
Doubly Metathetic NiCl2-Catalyzed Coupling Between Bis(2-oxazolines) and Aldehydes: A Novel Access to Bis(ester-imine) Derivatives

 , , ,
, , ,  , , ,
, , ,  , and
, and
Abstract

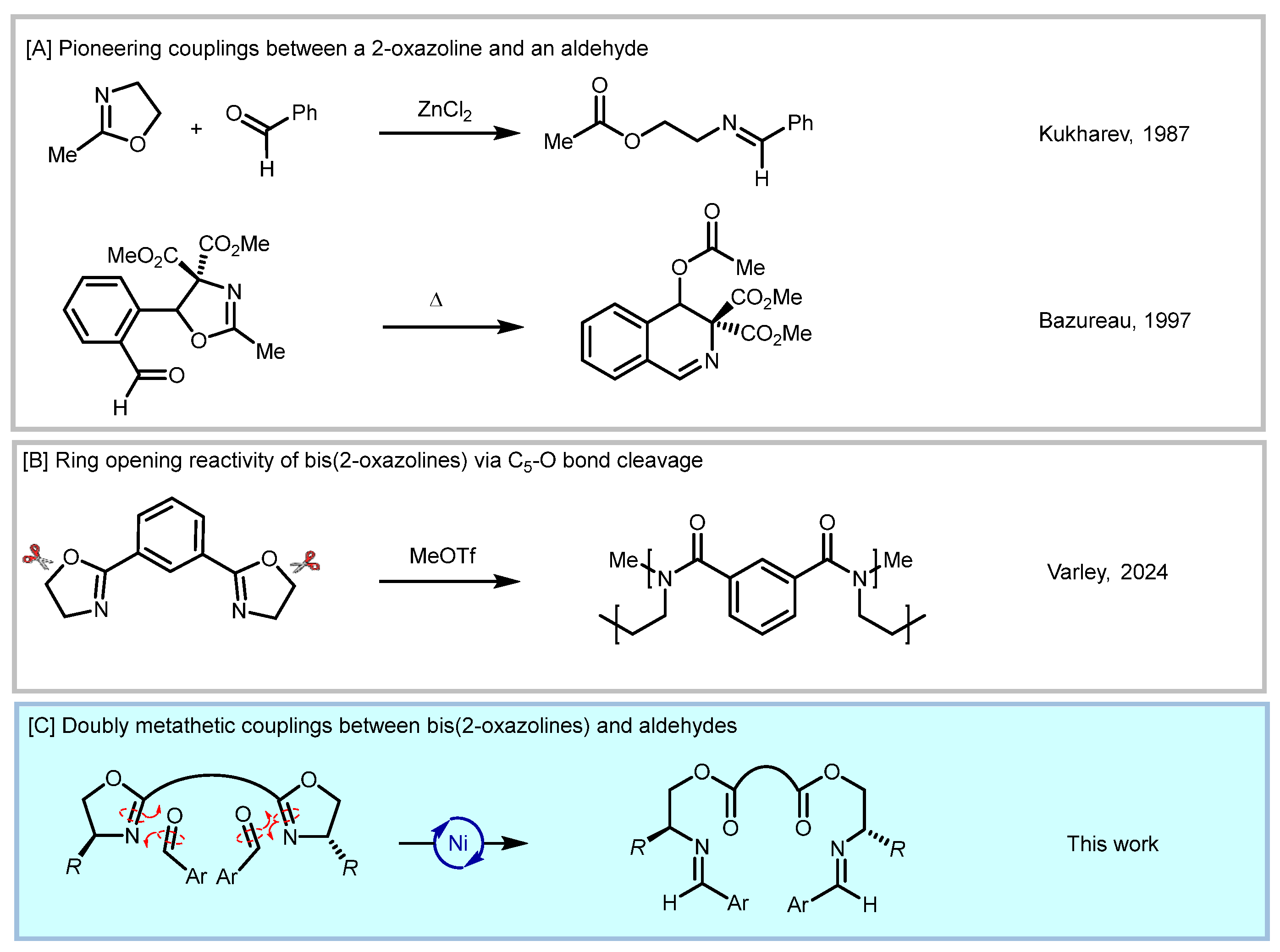
1. Introduction
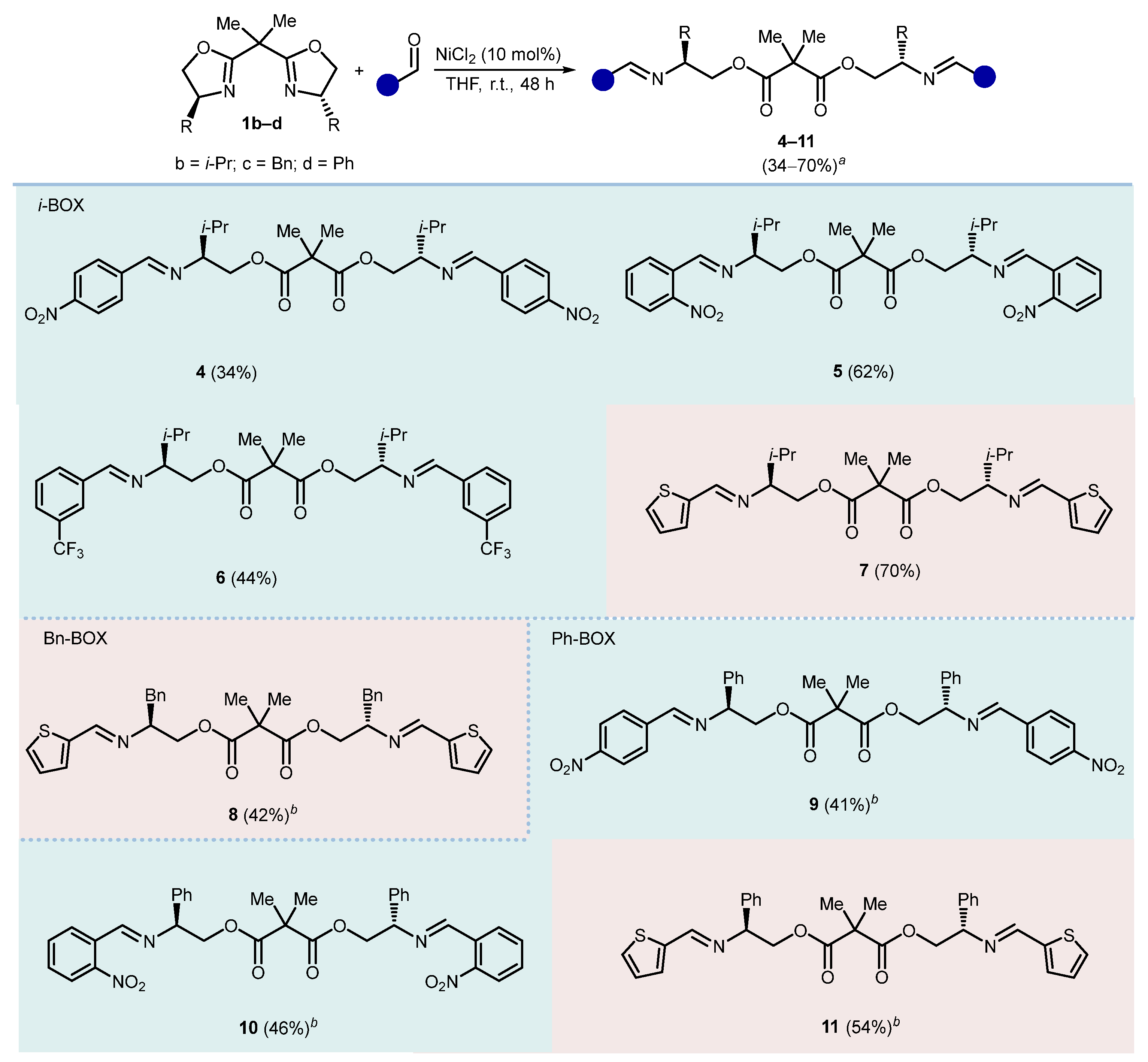
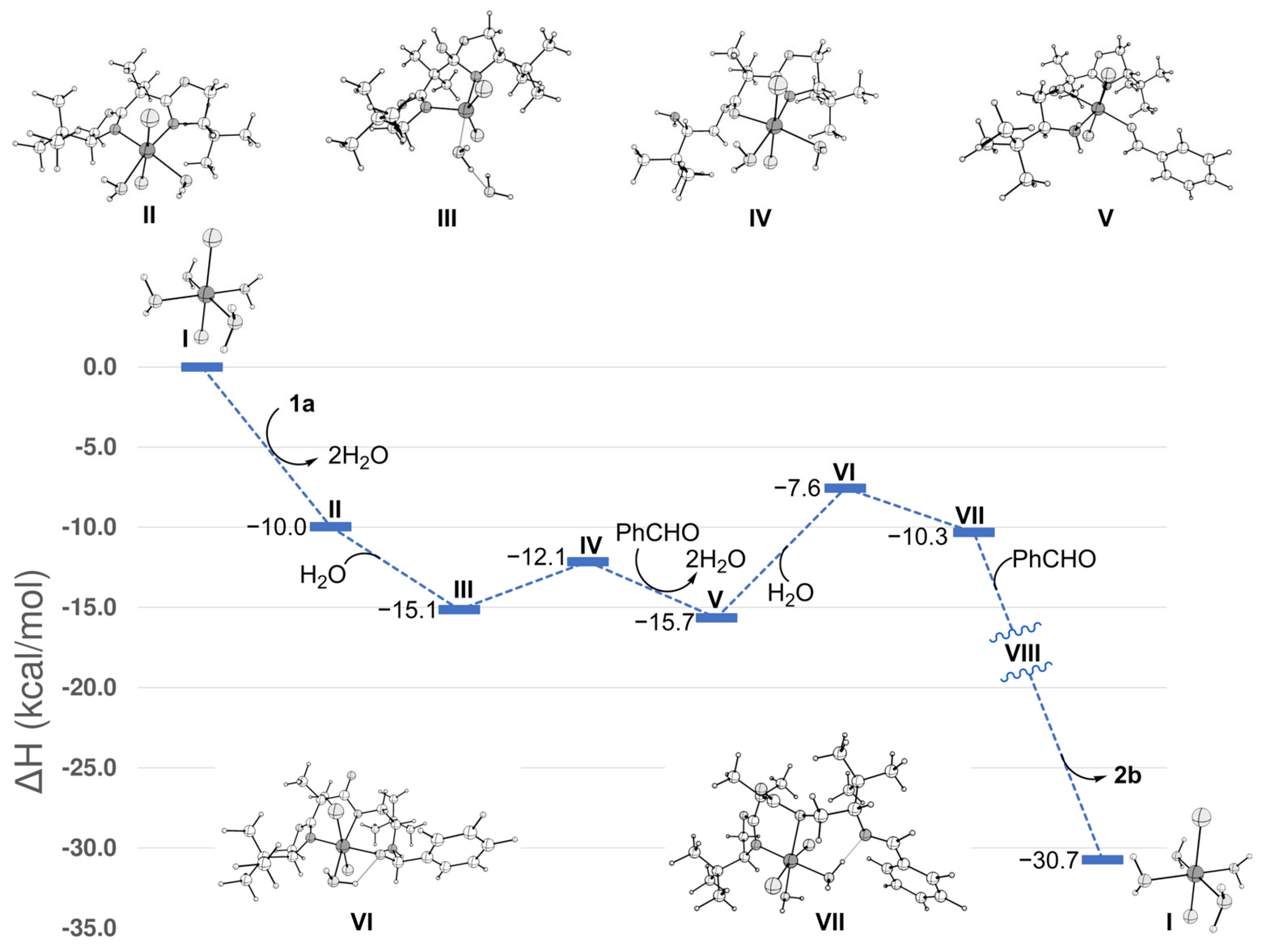
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Experimental Procedures
3.2.1. Experimental Procedure for Compounds 2a–k, 4–13, and 16
3.2.2. Experimental Procedure for Compound 15
3.3. Characterization Data of Synthesized Compounds
Characterization Data of Final Compounds 2a–k, 3–13 and 15–16
3.4. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dargaville, T.R.; Park, J.R.; Hoodenboom, R. Poly(2-oxazoline) hydrogels: State-of-the-art and emerging applications. Macromol. Biosci. 2018, 18, 1800070. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Onda, Y.; Masuda, Y.; Doi, T. Potent oxazoline analog of Apratoxin C; synthesis, biological evaluation, and conformational analysis. PeptideScience 2016, 106, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.N.; Beveridge, R.E.; Blay, J.; Boyd, A.R.; Chojnacka, M.W.; Decken, A.; Deshpande, A.A.; Gardiner, M.G.; Hambley, T.W.; Hughes, M.J.; et al. Platinum-oxazole complexes as anti-cancer agents: Syntheses, characterization and initial biological studies. Med. Chem. Commun. 2011, 2, 274–277. [Google Scholar] [CrossRef]
- Connon, R.; Roche, B.; Rokadem, B.V.; Guiry, P.J. Further developments and application of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev. 2021, 121, 6373–6521. [Google Scholar] [CrossRef]
