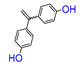
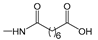
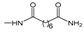
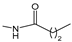
2.1. Ferrocenophanes Derived from Ferrocifene Derivatives
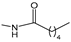
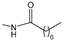
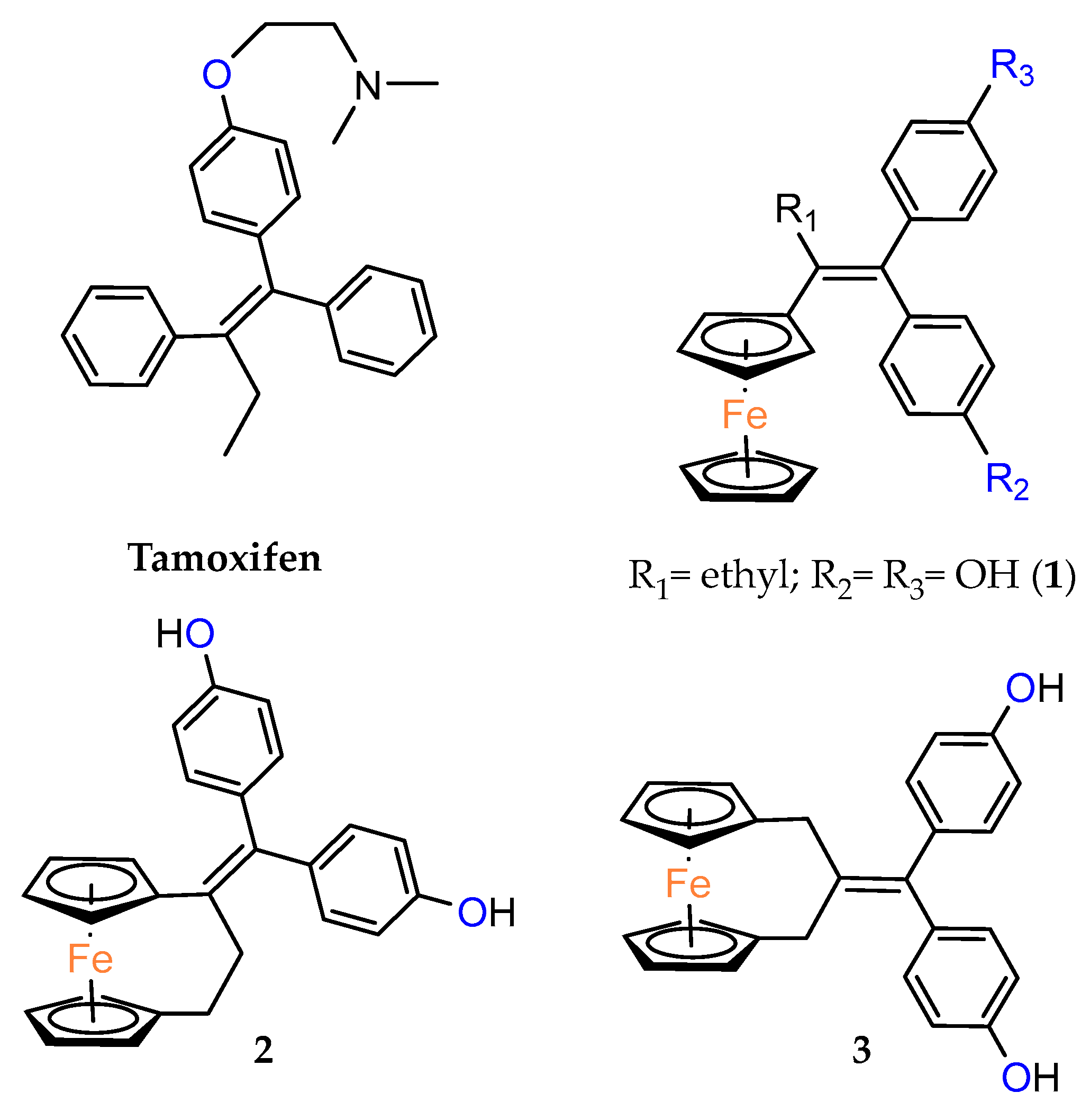
Ferrocenophane phenols are a type of ferrocene named after the first published examples of hybrid compounds containing both ferrocene fragments and the anticancer drug tamoxifen [
20]. Tamoxifen is a nonsteroidal selective estrogen receptor modulator (SERM) used to treat all stages of hormone receptor–positive breast cancer [
21]. Ferrocenyl hydroxytamoxifen
1 as a prototype for a new range of cytostatic agents targeting the estradiol receptor site was published in 1996 (
Figure 1) [
8]. Ferrocifenes were the first examples demonstrating that the modification of polyphenols with a ferrocenyl substituent can significantly increase cytotoxicity in vitro [
22,
23]. Although the compounds were designed to target delivery of the cytotoxic ferrocene group to cells overexpressing estrogen receptors, they showed significant in vitro activity against both hormone–dependent MC–7 cells and triple–negative MDA–MB–23 breast cancer cells (IC
50 = 0.7 μM and IC
50 = 0.6 for
1, respectively). These observed antiproliferative effects were much stronger than those of 4–hydroxytamoxifen, the reference antiestrogen and ferrocene alone, which was inactive against these breast cancer lines. However, the recent cytotoxicity values for
1 have been inconsistent with those previously published [
24]. The IC
50s determined were only 43.3 μM for MCF–7 cells and 26.3 μM for MDA–MB–231 cells after 72 h of incubation. In this study, compound
1 also showed activity against PANC1 pancreatic cancer cells with an IC
50 of 12.5 μM, twice that of tamoxifen, and significantly increased cellular oxidative stress compared to the reference compound. It is noteworthy that, while promising structures for pancreatic cancer therapy are still being investigated in the case of ferrocene derivatives (see, for example, ferronucleoside, which inhibits DNA replication in a panel of pancreatic cancer cells [
25]), no such reports have been published in the case of
ansa–ferrocenes.
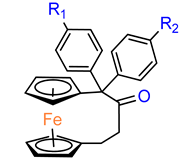
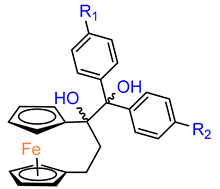
