
New Phytol Derivatives with Increased Cosmeceutical Potential





Abstract

1. Introduction
2. Results and Discussion
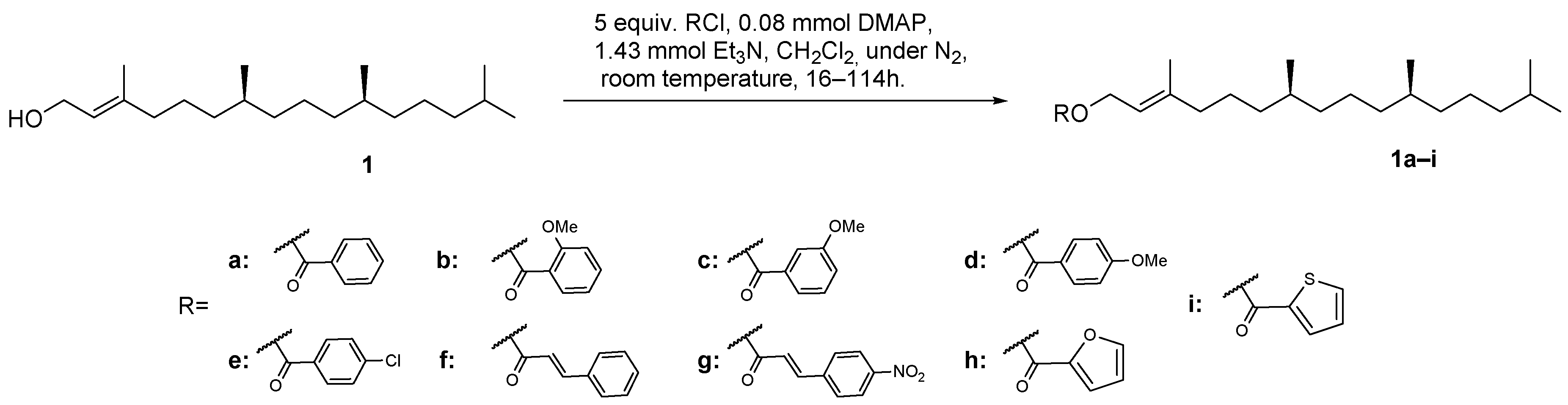
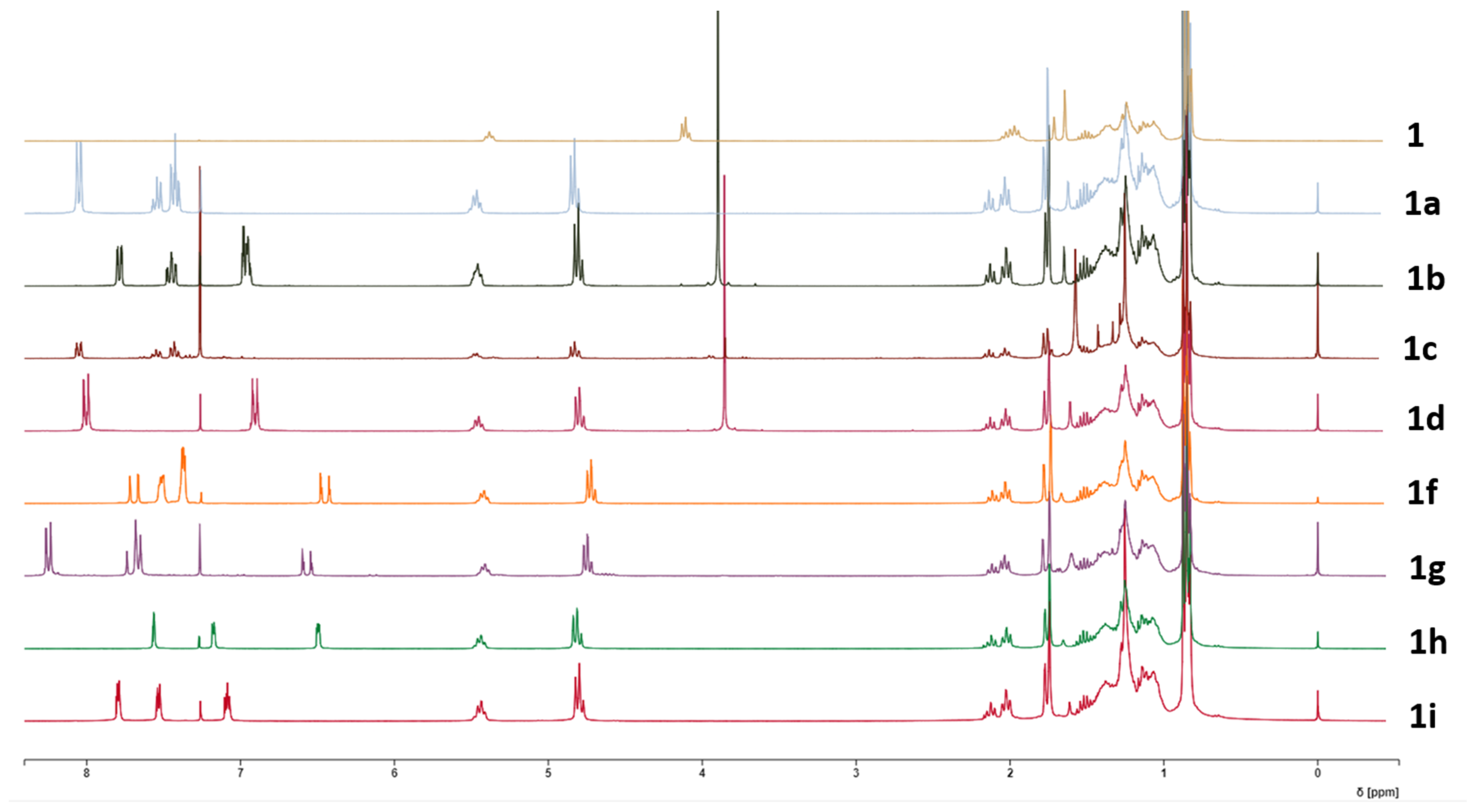
2.1. Chemistry
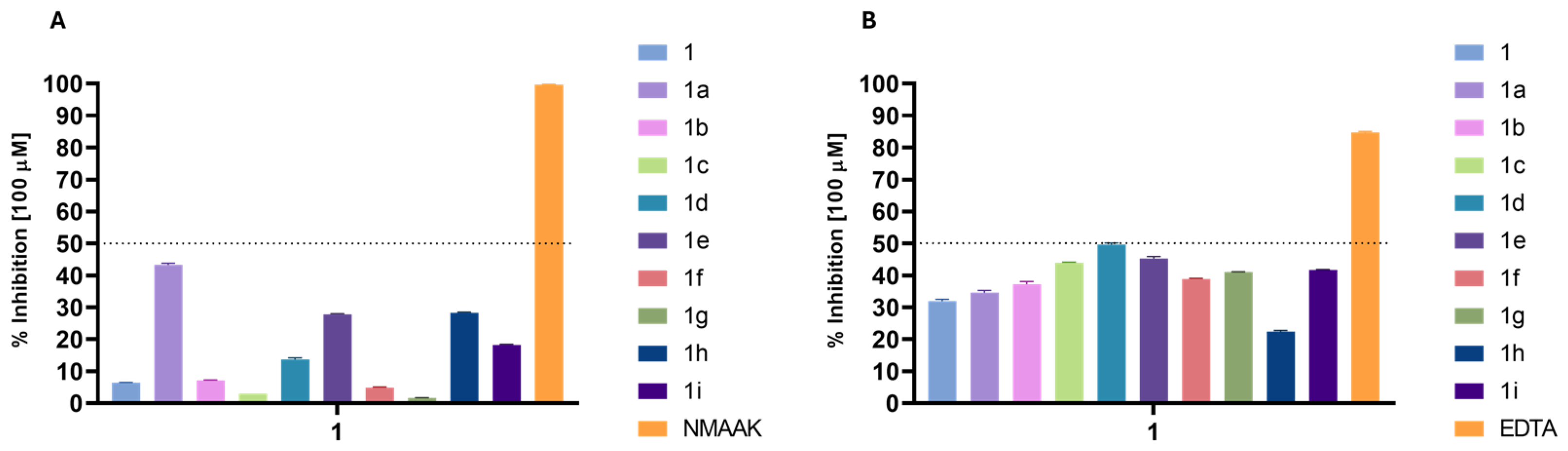
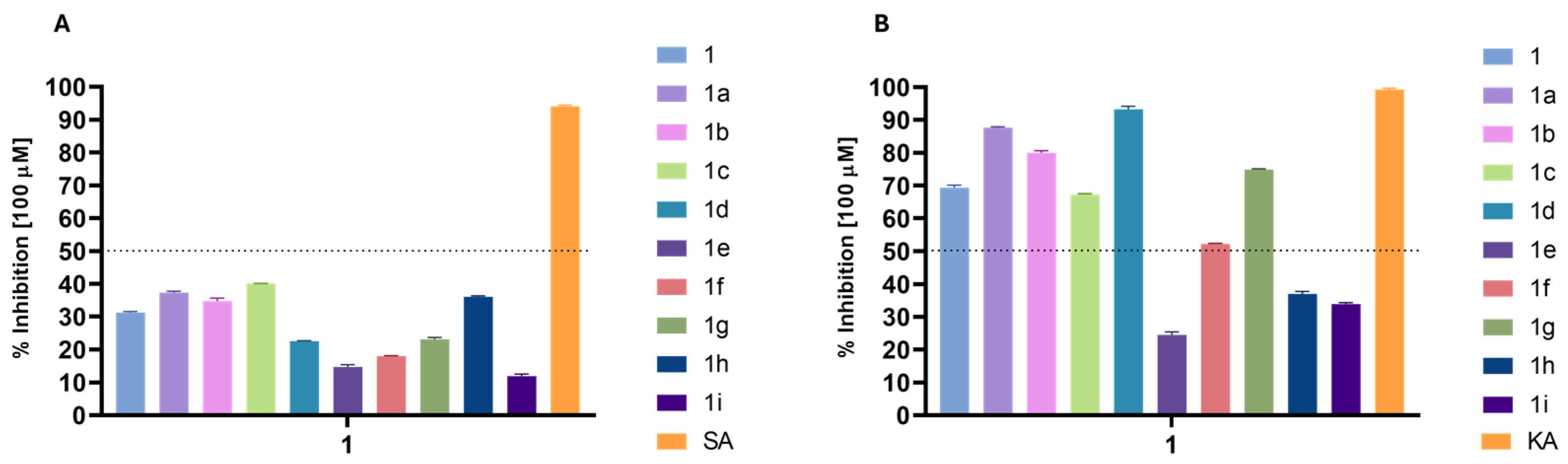
2.2. Biological Activities
2.2.1. Level of Activity
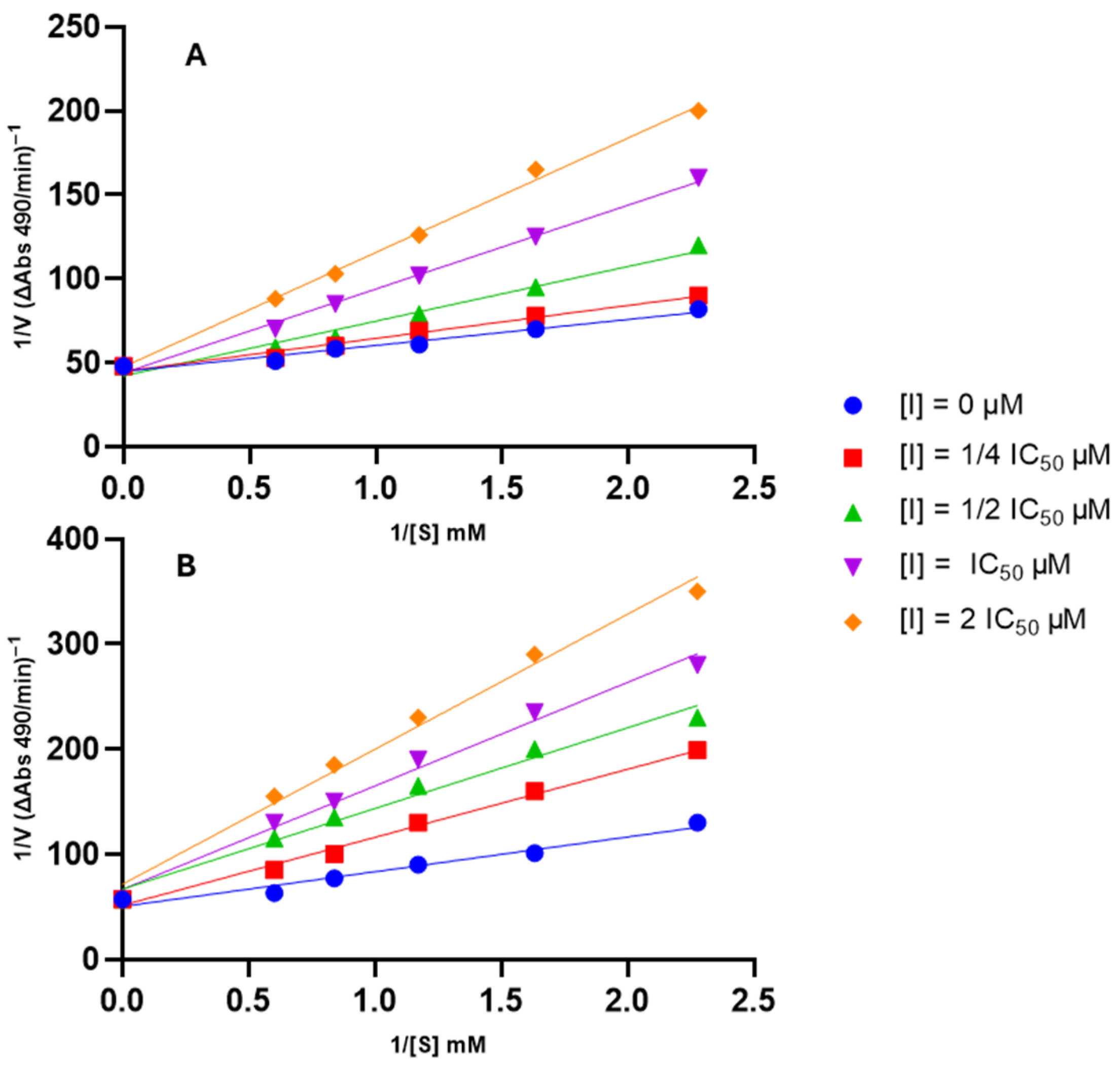
2.2.2. Mode of Inhibition
2.2.3. Molecular Docking
3. Materials and Methods
3.1. General
3.2. Standards and Reagents
3.3. Synthesis of Derivatives
3.4. Biological Activities
3.4.1. DPPH Radical Scavenging Activity
3.4.2. ABTS Radical Scavenging Assay
3.4.3. Ferrous Chelating Activity
3.4.4. Hyaluronidase Inhibition Assay
3.4.5. Tyrosinase Inhibition Assay
3.4.6. Elastase Inhibition Assay
3.4.7. Collagenase Inhibition Assay
3.5. Docking Simulations
3.6. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fernandes, A.; Rodrigues, P.M.; Pintado, M.; Tavaria, F.K. A Systematic Review of Natural Products for Skin Applications: Targeting Inflammation, Wound Healing, and Photo-Aging. Phytomedicine 2023, 115, 154824. [Google Scholar] [CrossRef]
- Sasounian, R.; Martinez, R.M.; Lopes, A.M.; Giarolla, J.; Rosado, C.; Magalhães, W.V.; Velasco, M.V.R.; Baby, A.R. Innovative Approaches to an Eco-Friendly Cosmetic Industry: A Review of Sustainable Ingredients. Clean Technol. 2024, 6, 176–198. [Google Scholar] [CrossRef]
- Kryczyk-Poprawa, A.; Kwiecień, A.; Opoka, W. Photostability of Topical Agents Applied to the Skin: A Review. Pharmaceutics 2020, 12, 10. [Google Scholar] [CrossRef]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern Approaches in the Discovery and Development of Plant-Based Natural Products and Their Analogues as Potential Therapeutic Agents. Molecules 2022, 27, 349. [Google Scholar] [CrossRef]