- McManus, A.H.; Guiry, P.J. Recent developments in the application of oxazoline-containing ligands in asymmetric catalysis. Chem. Rev. 2004, 104, 4151–4202. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Muller, G.; Rocamora, M. Coordination chemistry of oxazoline ligands. Coord. Chem. Rev. 1999, 193–195, 769–835. [Google Scholar] [CrossRef]
- Desimoni, G.; Faita, G.; Jørgensen, K.A. C2-Symmetric Chiral Bis (Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2006, 106, 3561–3651. [Google Scholar] [CrossRef]
- Deng, S.K.; Chen, H.C.; Liu, Y.-F.; Ji, H.T.; Shen, J. Rin-opening phosphanylation of oxazolines. Tetrahedron Lett. 2024, 134, 154866. [Google Scholar] [CrossRef]
- Hess, A.; Guelen, H.C.; Alandini, N.; Mourati, A.; Guersoy, Y.C.; Knochel, P. Preparation of polyfunctionalized aromatic nitriles from aryl oxazolines. Chem. Eur. J. 2022, 28, e202103700. [Google Scholar] [CrossRef]
- Lin, Q.; Zhang, S.; Li, B. KOt-Bu-Promoted selective ring-opening N-alkylation of 2-oxazolines to access 2-aminoethyl acetates and N-substituted thiazolidinones. Beilstein J. Org. Chem. 2020, 16, 492–501. [Google Scholar] [CrossRef]
- Yang, Z.; Jie, L.; Yao, Z.; Yang, Z.; Cui, X. Rhodium (III)-catalyzed synthesis of N-(2-acetoxyalkyl) isoquinolones from oxazolines and alkynes through C-N bond formation and ring-opening. Adv. Synth. Catal. 2019, 361, 214–218. [Google Scholar] [CrossRef]