Over the last three decades, intensive SAR research has led to the discovery of 300 ferrocifenes with various substitutions at the R
1, R
2 and R
3 positions (see
Figure 1). Some of these compounds have demonstrated potent anticancer properties against various MDR cancer cell lines, including glioma, metastatic melanoma, breast cancer and leukemia [
26]. One direction of synthetic modifications to improve the biological activity of ferrociphenols has been the introduction of carbon, or other atoms containing bridges, linking the two cyclopentadienyl rings in the structure.
Jaouen and coworkers, the leading research group in the synthesis and biological evaluation of ferrocifen analogues, first demonstrated the effect of stiffening structure
1 on biological activity. Two rigid analogues
2 and
3 with cyclopentadienyl rings connected by a three–carbon bridge were designed, synthesized and compared in bioassays with a flexible analogue [
27]. Studies of relative binding affinity (RBA) for the estrogen receptor (ER) and activity against breast cancer lines MCF–7 ER+ and MDA–MB–231 ER– and PC–3 prostate cancer cells showed the greatest difference in cytotoxicity of the compounds on hormone–independent MDA–MB–231 and PC–3 cells. (
Table 1) Rigid compound
2 was one order of magnitude more cytotoxic than flexible compound
1 or rigid
3, which lacks conjugation between the phenolic and ferrocenyl groups. Both rigid compounds had estimated cytotoxic activity comparable to
1 for MCF–7 cells. These results suggested that there may be a competition between the positive estrogenic effects and negative cytotoxic effects on this cell line, which could not be predicted from the simple binding values. Compound
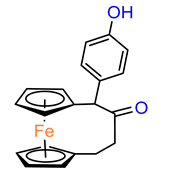
4, with one hydroxyl group,
5 with one amine group in the phenyl ring and
6 with an acetamide substituent showed similar activity against MDA–MB–231, acting less effectively than analogue
2 [
28]. Replacement of one hydroxyl in
2 by amine (
8) or acetamide (
10) substituents resulted in similarly high activities against MDA–MB–231 with IC
50 = 0.06 and 0.09 μM, respectively. Compound
11 with one amine group in each phenyl ring also showed high activity. It was in contrast to the result for its acetamide analogue
9, which acted two orders of magnitude weaker. Interestingly, acetylated prodrug analogues of
2, i.e., mono acetylated
12 or peracetylated
13, acted at the level of the leading structure against MD–MB–231 [
29,
30].