- Ding, Y.; Xue, X. Medicinal Chemistry Strategies for the Modification of Bioactive Natural Products. Molecules 2024, 29, 689. [Google Scholar] [CrossRef]
- Liu, J.K. Natural Products in Cosmetics. Nat. Prod. Bioprospect. 2022, 12, 40. [Google Scholar] [CrossRef]
- Goyal, N.; Jerold, F. Biocosmetics: Technological Advances and Future Outlook. Environ. Sci. Pollut. Res. Int. 2023, 30, 25148–25169. [Google Scholar] [CrossRef]
- Islam, M.T.; Ali, E.S.; Uddin, S.J.; Shaw, S.; Islam, M.A.; Ahmed, M.I.; Chandra Shill, M.; Karmakar, U.K.; Yarla, N.S.; Khan, I.N.; et al. Phytol: A Review of Biomedical Activities. Food Chem. Toxicol. 2018, 121, 82–94. [Google Scholar] [CrossRef]
- Durrett, T.P.; Welti, R. The Tail of Chlorophyll: Fates for Phytol. J. Biol. Chem. 2021, 296, 100802. [Google Scholar] [CrossRef]
- Santos, C.C.d.M.P.; Salvadori, M.S.; Mota, V.G.; Costa, L.M.; de Almeida, A.A.C.; de Oliveira, G.A.L.; Costa, J.P.; de Sousa, D.P.; de Freitas, R.M.; de Almeida, R.N. Antinociceptive and Antioxidant Activities of Phytol in Vivo and in Vitro Models. Neurosci. J. 2013, 2013, 949452. [Google Scholar] [CrossRef]
- Silva, R.O.; Sousa, F.B.M.; Damasceno, S.R.B.; Carvalho, N.S.; Silva, V.G.; Oliveira, F.R.M.A.; Sousa, D.P.; Aragão, K.S.; Barbosa, A.L.R.; Freitas, R.M.; et al. Phytol, a Diterpene Alcohol, Inhibits the Inflammatory Response by Reducing Cytokine Production and Oxidative Stress. Fundam. Clin. Pharmacol. 2014, 28, 455–464. [Google Scholar] [CrossRef]