- Guan, D.; Luan, H.X.; Patiguli, M.; Jiao, Q.J.; Yun, Q.Q.; Chen, Q.S.; Xu, C.J.; Nie, X.B.; Hu, F.P.; Huang, G.S. Metal–free Efficient Method for the Synthesis of N-(2-haloethyl)benzamides through the Ring-opening of 2-oxazolines. ChemistrySelect 2019, 4, 6668–6671. [Google Scholar] [CrossRef]
- Qiao, K.; Yuan, X.; Wan, L.; Zheng, M.-W.; Zhang, D.; Fan, B.B.; Di, Z.C.; Fang, Z.; Guo, K. Highly efficient synthesis of β-nitrate ester carboxamides through the ring-opening of 2-oxazolines. Green Chem. 2017, 19, 5789–5793. [Google Scholar] [CrossRef]
- Gutmann, B.; Roduit, J.P.; Roberge, D.; Kappe, C.O. A two-step continuous-flow synthesis of N-(2-aminoethyl) acylamides through ring-opening/hydrogenation of oxazolines. Chem. Eur. J. 2011, 17, 13146–13150. [Google Scholar] [CrossRef]
- Laitar, D.S.; Kramer, J.W.; Whiting, B.T.; Lobkovsky, E.B.; Coates, G.W. β-Amidoaldehydes via oxazoline hydroformylation. Chem. Commun. 2009, 14, 5704–5706. [Google Scholar] [CrossRef]
- Kukharev, B.F.; Stankevich, V.K.; Terent’eva, V.P.; Kukhareva, V.A. Reaction of oxazolines with aldehydes. Zh. Org. Khim. 1987, 23, 1336–1337. [Google Scholar]
- Lerestif, J.M.; Toupet, L.; Sindandhit, S.; Tonnard, F.; Bazureau, J.P.; Hamelin, J.A. New route to 2-oxazolines, bis-oxazolines, and 2-imidazoline-5-ones from imidates using solvent-free cycloadditions: Synthesis, chemical properties, and PM3 MO calculations. Tetrahedron 1997, 53, 6351–6364. [Google Scholar] [CrossRef]
- Maguet, M.; Poirier, Y.; Guglielmetti, R. Spiropyrannes et mérocyanines en séries azahétérocycliques saturées. Bull. Soc. Chem. Fr. 1978, 11–12. [Google Scholar]