Compound
5 in extended screening in vitro revealed better activity against human glioblastoma SF–295 (IC
50 = 1.0 µM), human ileocecal colorectal adenocarcinoma HCT–8 (IC
50 = 0.5 µM) and human promyelocytic leukemia HL–60 (IC
50 < 0.12 µM) than against melanoma cancer line MDA–MB–435 (IC
50 > 61.68 µM) derived from the M14 line [
32]. Moreover, it showed promising characteristics possessing of high antiproliferative activity and low hemolytic activity on mouse erythrocytes [
33].
Further study indicated that hybrids
2,
8,
10 and
12 could act as DNA alkylating agents or DNA antimetabolites [
11,
29]. Particularly, hybrid
2 (IC
50 range 48–580 nM) possessed a broad activity spectrum against a panel of 60 human cancer cell lines, derived from nine different cancer types: leukemia, lung, colon, CNS, melanoma, ovarian, renal, prostate and breast [
29,
30]. The highest sensitivity of
2 was observed in vitro on human SK–Mel28 melanoma cells (IC
50 = 1.2 μM) [
26,
34]. Compound
2 was also tested on three ovarian epithelial cancer cell lines, including A2780–Cis resistant to cisplatin (IC
50 = 0.359 µM for A2780; IC
50 = 0.165 µM for A2780–Cis; IC
50 = 1.910 µM for SK–OV–3). However,
2 showed a twofold higher selectivity factor (SF = 0.46) against A2780 than the cisplatin–resistant line, when compared to normal human pulmonary fibroblasts MRC5 [
35]. The hybrid
2 also showed acceptable acute toxicity in mice, with the maximum tolerated dose of 100 mg/kg. Compound
2 induces senescence in various cancer cell models associated with distinct sensitivity to pro–apoptotic stimuli [
36]. The high and broad–spectrum anticancer activity as well as low toxicity made this hybrid a useful starting point in drug development to combat various types of cancers.
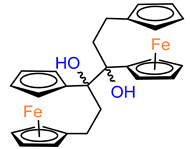
Another study was conducted to investigate the impact of the length of the carbon bridge connecting the cyclopentadienyl rings on the activity against MDA–MB–231 cells [
37]. The analogues of compound
2 with a four– (
19) and five–carbon bridge (
20) exhibited an order of magnitude lower activity than the original structure of
2. Furthermore, the introduction of an additional methyl substituent into the compound altered its activity. However, the R–isomer (
21) demonstrated activity similar to that of
2. The other compound of interest was the relatively bulky derivative of [
5]ferrocenophane with two –[bis–(4–hydroxyphenyl)]methylidene groups (
24). This relatively large compound was still active with an IC
50 of 2.7 μM [
37].