- Costa, J.P.; Islam, T.; Santos, P.S.; Ferreira, P.B.; Oliveira, G.L.S.; Alencar, M.V.O.B.; Paz, M.F.C.J.; Ferreira, E.L.F.; Feitosa, C.M.; Citó, A.M.G.L.; et al. Evaluation of Antioxidant Activity of Phytol Using Non- and Pre-Clinical Models. Curr. Pharm. Biotechnol. 2016, 17, 1278–1284. [Google Scholar] [CrossRef]
- Olofsson, P.; Hultqvist, M.; Hellgren, L.I.; Holmdahl, R. Phytol: A Chlorophyll Component with Anti-Inflammatory and Metabolic Properties. In Recent Advances in Redox Active Plant and Microbial Products; Jacob, C., Kirsch, G., Slusarenko, A., Winyard, P., Burkholz, T., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 345–359. [Google Scholar]
- Islam, M.T.; Ayatollahi, S.A.; Zihad, S.M.N.K.; Sifat, N.; Khan, M.R.; Paul, A.; Salehi, B.; Islam, T.; Mubarak, M.S.; Martins, N.; et al. Phytol Anti-Inflammatory Activity: Pre-Clinical Assessment and Possible Mechanism of Action Elucidation. Cell. Mol. Biol. 2020, 66, 264–269. [Google Scholar] [CrossRef]
- de Alencar, M.V.O.B.; Islam, M.T.; da Mata, A.M.O.F.; dos Reis, A.C.; de Lima, R.M.T.; de Oliveira Ferreira, J.R.; de Castro e Sousa, J.M.; Ferreira, P.M.P.; de Carvalho Melo-Cavalcante, A.A.; Rauf, A.; et al. Anticancer Effects of Phytol against Sarcoma (S-180) and Human Leukemic (HL-60) Cancer Cells. Environ. Sci. Pollut. Res. Int. 2023, 30, 80996–81007. [Google Scholar] [CrossRef]
- Bobe, G.; Zhang, Z.; Kopp, R.; Garzotto, M.; Shannon, J.; Takata, Y. Phytol and Its Metabolites Phytanic and Pristanic Acids for Risk of Cancer: Current Evidence and Future Directions. Eur. J. Cancer Prev. 2020, 29, 191–200. