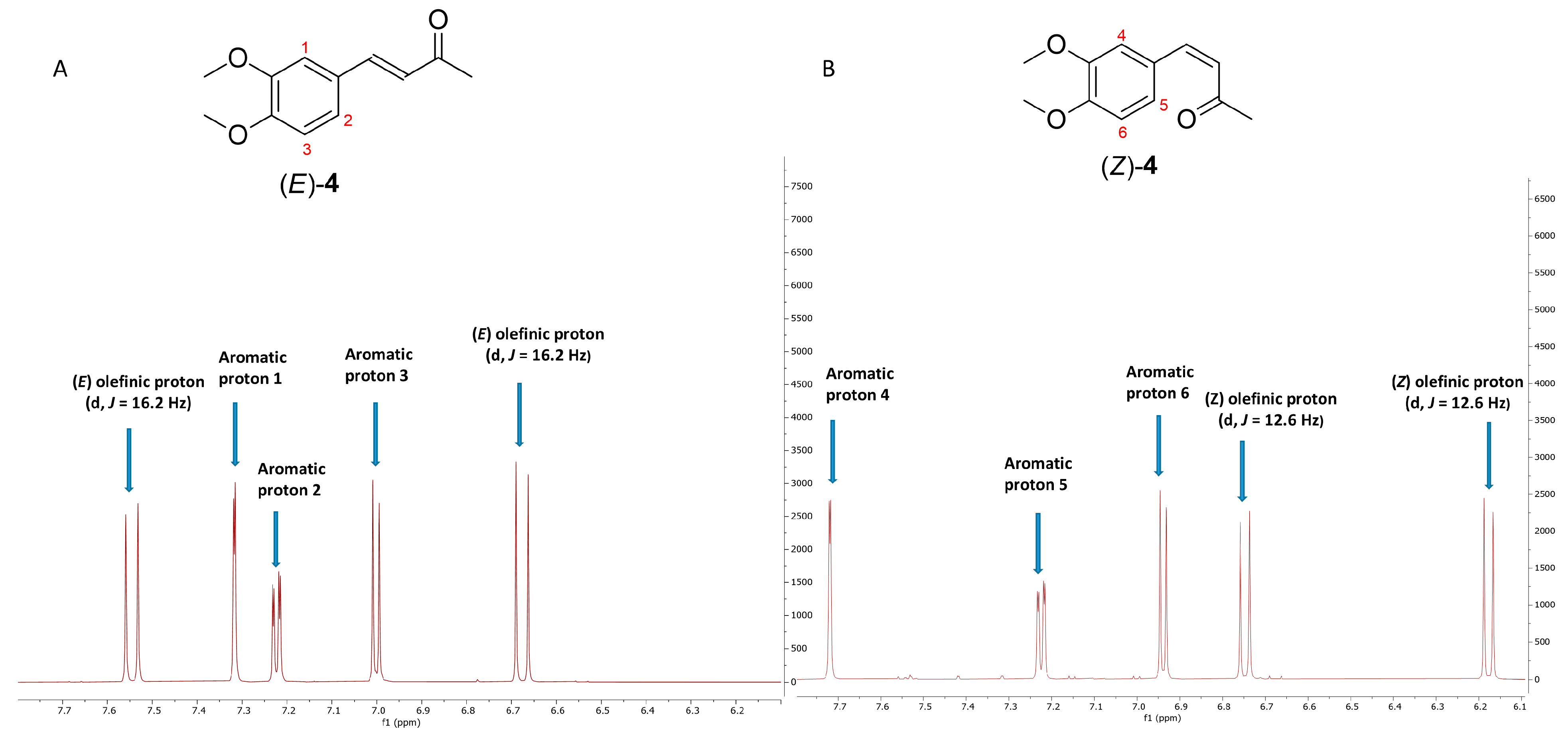
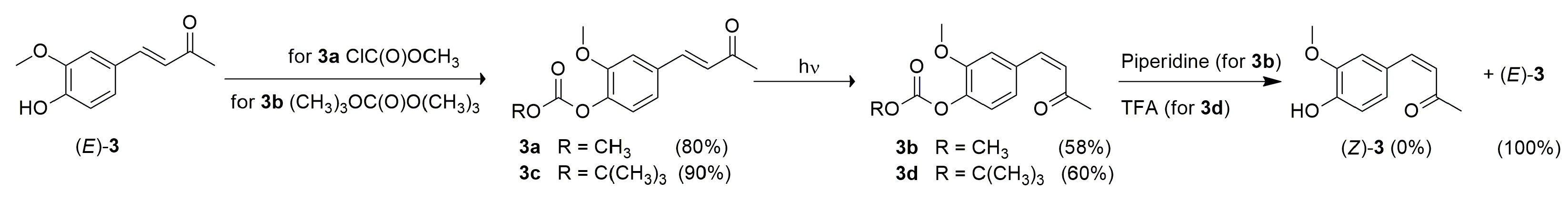
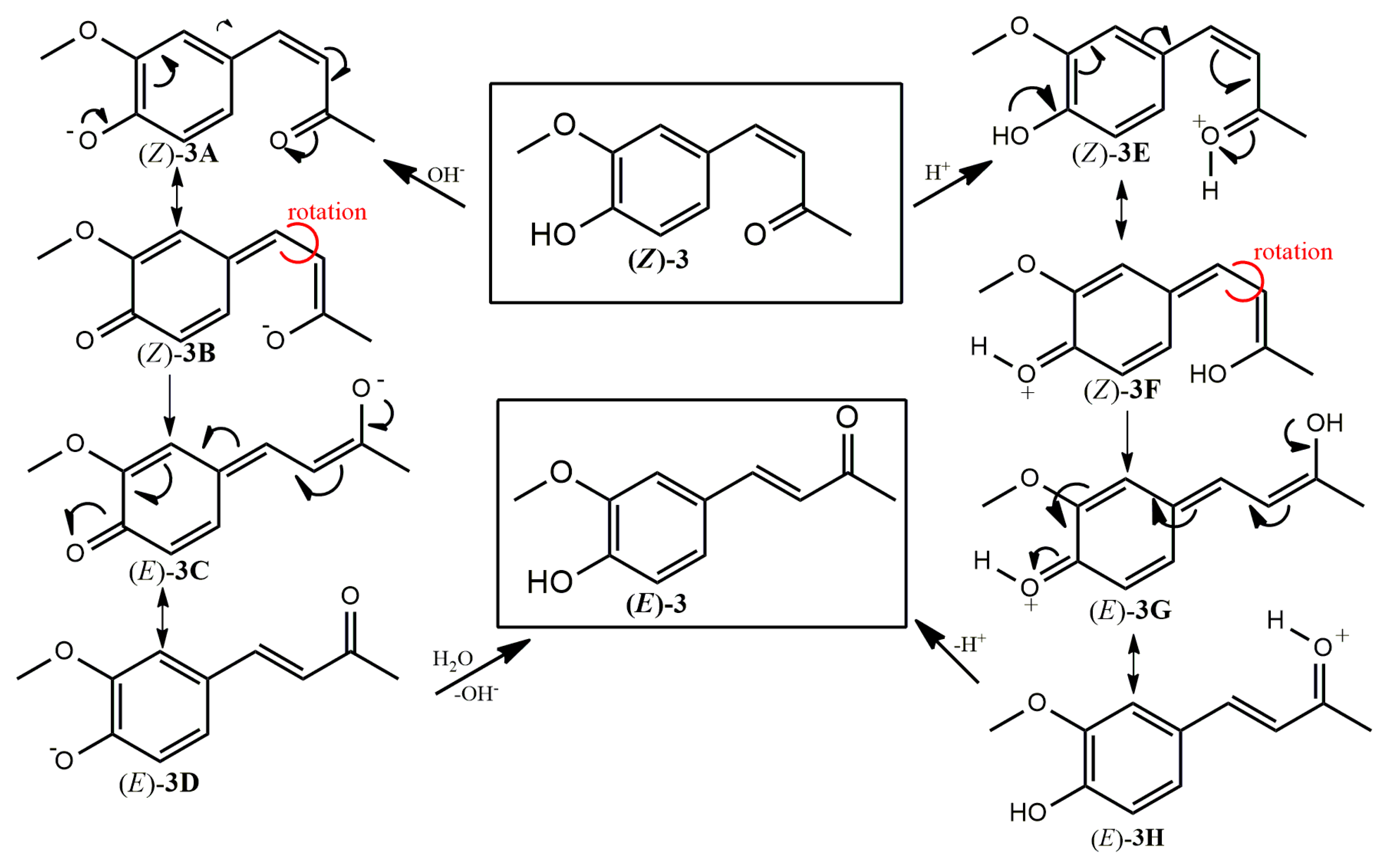
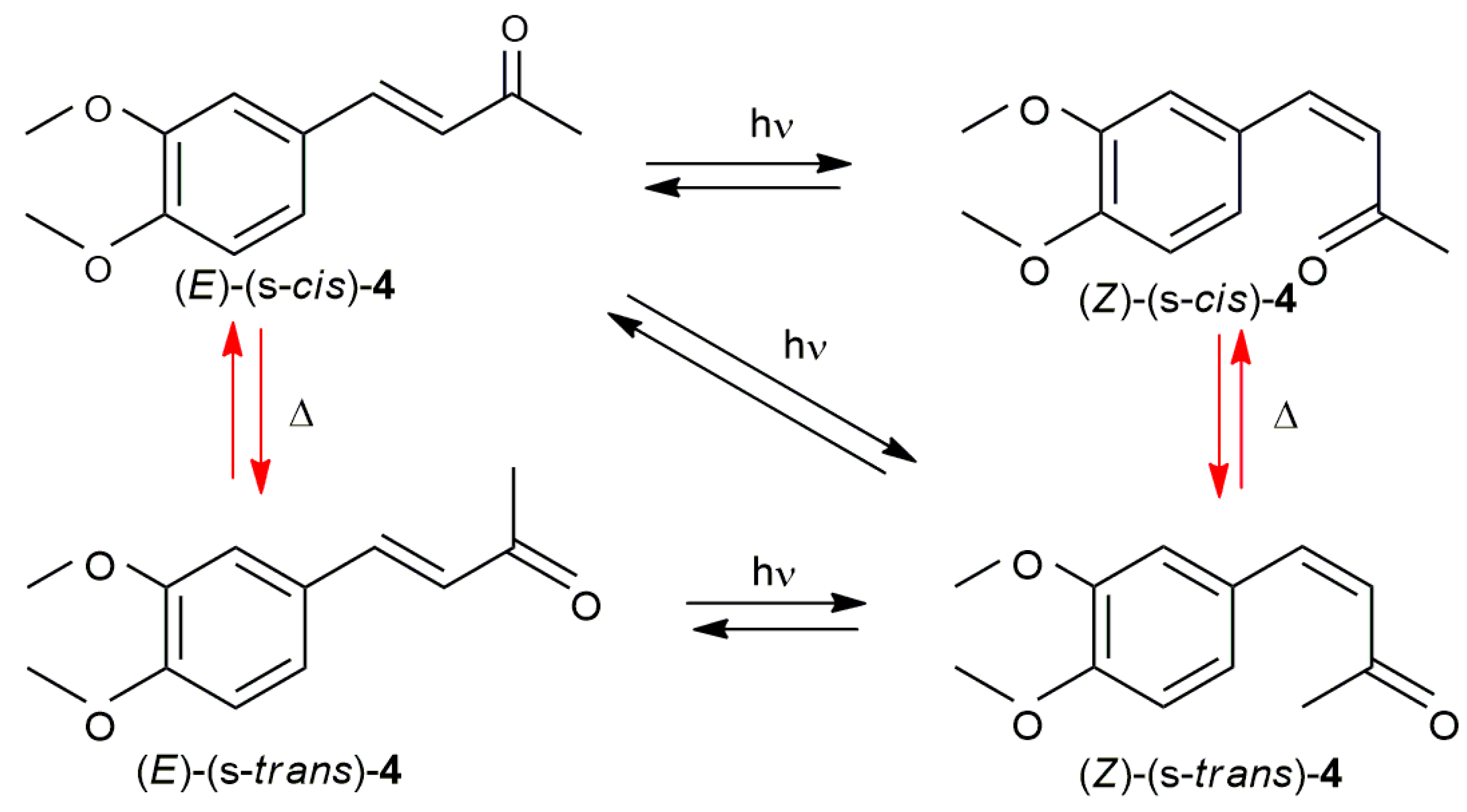
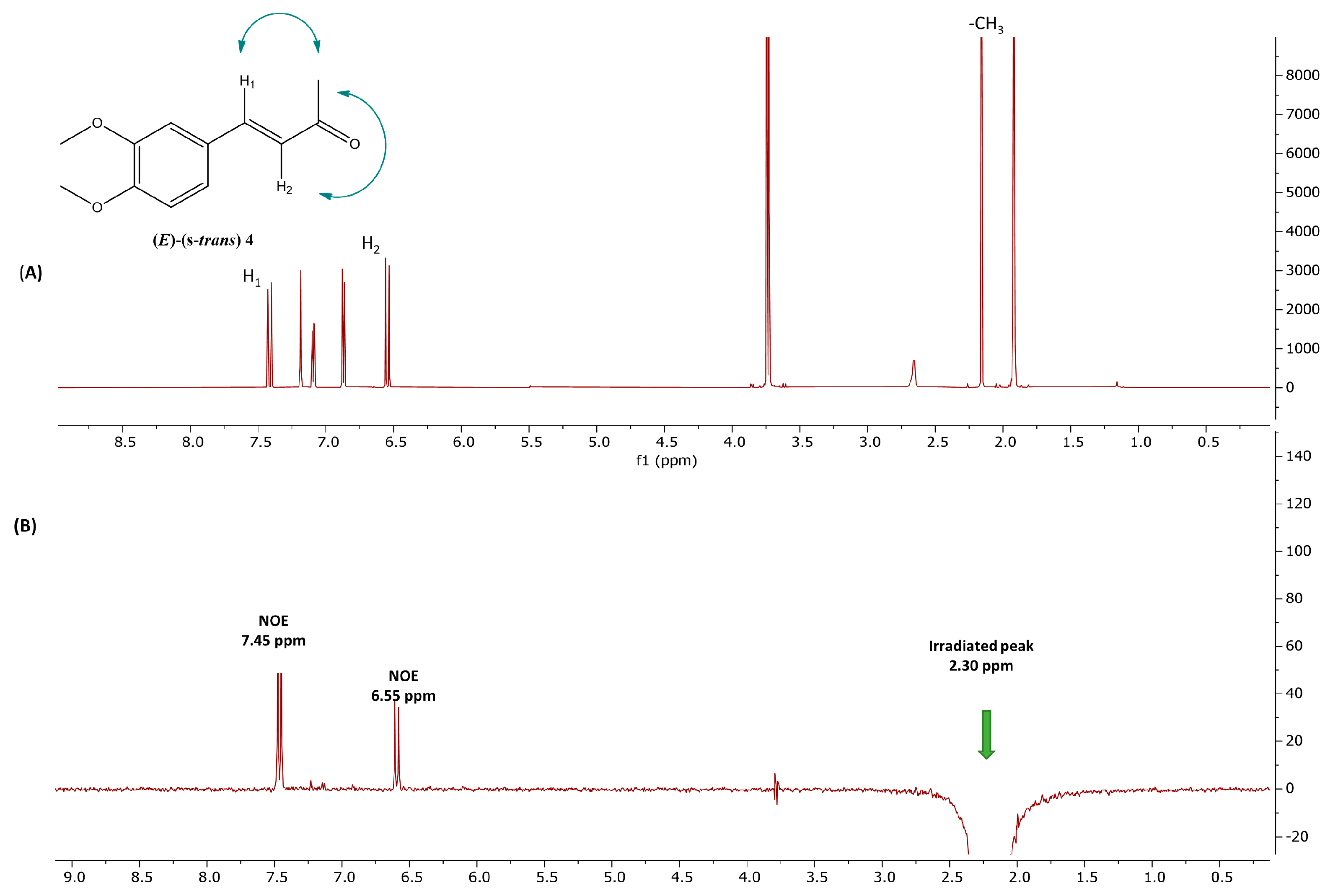
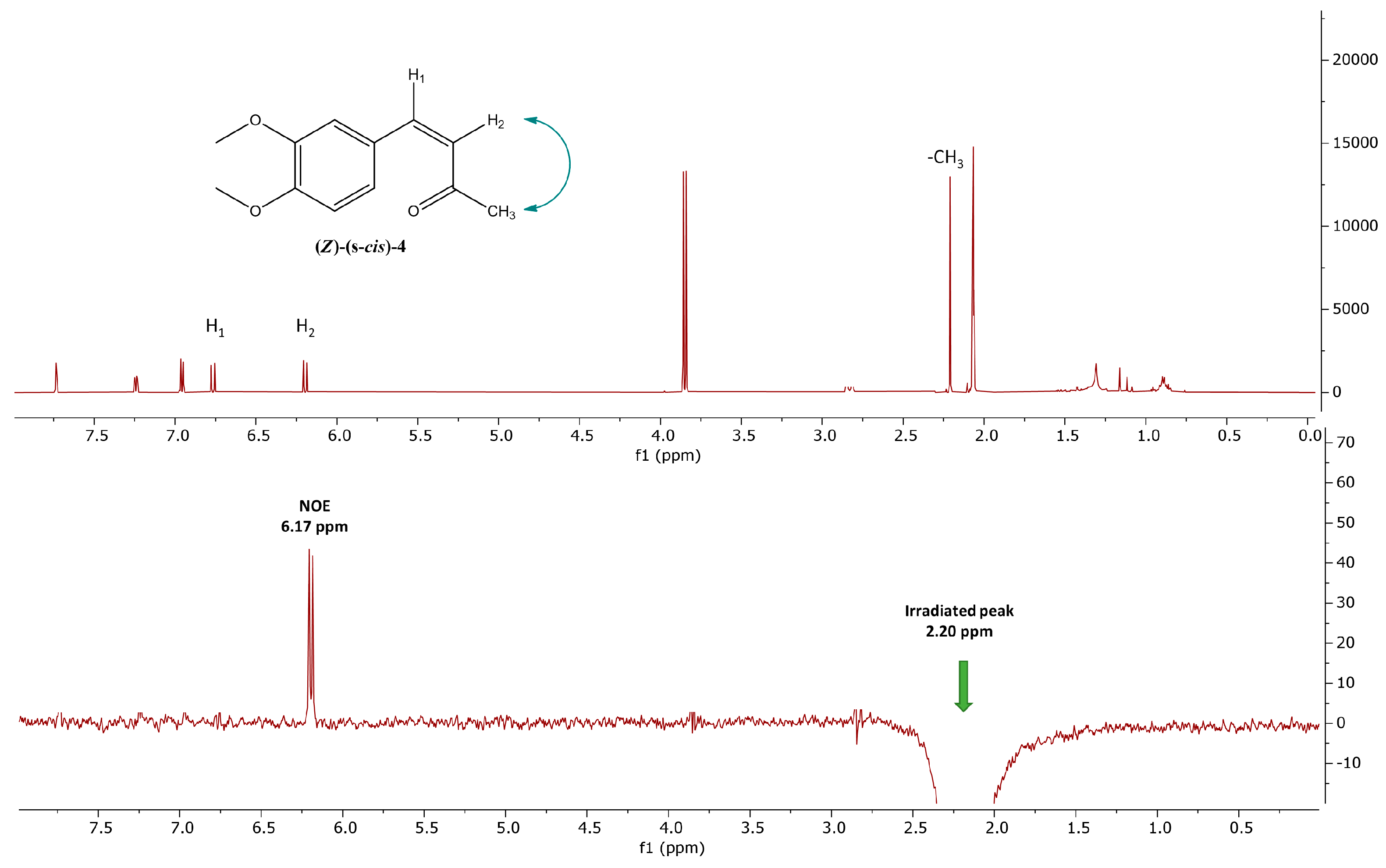
2.1. Photoisomerization Studies
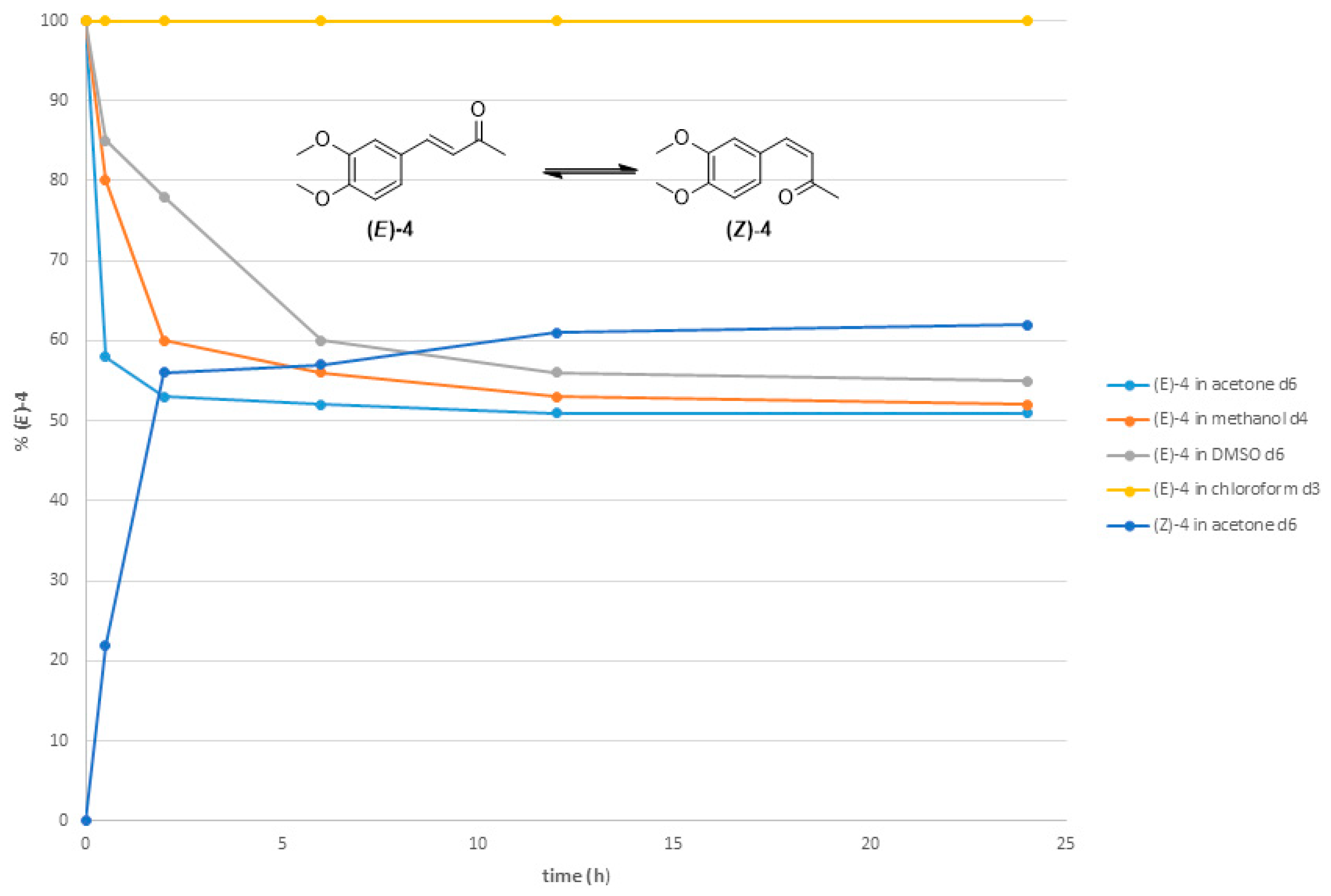
2.2. Thermal Stability of (Z) Isomers
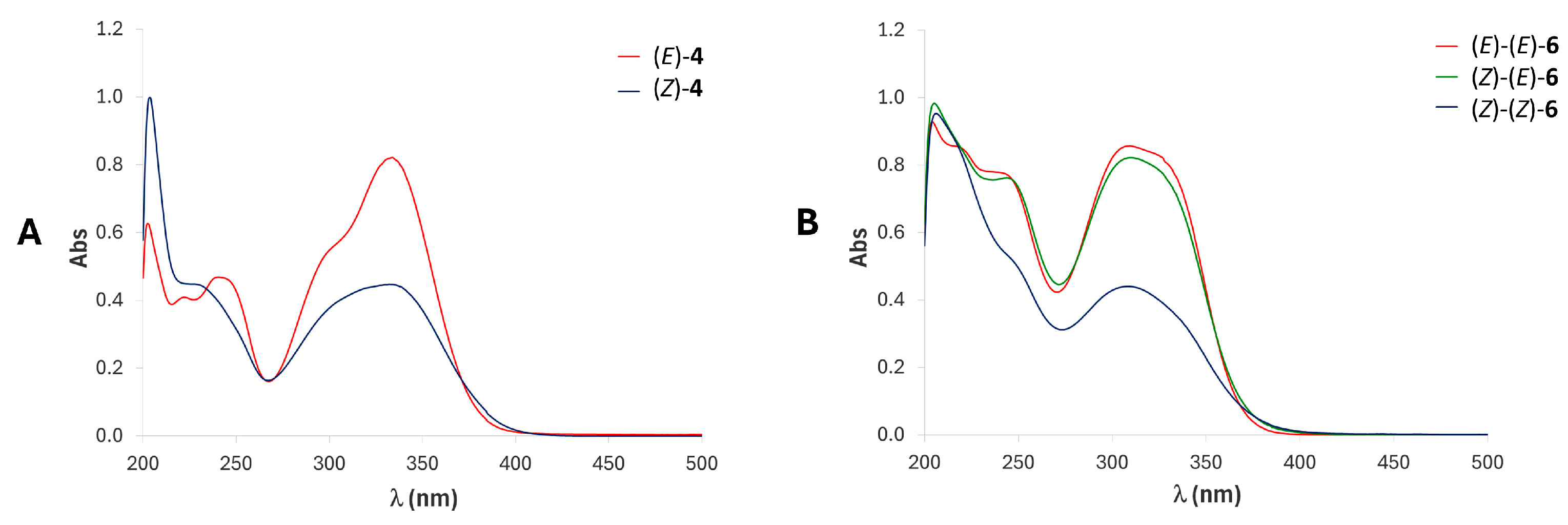
2.3. UV-Vis Studies
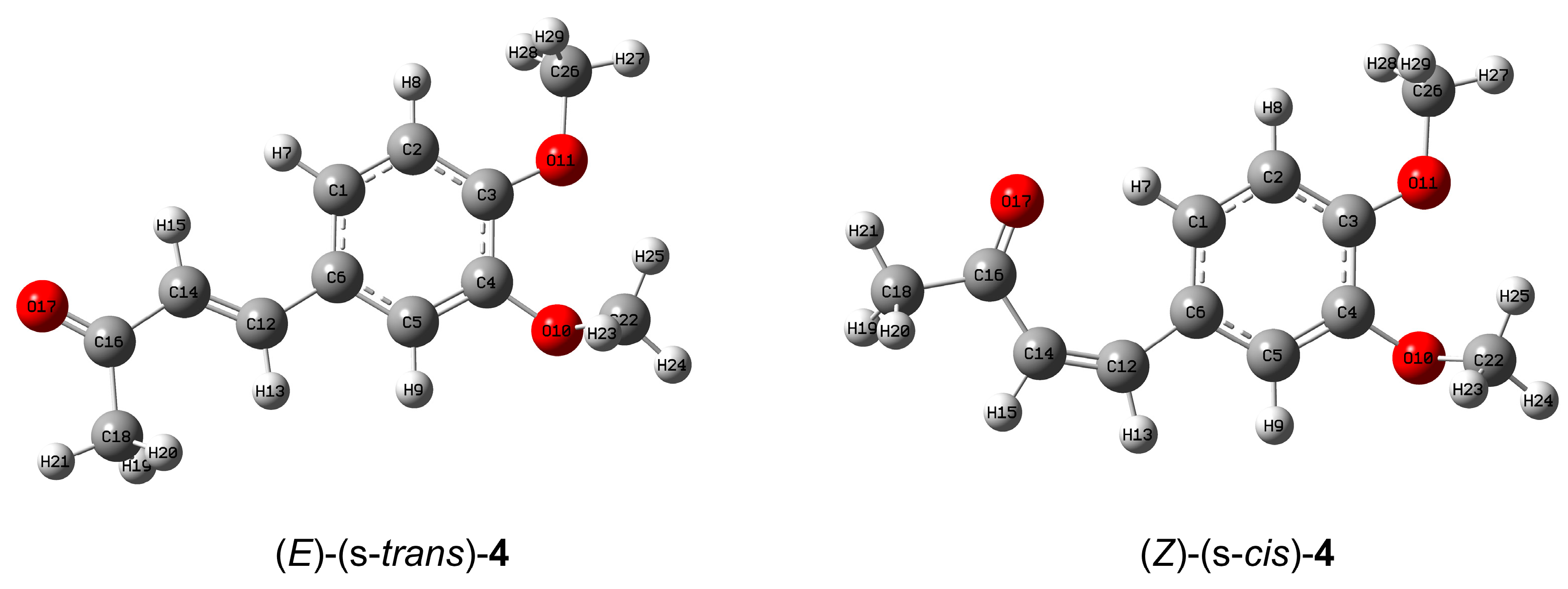
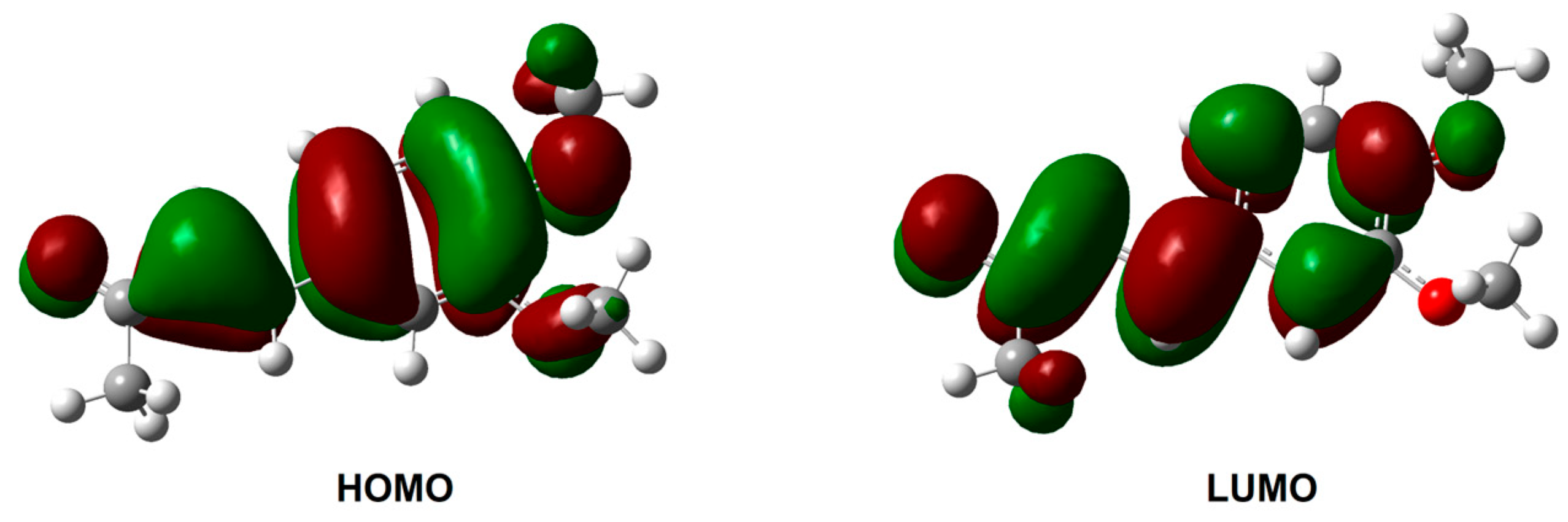
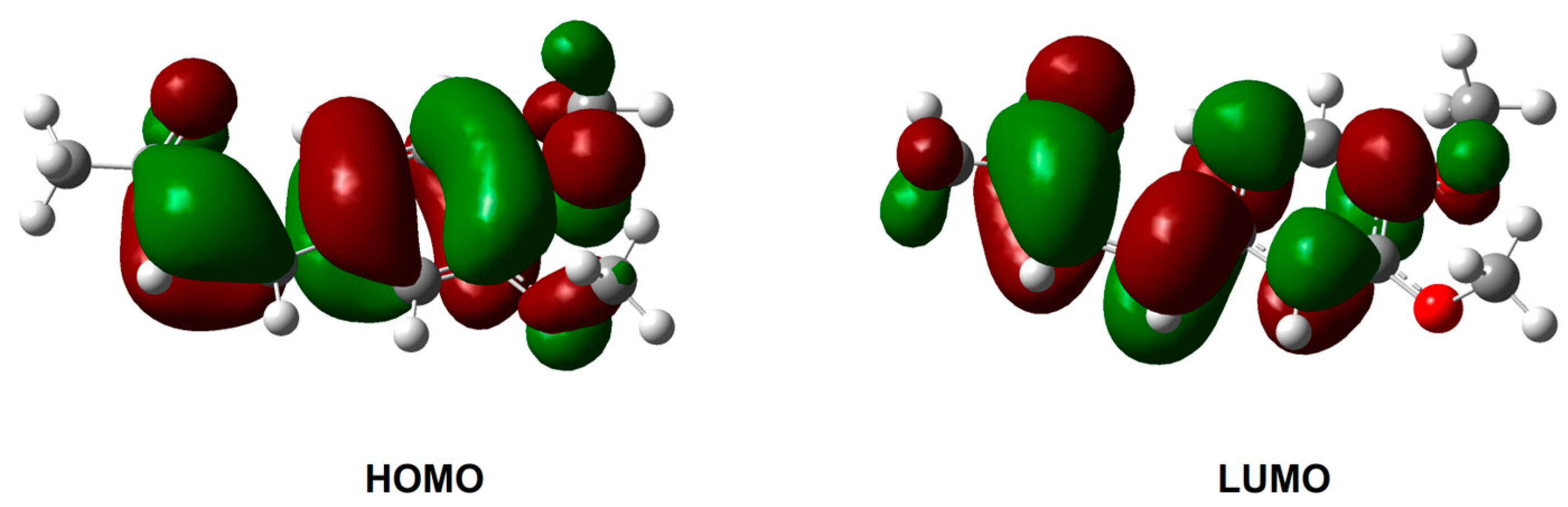
2.4. In Silico Investigation
3. Materials and Methods
3.1. General
3.2. General Procedure for Synthesis of (E)-2-Methoxy-4-(3-Oxobut-1-en-1-Yl) Phenyl Methyl Carbonate 3a and (E)-2-Methoxy-4-(3-Oxobut-1-en-1-Yl) Phenyl Tert-Butyl Carbonate 3c
3.3. General Procedure for Photoisomerization of Compounds (E)-3, 3a, 3c, and (E)-4
- (Z)-2-methoxy-4-(3-oxobut-1-en-1-yl) phenyl methyl carbonate 3b
- (Z)-2-methoxy-4-(3-oxobut-1-en-1-yl) phenyl tert-butyl carbonate 3d