The anticancer activity of hybrid derivatives of superoylanilidine hydroxamic acid (SAHA) having a [3]ferrocenophan–1–ylidene substituent was also evidenced [
38]. The compounds
25,
26 and
27 showed strong antiproliferative activity against triple–negative MDA–MB–231 cells, with IC
50 values in the 0.84–2.72 μM range. The primary amide
26 was found to be slightly more cytotoxic than its hydroxamide analogue
27. The cytotoxic effects of compounds
25–27 were also observed on hormone–dependent MCF–7 breast cancer cells (
Table 2), where all three compounds (
25,
26 and
27) showed antiproliferative activity with IC
50 values in the range of 0.87–4.05 μM. In contrary, their analogues lacking the organometallic moiety were unable to inhibit 50% of cell growth even at concentrations of 10 μM. The exception was SAHA (IC
50 = 1.04 μM), although ferrocenophane hydroxamide
27 (IC
50 = 0.87 μM) was again slightly more active. In general, incorporation of the ferrocenophane and ferrocene derivatives of hydroxytamoxifene into the suberamide structure enhanced the antiproliferative activity of the resulting compounds against MDA–MB–231 and MCF–7 cancer lines. Of the compounds tested, ferrocenophane derivatives showed the greatest cytotoxicity on MCF–7 breast cancer cells. The electrochemical behavior of these ferrocenophanic suberamides suggested that they undergo redox activation, which may contribute to their antiproliferative activity [
38].