- Lerestif, J.M.; Feuillet, S.; Bazureau, J.P.; Hamelin, J. Novel Synthesis of Protected Methyl 4-Hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylate via Cleavage of Functionalized Dihydrooxazoles (Oxazolines). J. Chem. Res. 1999, 1, 32–33. [Google Scholar] [CrossRef]
- Molteni, L.; Beccalli, E.M.; Castoldi, L.; Broggini, G.; Loro, C. Methanol as a C1 Source for the Synthesis of 1,3-Polyheterocyclic Systems. Eur. J. Org. Chem. 2023, 26, e202301106. [Google Scholar] [CrossRef]
- Loro, C.; Molteni, L.; Lo Presti, L.; Foschi, F.; Beccalli, E.M.; Broggini, G. Non-decarboxylative ruthenium-catalyzed rearrangement of 4-alkylidene-isoxazol-5-ones to pyrazole- and isoxazole-4-carboxylic acids. Org. Lett. 2022, 24, 3092–3096. [Google Scholar] [CrossRef] [PubMed]
- Molteni, L.; Loro, C.; Christodoulou, M.S.; Papis, M.; Foschi, F.; Beccalli, E.M.; Broggini, G. Ruthenium-catalyzed decarboxylative rearrangement of 4-alkenyl-isoxazol-5-ones to pyrrole derivatives. Eur. J. Org. Chem. 2022, 2022, e202200496. [Google Scholar] [CrossRef]
- Issazadeh, S.; Khan, M.J.; Gan, H.; Zhang, J.; Henderson, L.C.; Varley, R.J. Synthesis and cure kinetics of bisoxazoline monomers during cationic ring-opening polymerization. J. Appl. Polym. Sci. 2024, 141, e54900. [Google Scholar] [CrossRef]
- Taylan, E.; Küsefoğlu, S.H. Chain Extension Reactions of Unsaturated Polyesters with Bis(2-Oxazoline)s. J. Appl. Polym. Sci. 2012, 124, 3229. [Google Scholar] [CrossRef]
- Fry, E.M. Oxazolidine ring-opening. J. Org. Chem. 1950, 15, 802–806. [Google Scholar] [CrossRef]
