Abstract
The angular triquinane carbocyclic ring system is a component of many natural products found in numerous terrestrial and marine plants. A strategy for the synthesis of functionalized angular triquinanes utilizing two trimethylenemethane (TMM)-based [3+2] cycloaddition reactions is presented. This synthetic strategy employs the intermolecular dyil-trapping reaction to give eventual access to the bicyclo[3.3.0]oct-1-en-3-one system. A subsequent [3+2] cycloaddition with a TMM equivalent provides the angular triquinane carbocyclic framework.
1. Introduction
Triquinanes, or polycarbocyclic ring systems composed of three five-membered rings, occur in numerous terrestrial and marine plants. These naturally occurring quinanes have been the subject of synthetic and biological investigations for many decades [1,2]. The five-membered rings can be adjoined in a linear, angular, or propellane pattern. These triquinane molecules possess biological and medicinal value [3,4,5]. The family of angular triquinanes is characterized by three five-membered rings fused in an angular pattern so that all three rings share a common quaternary carbon in an all-cis-fused fashion. Sub-families of angular triquinanes are characterized by the position of the methyl groups on the carbocyclic skeleton. Specific angular triquinanes are characterized by their specific structure, including their stereochemistry, such as silphinene, pentalenene, and subergorgic acid. Natural angular triquinanes also serve as biosynthetic precursors to pentalenolactone antibiotics [6]. We are interested in angular triquinanes because we believe that synthetic access to unique derivatives has the potential to deliver new drug lead molecules. This belief leads us not toward the total synthesis of a particular natural product but to the development of a general synthetic methodology to angular triquinane-bearing functional groups readily usable for analog synthesis.
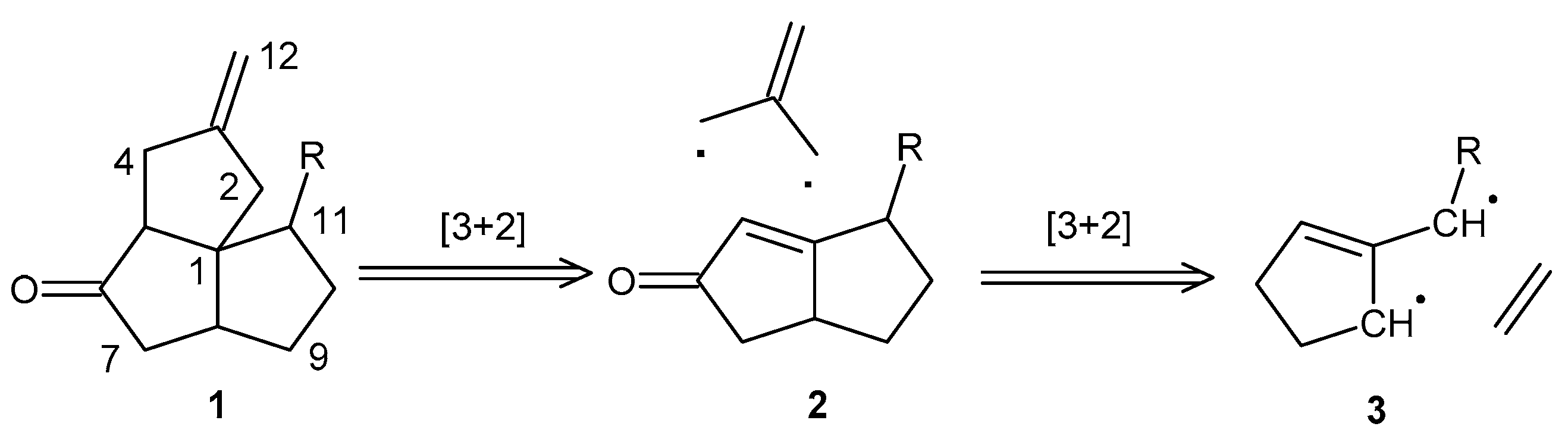
We envisioned a relatively quick access to the angular triquinane ring system, with each ring bearing a synthetically useful functionalization, by the direct [3+2] cycloaddition of a trimethylenemethane (TMM) unit with a functionalized bicyclo[3.3.0]oct-1-en-3-one. This ring system can be accessed by the [3+2] addition of a TMM-based diradical with an olefin (Scheme 1) [7,8,9]. Reports in the literature of the direct cyclopentyl-annulation of bicyclo[3.3.0]oct-1-en-3-ones toward angular triquinanes are limited to two-step annulations using conjugate-addition reactions with sulfinylallyl anions or bifunctional cuprates, sometimes as one-pot reactions [10,11,12,13]. The drawbacks of using bifunctional reagents are additional synthetic reactions, the use of organotin reagents, and potentially sensitive transmetallation steps. There are also examples of Danheiser annulation with similar substrates having been successful in giving access to angular tetra- and triquinanes [14,15,16]. The drawback of employing this approach is the use of a strong Lewis acid that may not be tolerated by some functional groups. There are no examples of the direct cyclopentyl-annulation of bicyclo[3.3.0]oct-1-en-3-one with a TMM equivalent. We are aware of one other TMM-based diradical strategy toward angular triquinanes that involves the intramolecular [3+2] cycloaddition of a diazene-derived TMM in route to the total synthesis of crinipellin A [17,18,19]. Our motivation for investigating this synthetic route is the production of a useful intermediate for elaboration into non-natural angular triquinane analogs for the eventual identification of new antimicrobial and anticancer drug lead molecules.