- (Z)-4-(3,4-dimethoxyphenyl) but-3-ene-2-one (Z)-4
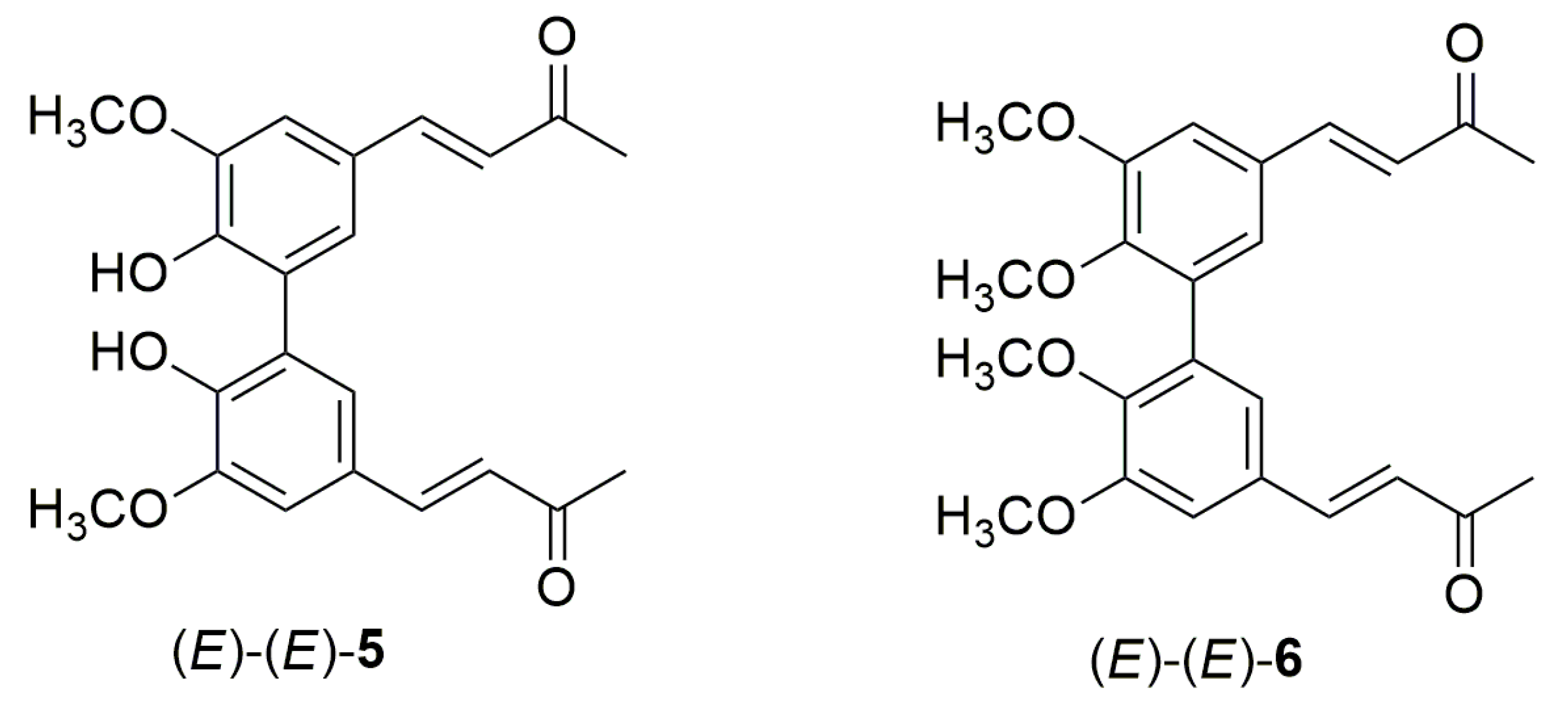
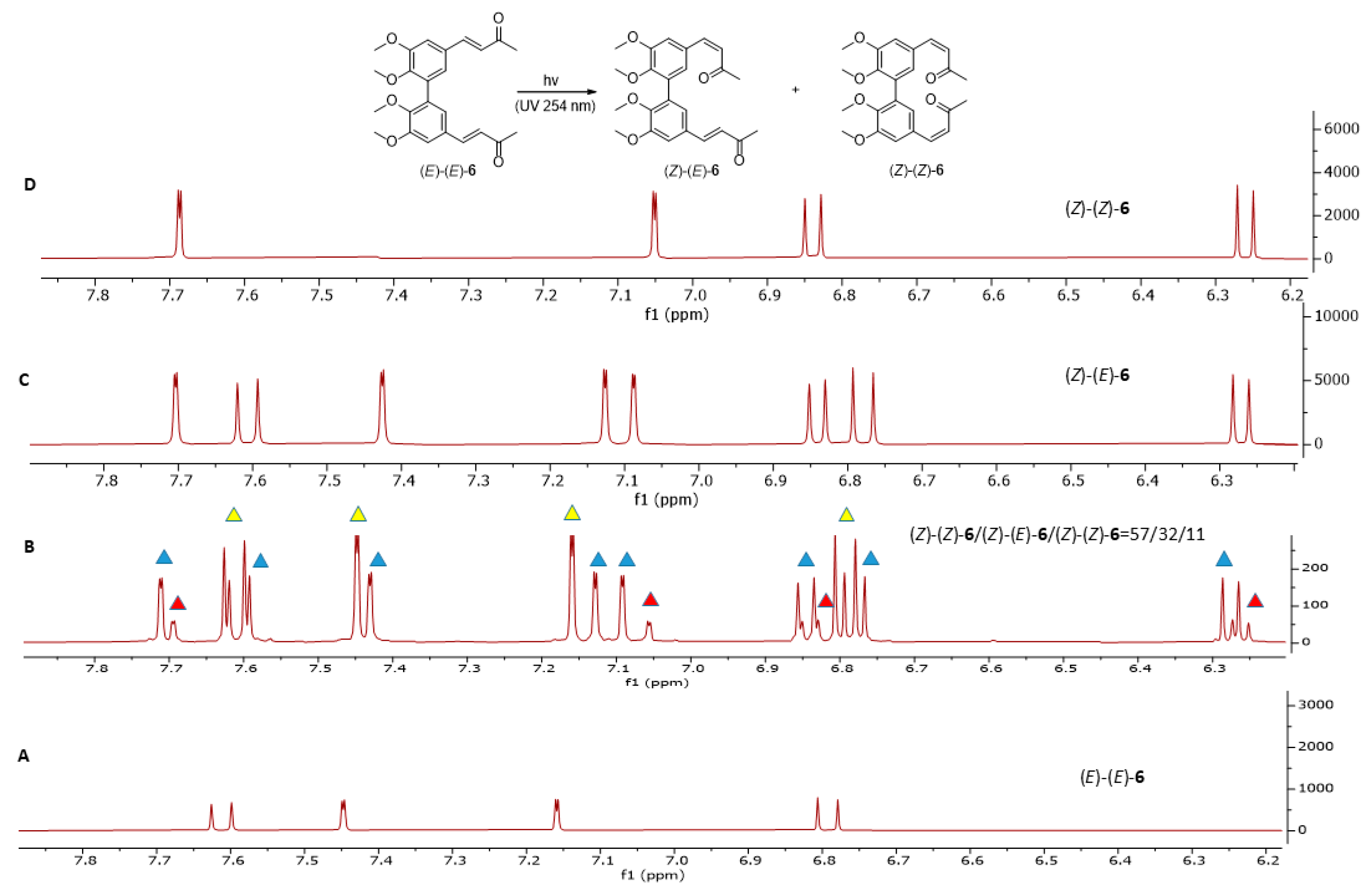
3.4. General Procedure for Photoisomerization of Compounds (E)-(E)-5 and (E)-(E)-6
- (3Z,3′E)-4,4′-(5,5′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(but-3-en-2-one) (Z)-(E)-6
- (3Z,3′Z)-4,4′-(5,5′,6,6′-tetramethoxy-[1,1′-biphenyl]-3,3′-diyl)bis(but-3-en-2-one) (Z)-(Z)-6
3.5. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yadav, N.; Deshmukh, R.; Mazumder, R. A Comprehensive Review on the Use of Traditional Chinese Medicine for Cancer Treatment. Pharmacol. Res. Mod. Chin. Med. 2024, 11, 100423. [Google Scholar] [CrossRef]
- Mahady, G.B.; Pendland, S.L.; Yun, G.; Lu, Z.Z. Turmeric (Curcuma Longa) and Curcumin Inhibit the Growth of Helicobacter Pylori, a Group 1 Carcinogen. Anticancer Res. 2002, 22, 4179–4181. [Google Scholar] [PubMed]