- Tschan, M.; Thomas, C.M.; Strub, H.; Carpentier, J.F. Copper(II) triflate as a source of triflic acid: Effective, green catalyst of hydroalkoxylation reactions. Adv. Synth. Catal. 2009, 351, 2496–2504. [Google Scholar] [CrossRef]
- Kang, Y.B.; Gade, L.H. The nature of the catalytically active species in olefin deoxygenation with PhI(OAc)2: Metal or proton? J. Am. Chem. Soc. 2011, 133, 3658–3667. [Google Scholar] [CrossRef]
- Dang, T.; Boeck, F.; Hinterman, L. Hidden Brønsted acid catalysis: Pathways of accidental or delicerate generation of triflic acid from metal triflates. J. Org. Chem. 2011, 76, 9353–9361. [Google Scholar] [CrossRef]
- Loro, C.; Oble, J.; Foschi, F.; Papis, M.; Beccalli, E.M.; Giofrè, S.; Poli, G.; Broggini, G. Acid-mediated decarboxylative C–H coupling between arenes and O-allyl carbamates. Org. Chem. Front. 2022, 9, 1711–1718. [Google Scholar] [CrossRef]
- Loro, C.; Papis, M.; Foschi, F.; Broggini, G.; Poli, G.; Oble, J. Copper(II)-Catalyzed Three-Component Arylation/Hydroamination Cascade from Allyl Alcohol: Access to 1-Aryl-2-sulfonylamino-propanes. J. Org. Chem. 2023, 88, 13995–14003. [Google Scholar] [CrossRef]
- Schwekendiek, K.; Glorius, F. Efficient Oxidative Synthesis of 2-Oxazolines. Synthesis 2006, 18, 2996–3002. [Google Scholar] [CrossRef]
- Soleymani Movahed, F.; Foo, S.W.; Mori, S.; Ogawa, S.; Saito, S. Phosphorus-Based Organocatalysis for the Dehydrative Cyclization of N-(2-Hydroxyethyl)amides into 2-Oxazolines. J. Org. Chem. 2022, 87, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE), Version 2022.02; Chemical Computing Group ULC: Montreal, QC, Canada, 2023.