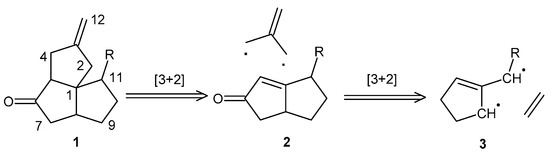
Scheme 1.
Retrosynthetic scheme depicting the TMM-based [3+2] cycloaddition strategy to access functionalized angular triquinanes.
2. Results and Discussion
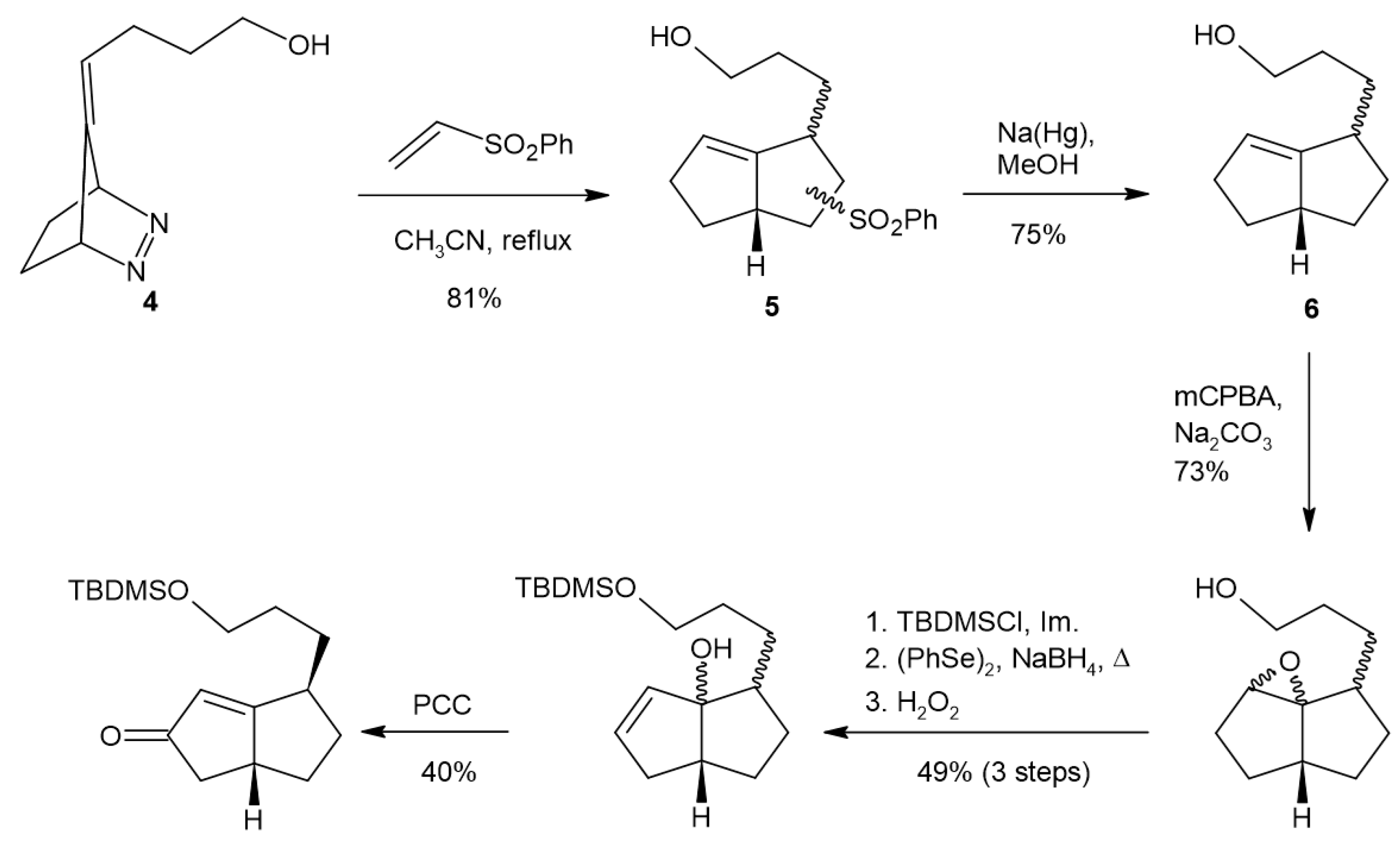
To test our proposed route to the creation of functionalized angular triquinanes, we used diazene 4 (Scheme 2) [20,21]. This compound provides the functionalization of one of the rings of the product in the form of a tether that could be used for the future attachment of various labeling moieties. Heating 4 in degassed acetonitrile to generate the corresponding TMM diyl, in the presence of phenylvinylsulfone as the diylophile, resulted in the formation of 5 as a mixture of isomers. Phenylvinylsulfone was chosen as the diylophile because of its ability to serve as an ethylene and a ketene equivalent [22,23,24]. The phenylsulfone group was removed under reducing conditions with sodium amalgam to provide compound 6 and reveal relative stereoselectivity in the diyl-trapping reaction. Purification by column chromatography provided the major isomer contaminated with a small amount of the minor isomer. The nuclear magnetic resonance (NMR) spectra (1H, 13C, TOCSY, HMQC, NOSEY) were consistent with the syn-relative stereochemistry of the predominant isomer. The vinyl and ring juncture methine 1H chemical shifts in the major isomer of 6 appeared downfield of the same resonances for the minor isomer. Additionally, the 13C resonances