- Shehzad, A.; Rehman, G.; Lee, Y.S. Curcumin in Inflammatory Diseases. BioFactors 2013, 39, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Pyun, C.; Kim, J.; Han, K.; Hong, G.; Lee, C. In Vivo Protective Effects of Dietary Curcumin and Capsaicin against Alcohol-induced Oxidative Stress. BioFactors 2014, 40, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.R.; Parks, R.J. Curcumin as an Antiviral Agent. Viruses 2020, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Gordon, O.N.; Edwards, R.L.; Luis, P.B. Degradation of Curcumin: From Mechanism to Biological Implications. J. Agric. Food Chem. 2015, 63, 7606–7614. [Google Scholar] [CrossRef] [PubMed]
- Dettori, M.A.; Carta, P.; Fabbri, D. Environmentally Friendly One-Pot Two-Step Sequential Synthesis of Biological Active Curcumin Analogues. Tetrahedron 2024, 153, 133867. [Google Scholar] [CrossRef]
- Marchiani, A.; Mammi, S.; Siligardi, G.; Hussain, R.; Tessari, I.; Bubacco, L.; Delogu, G.; Fabbri, D.; Dettori, M.A.; Sanna, D.; et al. Small Molecules Interacting with Alpha-Synuclein: Antiaggregating and Cytoprotective Properties. Amino Acids 2013, 45, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Kancheva, V.; Slavova-Kazakova, A.; Fabbri, D.; Angelova, S.; Dettori, M.A.; Nechev, J.; Delogu, G. Antiradical and Antioxidant Activities of New Natural-like Hydroxylated Biphenyls of Dehydrozingerone, Zingerone and Ferulic acid. C. R. Acad. Bulg. Sci. 2013, 66, 361–368. [Google Scholar] [CrossRef]
- Rozzo, C.; Fanciulli, M.; Fraumene, C.; Corrias, A.; Cubeddu, T.; Sassu, I.; Cossu, S.; Nieddu, V.; Galleri, G.; Azara, E.; et al. Molecular Changes Induced by the Curcumin Analogue D6 in Human Melanoma Cells. Mol. Cancer 2013, 12, 37. [Google Scholar] [CrossRef]
- Neveselý, T.; Wienhold, M.; Molloy, J.J.; Gilmour, R. Advances in the E → Z Isomerization of Alkenes Using Small Molecule Photocatalysts. Chem. Rev. 2022, 122, 2650–2694. [Google Scholar] [CrossRef] [PubMed]