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Ai, W.; Zhang, F.; Yang, K.; Wang, L.; Zhu, X.; Gao, P.; Shu, G.; Jiang, Q.; et al. Phytol Increases Adipocyte Number and Glucose Tolerance through Activation of PI3K/Akt Signaling Pathway in Mice Fed High-Fat and High-Fructose Diet. Biochem. Biophys. Res. Commun. 2017, 489, 432–438. [Google Scholar] [CrossRef]
- Kim, E.N.; Trang, N.M.; Kang, H.; Kim, K.H.; Jeong, G.S. Phytol Suppresses Osteoclast Differentiation and Oxidative Stress through Nrf2/HO-1 Regulation in RANKL-Induced RAW264.7 Cells. Cells 2022, 11, 3596. [Google Scholar] [CrossRef]
- Papaccio, F.; D’arino, A.; Caputo, S.; Bellei, B. Focus on the Contribution of Oxidative Stress in Skin Aging. Antioxidants 2022, 11, 1121. [Google Scholar] [CrossRef]
- Upadhyay, H.C.; Dwivedi, G.R.; Roy, S.; Sharma, A.; Darokar, M.P.; Srivastava, S.K. Phytol Derivatives as Drug Resistance Reversal Agents. ChemMedChem 2014, 9, 1860–1868. [Google Scholar] [CrossRef]
- Saxena, A.; Upadhyay, H.C.; Cheema, H.S.; Srivastava, S.K.; Darokar, M.P.; Bawankule, D.U. Antimalarial Activity of Phytol Derivatives: In Vitro and in Vivo Study. Med. Chem. Res. 2018, 27, 1345–1354. [Google Scholar] [CrossRef]
- Pinto, D.C.G.A.; Silva, A.M.S.; Cavaleiro, J.A.S. A Convenient Synthesis of New (E)-5-Hydroxy-2-Styrylchromones by Modifications of the Baker–Venkataraman Method. New, J. Chem. 2000, 24, 85–92. [Google Scholar] [CrossRef]
- Pinto, D.C.G.A.; Silva, A.M.S.; Almeida, L.M.P.M.; Cavaleiro, J.A.S.; Lévai, A.; Patonay, T. Synthesis of 4-Aryl-3-(2-chromonyl)-2-pyrazolines by the 1,3-dipolar Cycloaddition of 2-styrylchromones with Diazomethane. J. Heterocycl. Chem. 1998, 35, 217–224. [Google Scholar] [CrossRef]