of the ring juncture methine carbon and the ring juncture vinyl were downfield in the case of the major isomer, and the methinyl vinyl carbon was upfield compared to the minor isomer (Figures S4–S6). This type of stereoselectivity in the intermolecular diyl-trapping reaction has previously been observed [25,26,27,28]. Attempts at the allylic oxidation of TBDMS-protected 6 in order to access 9 using SeO2 or chromium oxide were unsuccessful. A multistep procedure through epoxide 7 and allylic alcohol 8 provided the desired enone. The epoxidation of 6 with m-chloroperbenzoic acid (mCPBA) exhibited a low facial selectivity and provided a mixture of diastereomers, presumably due to steric hinderance or hydrogen bonding effects of the tether moiety. The integration of the two epoxide methine proton resonances, present in the product 1H NMR spectra, indicated a 4:1 ratio of isomers. This result is consistent with the formation of both cis- and trans-fused products resulting from the epoxidation of similar systems [29]. Attempts to open the epoxide with a strong base to obtain allylic alcohol 8 were unsuccessful, but the phenylselenide ion facilitated nucleophilic opening of the epoxide, followed by oxidative elimination, producing allylic alcohol 8 [30,31]. Finally, oxidation of the allylic alcohols using pyridinum chlorochromate (PCC) provided the desired enone, 9 [32].
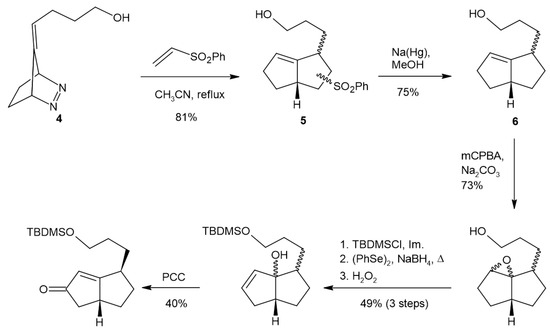
Scheme 2.
Synthesis of bicyclo[3.3.0]octeneone 9.