- Zähringer, T.J.B.; Wienhold, M.; Gilmour, R.; Kerzig, C. Direct Observation of Triplet States in the Isomerization of Alkenylboronates by Energy Transfer Catalysis. J. Am. Chem. Soc. 2023, 145, 21576–21586. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic Acid: Therapeutic Potential Through Its Antioxidant Property. J. Clin. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Ramphinwa, M.L.; Madala, N.E.; Mchau, G.R.A.; Ramabulana, A.T.; Mudau, F.N. Effect of UV-Induced Geometrical Isomerization of Hydroxyl-Cinnamic Acid-Containing Molecules of Bush Tea (Athrixia Phylicoides DC.) Using UHPLC-QTOF-MS. Sci. Hortic. 2022, 301, 111124. [Google Scholar] [CrossRef]
- Clifford, M.N.; Kirkpatrick, J.; Kuhnert, N.; Roozendaal, H.; Salgado, P.R. LC–MSn Analysis of the Cis Isomers of Chlorogenic Acids. Food Chem. 2008, 106, 379–385. [Google Scholar] [CrossRef]
- Wong, W.S.; Guo, D.; Wang, X.L.; Yin, Z.Q.; Xia, B.; Li, N. Study of Cis-Cinnamic Acid in Arabidopsis thaliana. Plant Physiol. Biochem. 2005, 43, 929–937. [Google Scholar] [CrossRef]