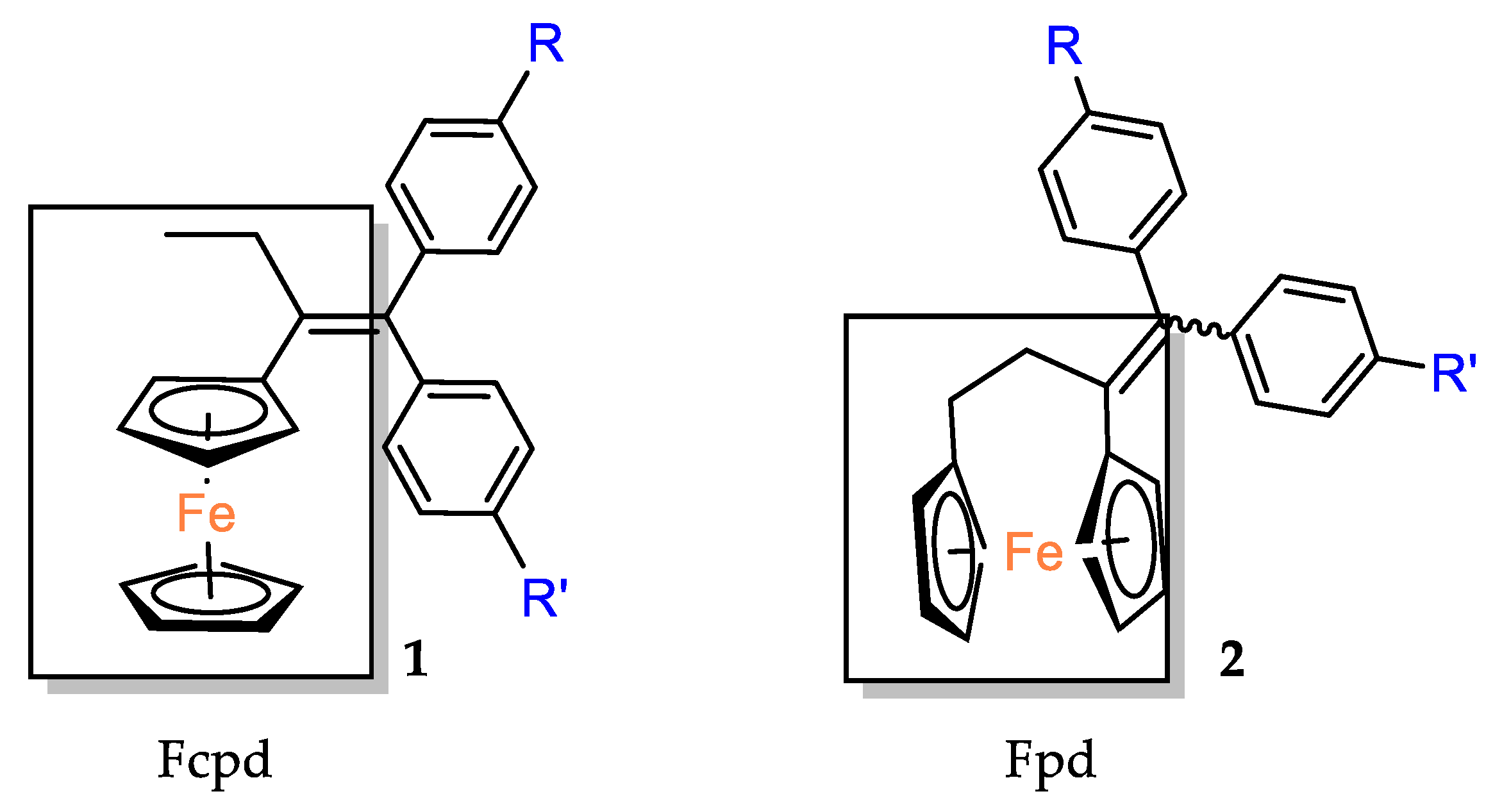
The cytotoxic activity of the [3]ferrocenophan–1–ylidene (Fpd) bis–aniline 11 (Fpd,
Figure 2) analogues, additionally substituted at the amine groups, was evaluated against hormone–resistant MDA–MB–231 breast cancer cells [
39]. The straight dose–dependent effects were not observed for compounds
28,
29 and
30. The percentage of cell growth inhibition at concentrations of 1 μM was found to be 27%, 23% and 18%, respectively. The decrease in activity observed for the analogues was accompanied by an increase in the length of the acyl carbon chains. At concentrations one order of magnitude higher (i.e., 10 μM), only compound
29 inhibited the growth of the cancer cell line by more than 50%, reaching 51%. The compounds obtained demonstrated reduced activity compared to the simple diacetanilide derivative
9 (IC
50 = 5.64 μM). Furthermore, it was demonstrated that modifying the substituents from a basic amine to an amide distinctly diminished the activity against MDA–MB–231 cells. All anilides obtained demonstrated a minimum of two orders of magnitude lower cell growth inhibition when compared to bis–aniline
11 (IC
50 = 0.05 μM) [
39]. The activation mechanism was found to be linked to the intramolecular electron transfer process previously observed for phenolic derivatives. The conversion of Fcpd compounds to ferrocene has been postulated to promote an increase in the ROS level, which can result in direct DNA damage and/or the activation of tumor suppressor genes, leading to apoptosis or senescence of cancer cells [