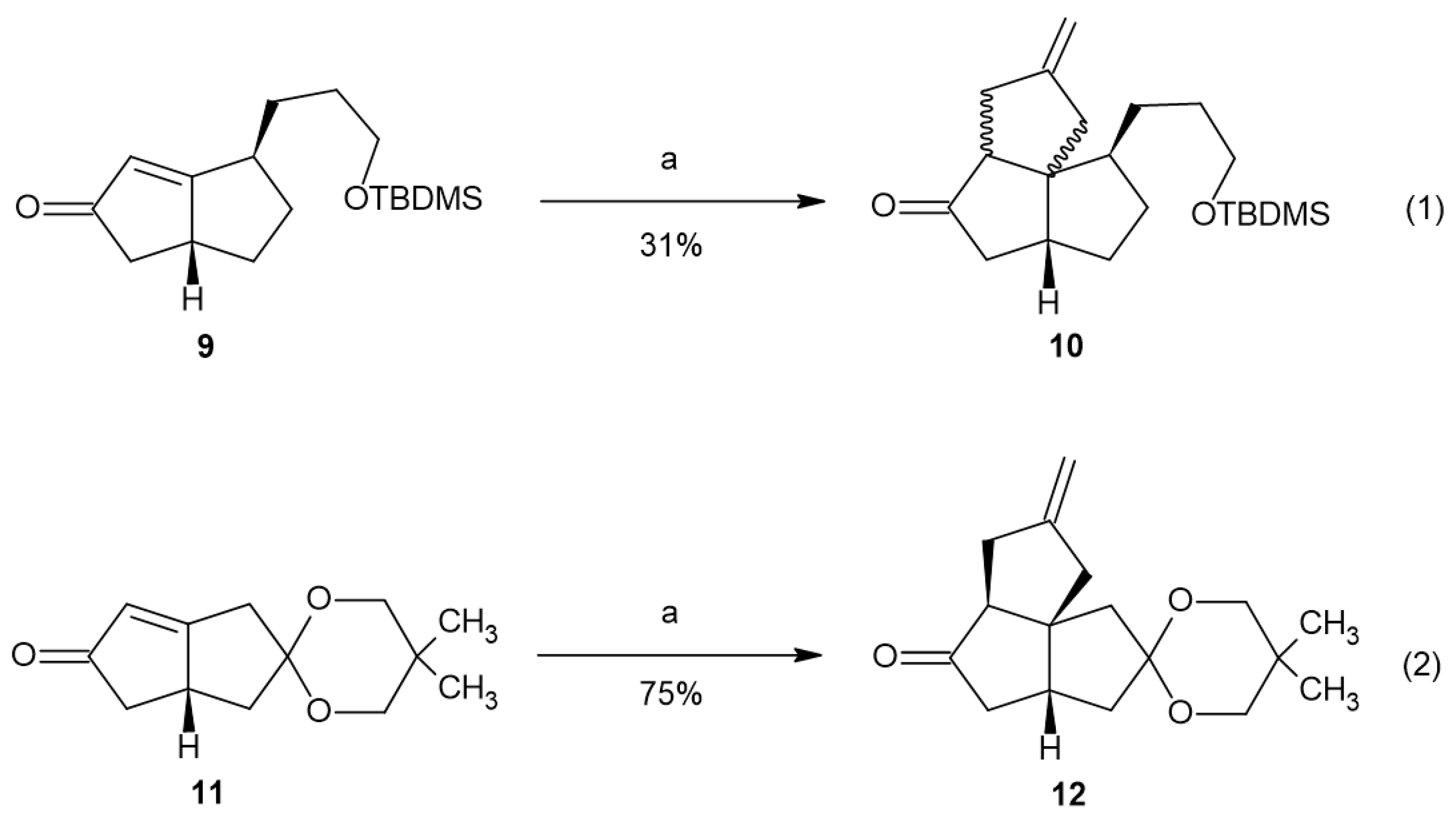
Enone 9 was subjected to [3+2] cycloaddition with Trost’s TMM precursor (Figure 1(1)) [33,34,35,36,37]. To our disappointment, a mixture of isomers was observed as the product. The expectation that the reaction of the bicyclo[3.3.0]oct-1-en-3-one system should exhibit facial selectivity to produce the thermodynamically more stable all-cis-fused angular triquinane product was not realized. The mixture of isomers consisting of the expected all-cis-fused product and the mixed cis–trans-fused product was not readily separable. Mass spectrometry confirmed the presence of 10. The 1H NMR of the product mixture showed two distinct pairs of methylene vinyl peaks in a 60:40 ratio, estimated by integration (Figure S17). This result is consistent with our initial assignment of the relative stereochemistry of a major isomer of 6 and its subsequent epoxidation result. We believe that the tether present in the allylic position of molecule 9 presents a significant steric effect to the approach of the palladium–TMM complex.