- Platzer, M.; Kiese, S.; Herfellner, T.; Schweiggert-Weisz, U.; Miesbauer, O.; Eisner, P. Common Trends and Differences in Antioxidant Activity Analysis of Phenolic Substances Using Single Electron Transfer Based Assays. Molecules 2021, 26, 1244. [Google Scholar] [CrossRef]
- Mechqoq, H.; Hourfane, S.; El Yaagoubi, M.; El Hamdaoui, A.; da Silva Almeida, J.R.G.; Rocha, J.M.; El Aouad, N. Molecular Docking, Tyrosinase, Collagenase, and Elastase Inhibition Activities of Argan By-Products. Cosmetics 2022, 9, 24. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant Determinations by the Use of a Stable Free Radical. Nature 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant Activity Applying an Improved ABTS Radical Cation Decolorization Assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Decker, E.A.; Welch, B. Role of Ferritin as a Lipid Oxidation Catalyst in Muscle Food. J. Agric. Food Chem. 1990, 38, 674–677. [Google Scholar] [CrossRef]
- Ling, S.-K.; Tanaka, T.; Kouno, I. Effects of Iridoids on Lipoxygenase and Hyaluronidase Activities and Their Activation by Beta.-Glucosidase in the Presence of Amino Acids. Biol. Pharm. Bull. 2003, 26, 352–356. [Google Scholar] [CrossRef]
- Shimizu, K.; Kondo, R.; Sakai, K.; Lee, S.-H.; Sato, H. The Inhibitory Components from Artocarpus incisus on Melanin Biosynthesis. Planta Med. 1998, 64, 408–412. [Google Scholar] [CrossRef]
- Manosroi, A.; Jantrawut, P.; Akihisa, T.; Manosroi, W.; Manosroi, J. In Vitro Anti-Aging Activities of Terminalia chebula Gall Extract. Pharm. Biol. 2010, 48, 469–481. [Google Scholar] [CrossRef]