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Papis, M.; Bucci, R.; Contini, A.; Gelmi, M.L.; Lo Presti, L.; Poli, G.; Broggini, G.; Loro, C. Phosphine-Catalyzed Domino Regio- and Stereo-Selective Hexamerization of 2-(Bromomethyl)acrylates to 1,2-Bis(cyclohexenyl)ethenyl Derivatives. Org. Lett. 2023, 25, 7380–7384. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadrupole zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A. 03; Gaussian. Inc.: Wallingford, UK, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of Shelx. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B 2013, 69, 249–259. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temp. °C | Product (Yield) b |
| 1 | NiCl2 | MeCN | r.t. | 2a (53%) |
| 2 | NiCl2 | THF | r.t. | 2a (81%) |
| 3 c | NiCl2 | THF | r.t. | 2a (36%) + 3 (42%) |
| 4 | NiCl2 | CH2Cl2 | r.t. | 2a (38%) + 3 (23%) |
| 5 | NiCl2 | Toluene | r.t. | 2a (28%) + 3 (32%) |
| 6 d | NiCl2 | THF | reflux | 2a (68%) |
| 7 | Ni(acac)2 | THF | r.t. | not detected |
| 8 | Ni(OTf)2 | THF | r.t | 2a (25%) + 3 (38%) |
| 9 | PdCl2 | THF | r.t. | not detected |
| 10 d | PdCl2 | THF | reflux | complex mixture |
| 11 | ZnCl2 | THF | r.t. | not detected |
| 12 d | ZnCl2 | THF | reflux | complex mixture |
| 13 | Cu(OTf)2 | THF | r.t. | complex mixture |
| 14 | TMSOTf | THF | r.t. | 2a (45%) |
| 15 | - | THF | r.t. | not detected |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colombo, S.; Oble, J.; Poli, G.; Lo Presti, L.; Macetti, G.; Contini, A.; Broggini, G.; Papis, M.; Loro, C. Doubly Metathetic NiCl2-Catalyzed Coupling Between Bis(2-oxazolines) and Aldehydes: A Novel Access to Bis(ester-imine) Derivatives. Molecules 2024, 29, 5756. https://doi.org/10.3390/molecules29235756
Colombo S, Oble J, Poli G, Lo Presti L, Macetti G, Contini A, Broggini G, Papis M, Loro C. Doubly Metathetic NiCl2-Catalyzed Coupling Between Bis(2-oxazolines) and Aldehydes: A Novel Access to Bis(ester-imine) Derivatives. Molecules. 2024; 29(23):5756. https://doi.org/10.3390/molecules29235756
Chicago/Turabian StyleColombo, Sara, Julie Oble, Giovanni Poli, Leonardo Lo Presti, Giovanni Macetti, Alessandro Contini, Gianluigi Broggini, Marta Papis, and Camilla Loro. 2024. "Doubly Metathetic NiCl2-Catalyzed Coupling Between Bis(2-oxazolines) and Aldehydes: A Novel Access to Bis(ester-imine) Derivatives" Molecules 29, no. 23: 5756. https://doi.org/10.3390/molecules29235756
APA StyleColombo, S., Oble, J., Poli, G., Lo Presti, L., Macetti, G., Contini, A., Broggini, G., Papis, M., & Loro, C. (2024). Doubly Metathetic NiCl2-Catalyzed Coupling Between Bis(2-oxazolines) and Aldehydes: A Novel Access to Bis(ester-imine) Derivatives. Molecules, 29(23), 5756. https://doi.org/10.3390/molecules29235756