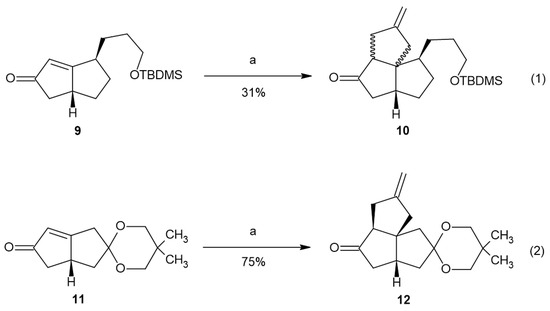
Figure 1.
The [3+2] cycloaddition of 9 and 11. Conditions: a—TMSCH2C(CH2)CH2OAc, Pd(Ph3P)4, dppe, THF, and reflux.
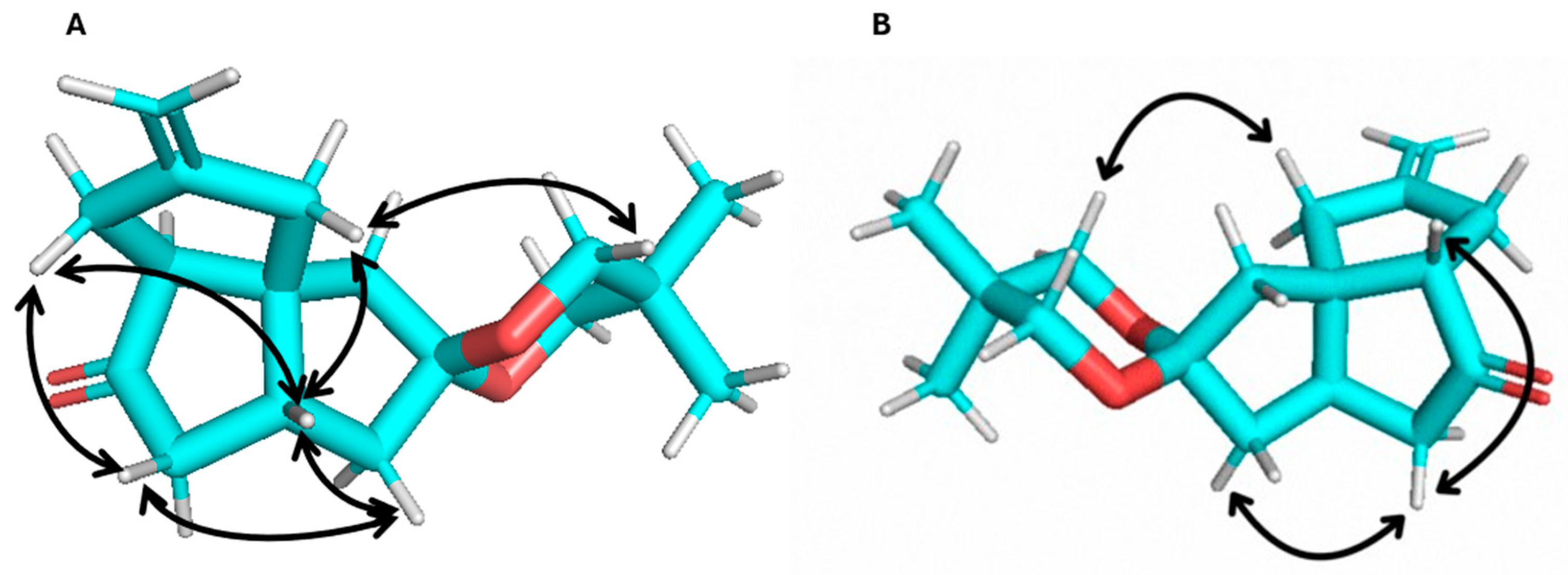
To test whether the tether present in the allylic position was the cause of the creation of the isomeric mixture in the [3+2] cycloaddition reaction, we subjected enone 11 to the same reaction conditions. Only one major isomer, 12, was isolated from the reaction (Figure 1(2)). It is likely that another isomer was also formed. The NMR (1H, 13C, TOCSY, NOESY) data support product 12 as the all-cis-fused angular triquinane. The NMR chemical shifts were consistent with similar angular triquinanes [13,38]. Notable nuclear Overhauser effects (NOEs) were observed between C2Hβ and C8H and between the ketal methylene protons and both C2Hα and C5H (Figure 2). It should be noted here that 12 possesses a carbonyl or carbonyl equivalent in each of the cyclopentane rings, thus having the synthetic potential to further functionalize all but the quaternary carbon atom.