- Ndlovu, G.; Fouche, G.; Tselanyane, M.; Cordier, W.; Steenkamp, V. In Vitro Determination of the Anti-Aging Potential of Four Southern African Medicinal Plants. BMC Complement. Altern. Med. 2013, 13, 304. [Google Scholar] [CrossRef]
- Mandl, I.; MacLennan, J.D.; Howes, E.L.; DeBellis, R.H.; Sohler, A. Isolation and Characterization of Proteinase and Collagenase from CL. histolyticum 12. J. Clin. Investig. 1953, 32, 1323–1329. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Yu, Q.; Fan, L.; Duan, Z. Five Individual Polyphenols as Tyrosinase Inhibitors: Inhibitory Activity, Synergistic Effect, Action Mechanism, and Molecular Docking. Food Chem. 2019, 297, 124910. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. Open. J. Stat. 2023, 13. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phytol Derivatives | Reaction Time (h) | Yield (%) |
|---|---|---|
| 1a | 24 | 87 |
| 1b | 24 | 91 |
| 1c | 20 | 5 |
| 1d | 17 | 79 |
| 1e | 21 | 69 |
| 1f | 22 | 89 |
| 1g | 114 | 25 |
| 1h | 16 | 85 |
| 1i | 16 | 90 |
| No | Carbon Type | 1H (ppm) | 13C |
|---|---|---|---|
| 1 | CH2 | 4.11 (dd) | 59.2 |
| 2 | CH | 5.38 (m) | 123.3 |
| 3 | C | 139.7 | |
| 4 | CH2 | 1.92–2.05 (m) | 39.9 |
| 5 | CH2 | 1.36–1.40 (m) | 25.2 |
| 6 | CH2 | 1.25–1.30 (m) | 37.4 |
| 7 | CH | 1.30–1.45 (m) | 32.7 |
| 8 | CH2 | 1.25–1.30 (m) | 37.3 |
| 9 | CH2 | 1.36–1.40 (m) | 24.5 |
| 10 | CH2 | 1.25–1.30 (m) | 36.7 |
| 11 | CH | 1.30–1.45 (m) | 32.8 |
| 12 | CH2 | 1.25–1.30 (m) | 37.3 |
| 13 | CH2 | 1.36–1.40 (m) | 24.8 |
| 14 | CH2 | 1.10–1.15 (m) | 39.4 |
| 15 | CH | 1.50–1.55 (m) | 28.0 |
| 16 | CH3 | 0.84 (d) | 22.6 |