39].
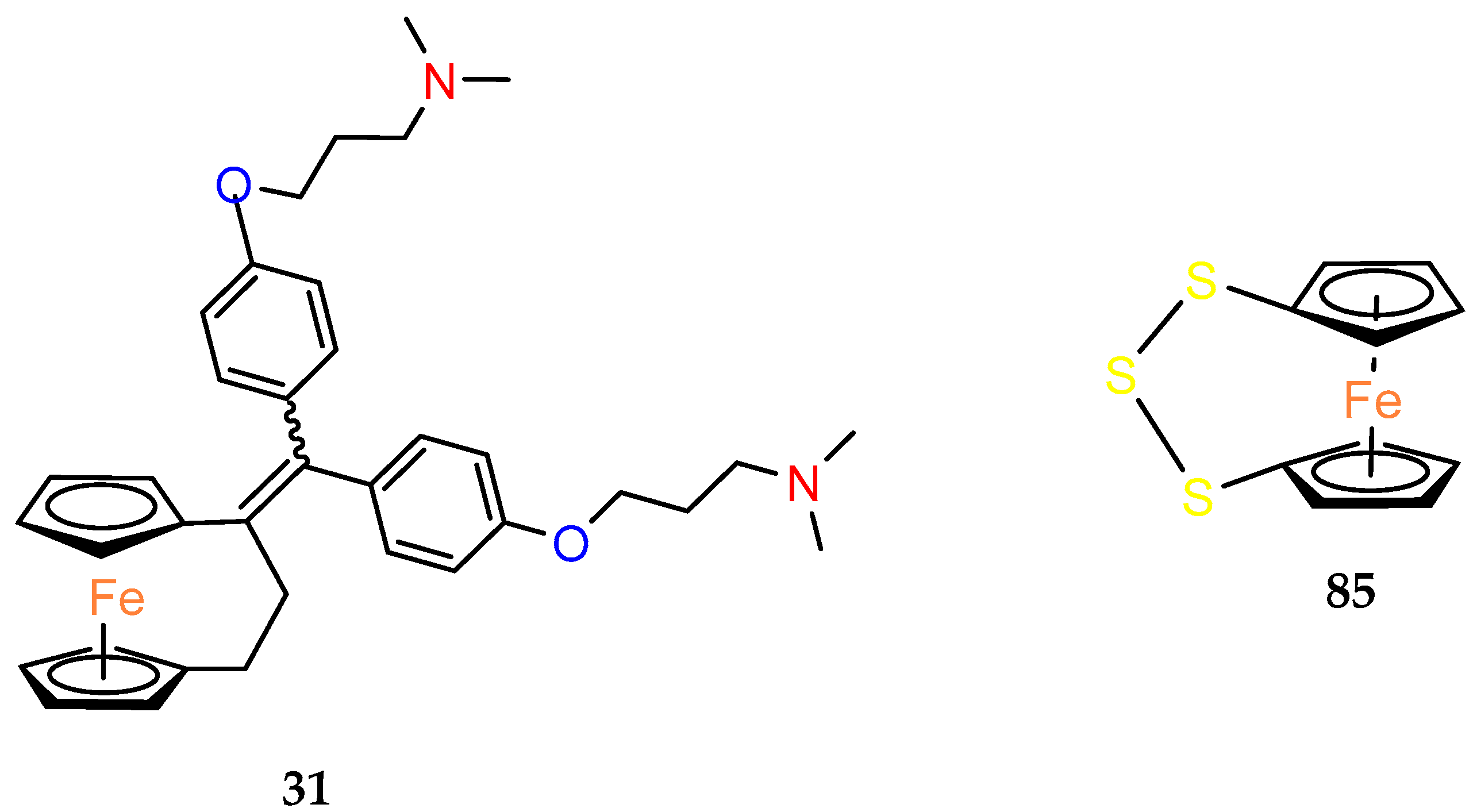
Replacement of the OH group in
2 by two dimethylaminoalkyloxy chains decreased the cytotoxicity effect on MDA–MB–231 in compound
31 [
40]. Against MCF–7 cells, compound
31 showed a slight, but reproducible, proliferative estrogenic effect at low concentrations (1 × 10
−8 and 1 × 10
−7 M). Following observation on MDA–MB–231 cells, it was very cytotoxic against MCF–7 at higher concentrations (between 1 × 10
−7 M and 1 × 10
−6 M). According to the binding affinity biochemical studies,
31 appeared highly recognized by the alpha form of ER. While the ability to generate quinone methide formation was blocked in the structure of
31, an alternative mechanism of its cytotoxicity was investigated in in silico studies. The estimated affinity of the compound to Zn
2+ and Ca
2+ indicated the complexing role of two aminoalkyl chains on these cations as a potential mechanism of action [
40].
The introduction of an additional hydroxyl group in the
meta position of the
para–hydroxyphenyl ring in compound
4 led to a compound
32 with similar biological activity to that of compounds
4,
6 and
31 (MDA–MB–231 breast cancer cells). The high antiproliferative activity of compound
31 was attributed to the narrower HOMO–LUMO gap present in the oxidized form of [3]ferrocenophane moieties, resulting in more reactive species [
41].