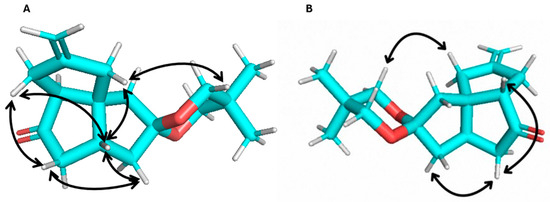
Figure 2.
Observed NOEs for compound 12: (A) β-face and (B) α-face views.
3. Conclusions
In conclusion, we have demonstrated that the intermolecular trapping of a diazene-derived TMM-based diyl may be used in route, via [3+2] cycloaddition with an olefin, to the bicyclo[3.3.0]oct-1-en-3-one system. The subsequent [3+2] cyclopentyl annulation of this ring system with a TMM equivalent can give access to the angular triquinane carbocyclic framework. The synthesis of 9 was plagued by the persistence of a minor isomer and low facial selectivity in the epoxidation step. Low facial selectivity was again observed in the [3+2] cycloaddition of 9 to give a mixture of two major isomers. To test the hypothesis that the allylic substituent in 9 was the cause of the formation of significant amounts of the undesired isomer, 11 was subjected to the reaction conditions. The reaction formed one major isomer of 12, and this result supports the hypothesis. The advantage of using this TMM equivalent strategy is that the TMM precursor is commercially available and provides a masked carbonyl in the product. The route presented here complements previous approaches but possesses the drawback of sensitivity to allylic substitution in the bicyclo[3.3.0]oct-1-en-3-one substrate, which reduces facial selectivity, resulting in the formation of significant amounts of undesired isomers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29225358/s1, Experimental procedures and compound characterization data.
Author Contributions
Conceptualization and manuscript preparation, W.A.R.; experiment execution and compound characterization, S.M.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors acknowledge the support of the FRC at the University of the Pacific in the form of an SAAG grant. The authors also thank Andreas Franze for recording the 2D NMR data and extend their sincere gratitude to the reviewers and editors for their comments and suggestions, which have improved this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Qiu, Y.; Lan, H.-J.; Chen, L.-P. Linear Triquinane Sesquiterpenoids: Their Isolation, Structures, Biological Activities, and Chemical Synthesis. Molecules 2018, 23, 2095. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Srikrishna, A. Synthesis of Polyquinane Natural Products: An Update. Chem. Rev. 1997, 97, 671–719. [Google Scholar] [CrossRef] [PubMed]
- Hudlicky, T.; Price, J.D. Anionic Approaches to the Construction of Cyclopentanoids. Chem. Rev. 1989, 89, 1467–1486. [Google Scholar] [CrossRef]
- Jeon, H.; Winkler, J.D. Synthesis of Cyclohexane-Angularly-Fused Triquinanes. Synthesis 2021, 53, 475–488. [Google Scholar] [CrossRef] [PubMed]
- González-Coloma, A.; Gutiérrez, C.; Cabrera, R.; Reina, M. Silphinene Derivatives: Their Effects and Modes of Action on Colorado Potato Beetle. J. Agric. Food Chem. 1997, 45, 946–950. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Chen, S.; Wu, W.; Sun, P. Isolation and Identification of Pentalenolactone Analogs from Steptomyces sp. NRRL S-4. Molecules 2021, 26, 7377. [Google Scholar] [CrossRef]
- Dowd, P. Trimethylenemethane. J. Am. Chem. Soc. 1966, 88, 2587–2589. [Google Scholar] [CrossRef]