- Song, K.-K.; Huang, H.; Han, P.; Zhang, C.-L.; Shi, Y.; Chen, Q.-X. Inhibitory Effects of Cis- and Trans-Isomers of 3,5-Dihydroxystilbene on the Activity of Mushroom Tyrosinase. Biochem. Biophys. Res. Commun. 2006, 342, 1147–1151. [Google Scholar] [CrossRef]
- Moni, L.; Banfi, L.; Basso, A.; Mori, A.; Risso, F.; Riva, R.; Lambruschini, C. A Thorough Study on the Photoisomerization of Ferulic Acid Derivatives. Eur. J. Org. Chem. 2021, 2021, 1737–1749. [Google Scholar] [CrossRef]
- Ward, R.S. Lignans, Neolignans and Related Compounds. Nat. Prod. Rep. 1999, 16, 75–96. [Google Scholar] [CrossRef]
- Singh, S.; Geetha, P.; Ramajayam, R. Isolation, Synthesis and Medicinal Chemistry of Biphenyl Analogs—A Review. Results Chem. 2023, 6, 101135. [Google Scholar] [CrossRef]
- Pisano, M.; Pagnan, G.; Dettori, M.A.; Cossu, S.; Caffa, I.; Sassu, I.; Emionite, L.; Fabbri, D.; Cilli, M.; Pastorino, F.; et al. Enhanced Anti-Tumor Activity of a New Curcumin-Related