A new series of compounds with various aminoalkyloxy side–chains (O(CH
2)
3NMe
2, O(CH
2)
3piperidine, O(CH
2)
3–pyrrolidine, NHCO(CH
2)
2NMe
2) were also characterized in terms of their antiproliferative activity against the MDA–MB–231 cancer cells. The IC
50 values of compounds
33,
34 and
36 were between 0.17 and 0.19 μM. These values were twice as weak as for the model diphenol
2 (0.09 μM). Nevertheless, these derivatives were among the most efficacious compounds against MDA–MB–23 ever obtained. The lipophilicity of these compounds did not appear to play an important role in their activity. Once again, it was demonstrated that systems lacking an OH group, as in
35 and
39, are less active. Although, the substitution of the aminoalkoxy chain –O(CH
2)
3NMe
2 by the amido chain NHCO(CH
2)
2NMe
2 may produce different effects, the IC
50 values of compounds
35 and
39 were very similar. In contrast, amido compound
38 exhibited three times better cytotoxicity than
33 (IC
50 = 0.05 μM vs. 0.18 μM), belonging to the most active
ansa–derivatives ever studied against the MDA–MB–231 cancer cell line. For comparison, molecules
26 and
27 were slightly less effective than compounds
35 and
39. The lengthening of the amido chain may impact their diminished activity [
42]. The in vitro effect of the new complexes
26,
27 and
33–39 on the growth of MCF–7 hormone–dependent cells was then studied concerning their estrogenic and antiestrogenic properties. The antiestrogenic effect was not observed in the ferrocenophane series
26,
27 and
33–39, although a slight estrogenic effect was observed for compounds
33,
34, 38 and
39. The compounds, in general, preserved an affinity for the estrogen receptor. Thus, the estrogenic or antiestrogenic effects were then analyzed exhaustively on the MCF–7 cells for two compounds,
33 and
40, which differed only in the length of the aminoalkoxy chain. It was investigated at three concentrations (1, 10, 100 nM). Compound
33 triggered an estrogenic effect at 1 nM, which was inverted at a higher concentration of 100 nM due to the appearance of a cytotoxic effect. In contrast, for compound
40, the antiestrogenic effect started immediately from 1 nM. Therefore, the authors proposed that this phenomenon could be attributed to the length of the aminoalkyl side chain, which may change the estrogenic effect into an antiestrogenic, as was confirmed in the modeling study [
42].
Furthermore, the same research group also studied the MDA–MB–231 cell line inhibition growth produced by compounds slightly more different in structure than typical ferrocenophane phenol derivatives (
Table 3) [
42]. Compounds
48 and
49, containing a 1,2–diol moiety in the place of a double bond present in model compound
2, showed the best antitumor properties in the obtained group. The results were comparable to these for their analogues
4 and
7, i.e., at the 0.1–0.01 μM level of IC
50. Moreover, it is interesting to note that two diastereoisomers
49 and
50 differ slightly in their activities.
2.2. Other Ferrocenophanes with All Carbon Bridges
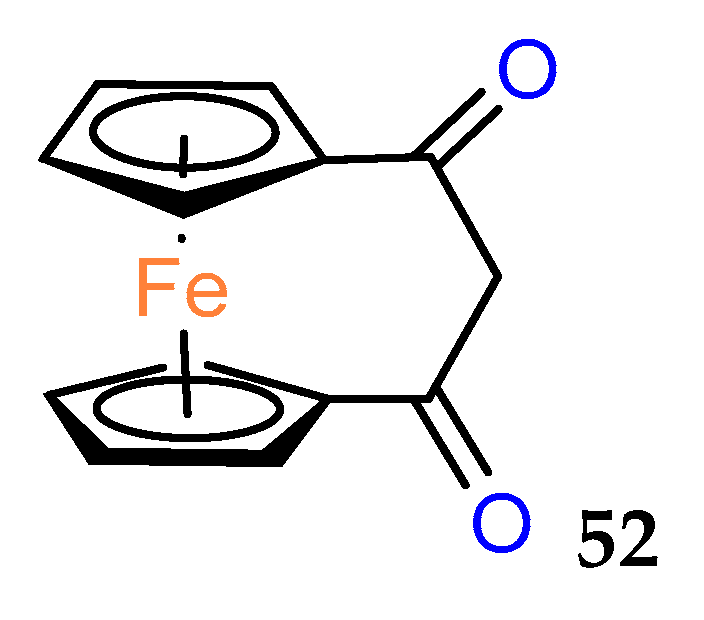
The history of biologically active ferrocenophanes is mainly based on the ferrociphenol derivatives. However, as early as 1980, a simple [3]ferrocenophane–1,3–dione
52 (
Figure 3) was tested in vivo against epithelial carcinoma and was found to be inactive [
43].