- Corwin, L.R.; McDaniel, D.M.; Bushby, R.J.; Berson, J.A. Dimerization and cycloaddition reactions of a trimethylenemethane derivative, 2-isoprpylidenecyclopenta-1,3-diyl. Mechanistic separation of triplet and singlet reactions. J. Am. Chem. Soc. 1980, 102, 276–287. [Google Scholar] [CrossRef]
- Little, R.D. Diyl Trapping and Electroreductive Cyclization Reactions. Chem. Rev. 1996, 96, 93–114. [Google Scholar] [CrossRef]
- Hua, D.H. Asymmetric Total Synthesis of (+)-Pentalenene via Chiral Sulfinylallyl Anions. Hydrolytic Ring Closure of Enol Thioether Ketones. J. Am. Chem. Soc. 1986, 108, 3835–3837. [Google Scholar] [CrossRef]
- Piers, E.; Karunaratne, V. 4-Chloro-2-lithio-1-butene, a Novel Donor-Acceptor Conjunctive Reagent. J. Org. Chem. 1983, 48, 1774–1776. [Google Scholar] [CrossRef]
- Piers, E.; Karunaratne, V. Organotin-based reagents: 4-Chloro-2-lithio-1-butene and related sustances. Methylenecyclopentane annulations. Total synthesis of (±)-δ9(12)-capnellene. Tetrahedron 1989, 45, 1089–1104. [Google Scholar] [CrossRef]
- Dragojlovic, V. Synthesis of a Highly Functionalized Triquinane: Studies Towards a Total Synthesis of Subergorgic Acid and Its Analogues. Molecules 2000, 5, 674–698. [Google Scholar] [CrossRef]
- Schmidt, A.W.; Olpp, T.; Baum, E.; Stiffle, T.; Knölker, H.-J. Organosilicon-Mediated Total Synthesis of the Triquinane Sesquiterpenes (±)-β-Isocomene and (±)-Isocomene. Org. Biomol. Chem. 2010, 8, 4562. [Google Scholar] [CrossRef] [PubMed]
- Danheiser, R.L.; Carini, D.J.; Basak, A. (Trimethylsilyl)cyclopentene Annulation: A Regiocontrolled Approach to the Synthesis of Five-Membered Rings. J. Am. Chem. Soc. 1981, 103, 1604–1606. [Google Scholar] [CrossRef]
- Xu, B.; Xun, W.; Su, S.; Zhai, H. Total Synthesis of (−)-Conidiogenone B, (−)-Conidiogenone, and (−)-Conidiogenol. Angew. Chem. 2020, 132, 16617–16621. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Jung, Y.; Yoon, Y.; Kim, B.G.; Kim, Y. Angularly Fused Triquinanes from Linear Substrates through Trimethylenemethane Diyl [2+3] Cycloaddition Reaction. Org. Lett. 2010, 12, 2672–2674. [Google Scholar] [CrossRef]
- Kang, T.; Kim, W.-Y.; Yoon, Y.; Kim, B.G.; Lee, H.-Y. Tandem Cycloaddition Reactions of Allenyl Diazo Compounds Forming Triquinanes via Trimethylenemethane Diyls. J. Am. Chem. Soc. 2011, 133, 18050–18053. [Google Scholar] [CrossRef]
- Kang, T.; Song, S.B.; Kim, W.-Y.; Kim, B.G.; Lee, H.-Y. Total Synthesi of (−)-Crinipellin A. J. Am. Chem. Soc. 2014, 136, 10274–10276. [Google Scholar] [CrossRef]
- Campopiano, O.; Little, R.D.; Petersen, J.L. Evidence for Hydrogen Atom Abstraction and Loss of Diylophile Stereochemistry in an Intramolecular 1,3-Diyl Trapping Reaction. J. Am. Chem. Soc. 1985, 107, 3721–3722. [Google Scholar] [CrossRef]
- Masjedizadeh, M.R.; Fite, C.; Little, R.D. Direct observation of intermediates involved in the intramolecular diyl trapping reaction. Tetrahedron Lett. 1990, 31, 1229. [Google Scholar] [CrossRef]
- Carr, R.V.C.; Paquette, L.A. An Ethylene and Terminal Olefin Equivalent in [4+2] π Cycloadditions. General Synthetic Application of Phenyl Vinyl Sulfone to the Construction of Functionalized Six-Membered Rings. J. Am. Chem. Soc. 1980, 102, 853–855. [Google Scholar] [CrossRef]