Compound against Melanoma and Neuroblastoma Cells. Mol. Cancer 2010, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Obregón-Mendoza, M.A.; Arias-Olguín, I.I.; Meza-Morales, W.; Alvarez-Ricardo, Y.; Chávez, M.I.; Toscano, R.A.; Cassani, J.; Enríquez, R.G. Expected and Unexpected Products in Half Curcuminoid Synthesis: Crystal Structures of But-3-En-2-Ones and 3-Methylcyclohex-2-Enones. Crystals 2021, 11, 404. [Google Scholar] [CrossRef]
- Park, S.; Kim, H.; Bang, M.; Um, B.-H.; Cha, J.W. A Study on the Photoisomerization of Phenylpropanoids and the Differences in Their Radical Scavenging Activity Using In-Situ NMR Spectroscopy and on-Line Radical Scavenging Activity Analysis. Appl. Biol. Chem. 2024, 67, 69. [Google Scholar] [CrossRef]
- Bisceglia, J.A.; Orelli, L.R. Recent Progress in the Horner-Wadsworth-Emmons Reaction. COC 2015, 19, 744–775. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dettori, M.A.; Ugone, V.; Fabbri, D.; Carta, P. A Study on the Photoisomerization of (E)-Dehydrozingerone, Its (E)-(E)-C₂ Symmetric Dimer, and Their O-Methylated Derivatives. Molecules 2024, 29, 5901. https://doi.org/10.3390/molecules29245901
Dettori MA, Ugone V, Fabbri D, Carta P. A Study on the Photoisomerization of (E)-Dehydrozingerone, Its (E)-(E)-C₂ Symmetric Dimer, and Their O-Methylated Derivatives. Molecules. 2024; 29(24):5901. https://doi.org/10.3390/molecules29245901
Chicago/Turabian StyleDettori, Maria Antonietta, Valeria Ugone, Davide Fabbri, and Paola Carta. 2024. "A Study on the Photoisomerization of (E)-Dehydrozingerone, Its (E)-(E)-C₂ Symmetric Dimer, and Their O-Methylated Derivatives" Molecules 29, no. 24: 5901. https://doi.org/10.3390/molecules29245901
APA StyleDettori, M. A., Ugone, V., Fabbri, D., & Carta, P. (2024). A Study on the Photoisomerization of (E)-Dehydrozingerone, Its (E)-(E)-C₂ Symmetric Dimer, and Their O-Methylated Derivatives. Molecules, 29(24), 5901. https://doi.org/10.3390/molecules29245901