| 17 | CH3 | 0.84 (d) | 22.7 |
| 18 | CH3 | 0.85 (d) | 19.7 |
| 19 | CH3 | 0.85 (d) | 19.7 |
| 20 | CH3 | 1.64 (s) | 16.1 |
| Compound | IC50 (µM) |
|---|---|
| 1 | 77.47 ± 0.80 a |
| 1a | 34.73 ± 0.10 b |
| 1b | 59.62 ± 0.51 c |
| 1c | 78.96 ± 0.87 a |
| 1d | 27.94 ± 0.63 d |
| 1e | >100 e |
| 1f | 95.68 ± 0.94 f |
| 1g | 64.11 ± 0.67 g |
| 1h | >100 e |
| 1i | >100 e |
| Kojic acid | 12.30 ± 0.10 h |
| Compound | Binding Score (Kcal/mol) | Binding Residues |
|---|---|---|
| 1 | −3.1 | His85, His263, Met280 |
| 1a | −3.8 | His65, His85, His 263, Arg268, Met280 |
| 1b | −3.3 | His85, His 263, Arg268, Met280 |
| 1c | −3.1 | His 263, Arg268, Met280 |
| 1d | −4.1 | His65, His85, Asn260, His 263, Arg268, Met280 |
| Kojic acid | −5.5 | His85, His259, His263, Met280 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosa, G.P.; Seca, A.M.L.; Pinto, D.C.G.A.; Barreto, M.C. New Phytol Derivatives with Increased Cosmeceutical Potential. Molecules 2024, 29, 4917. https://doi.org/10.3390/molecules29204917
Rosa GP, Seca AML, Pinto DCGA, Barreto MC. New Phytol Derivatives with Increased Cosmeceutical Potential. Molecules. 2024; 29(20):4917. https://doi.org/10.3390/molecules29204917
Chicago/Turabian StyleRosa, Gonçalo P., Ana M. L. Seca, Diana. C. G. A. Pinto, and M. Carmo Barreto. 2024. "New Phytol Derivatives with Increased Cosmeceutical Potential" Molecules 29, no. 20: 4917. https://doi.org/10.3390/molecules29204917
APA StyleRosa, G. P., Seca, A. M. L., Pinto, D. C. G. A., & Barreto, M. C. (2024). New Phytol Derivatives with Increased Cosmeceutical Potential. Molecules, 29(20), 4917. https://doi.org/10.3390/molecules29204917