- Little, R.D.; Brown, L. Facile Construction of C10 Modified Prostaglandin Precursors. Diyl Trapping Reactions Using Phenyl Vinyl Sulfoxide and Phenyl Vinyl Sulfone. Tetrahedron Lett. 1980, 21, 2303–2304. [Google Scholar] [CrossRef]
- Little, R.D.; Myong, S.O. Oxidative desulfonylation. Phenyl vinyl sulfone as a ketene synthetic equivalent. Tetrahedron Lett. 1980, 21, 3339–3342. [Google Scholar] [CrossRef]
- Little, R.D.; Bukhari, A.; Venegas, M.G. A new route to linearly fused tricyclopentanoids. Diyl trapping reactions in organic synthesis. Tetrahedron Lett. 1979, 20, 305–308. [Google Scholar] [CrossRef]
- Venegas, M.G.; Little, R.D. Carbon-13 chemical shifts in tricyclo[6.3.0.03,7] undecanes (linearly fused tricyclopentanoids). Tetrahedron Lett. 1979, 20, 309–312. [Google Scholar] [CrossRef]
- Little, R.D.; Carroll, G.L. Elecrochemical Generation of the Azo Linkage. Synthesis of Bicyclic Azo Compounds, Precursors of 1,3-Diyls. J. Org. Chem. 1979, 44, 4720–4722. [Google Scholar] [CrossRef]
- Whitesell, J.K.; Matthews, R.S. Carbon-13 Chemical Shifts in Bicyclo[3.3.0]octanes. J. Org. Chem. 1977, 24, 3878–3882. [Google Scholar] [CrossRef]
- Van Hijfte, L.; Little, R.D.; Petersen, J.L.; Moeller, K.D. Intramolecular 1,3-Diyl Trapping Reactions. Total Synthesis of (±)-Hypnophilin and (±)-Coriolin. Formation of the Trans-Fused Bicyclo[3.3.0]octane Ring System. J. Org. Chem. 1987, 52, 4647–4661. [Google Scholar] [CrossRef]
- Kissel, C.L.; Rickborn, B. The Base-Induced Rearrangement of Epoxides. IV. Reaction of Cyclohexene Oxide wih Various Lithium Alkylamides. J. Org. Chem. 1972, 37, 2060–2063. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Lauer, R.F. A Mild Method for the Conversion of Epoxides to Allylic Alcohols. The First Organoselenium Reagent. J. Am. Chem. Soc. 1973, 95, 2697–2699. [Google Scholar] [CrossRef]
- Dauben, W.G.; Michno, D.M. Direct oxidation of tertiary allylic alcohols. A simple and effective method for alkylative carbonyl transposition. J. Org. Chem. 1977, 42, 682–685. [Google Scholar] [CrossRef]
- Trost, B.M.; Chan, D.M.T. Regiochemistry of the Cycloaddition of a Substituted Trimethylenemethanepalladium Complex. J. Am. Chem. Soc. 1981, 103, 5972–5974. [Google Scholar] [CrossRef]
- Trost, B.M.; Chan, D.M.T. Intramolecular Carbocyclic [3+2] Cycloaddition via Organopalladium Intermediates. J. Am. Chem. Soc. 1982, 104, 3733–3735. [Google Scholar] [CrossRef]
- Trost, B.M.; Chan, D.M.T. Palladium-Mediated Cycloaddition Approach to Cyclopentanoids. Introduction and Initial Studies. J. Am. Chem. Soc. 1983, 105, 2315–2325. [Google Scholar] [CrossRef]
- Trost, B.M.; Chan, D.M.T. Palladium-Mediated Cycloaddition Approach to Cyclopentanoids. Mechanistic Studies. J. Am. Chem. Soc. 1983, 105, 2326–2335. [Google Scholar] [CrossRef]
- Nanninga, T.N.; Trost, B.M. Palladium-Mediated Cycloaddition Approach to Loganin Aglucon. J. Am. Chem. Soc. 1985, 107, 1293–1299. [Google Scholar]
- Rowley, E.G.; Schore, N.E. The Pauson-Khand Reaction in Triquinane Synthesis: Approaches to Pentalenene, Pentalenic Acid, and Silphinene. J. Org. Chem. 1992, 57, 6853–6861. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).