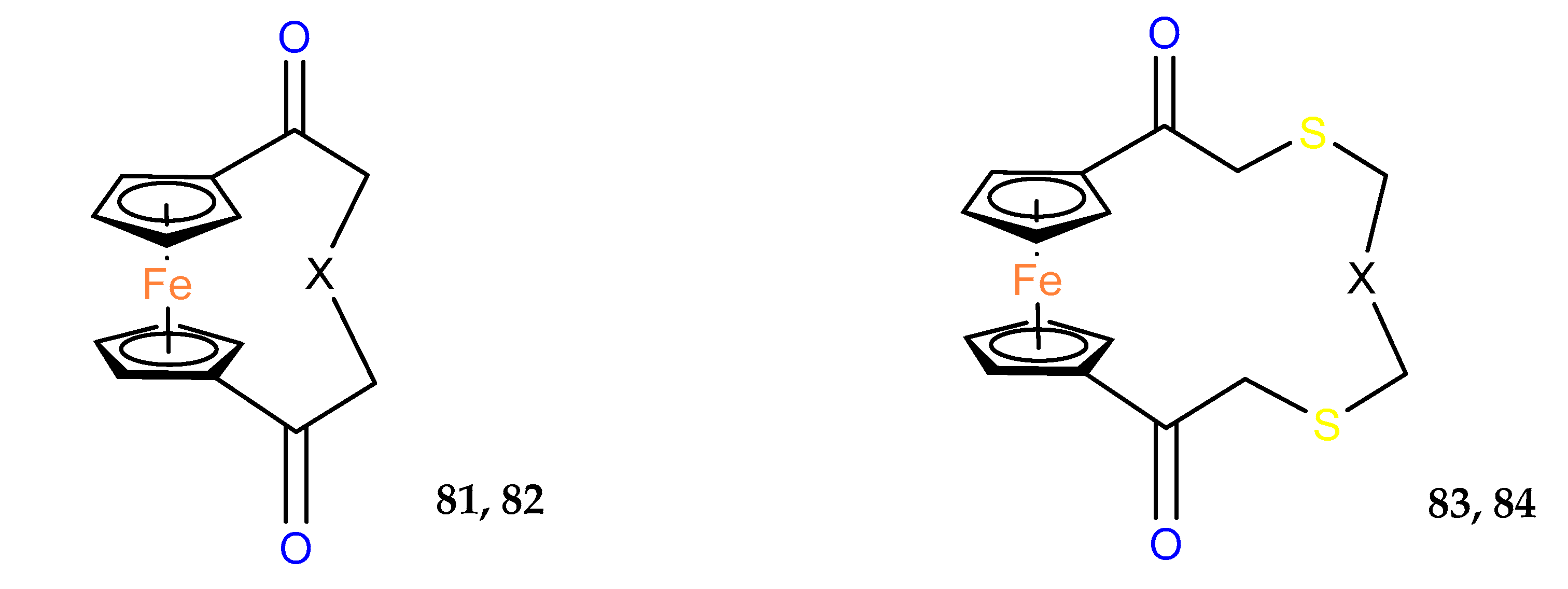
Another interesting group of compounds was designed to enhance the activity of the antibiotic platensimycin. Although the obtained
ansa–ferrocene derivatives did not show antimicrobial properties, their intermediates
54 and
55 with protective groups on carboxylic substituents and alternatively on hydroxyl substituents showed in vitro anticancer activity (
Table 4). In particular, compound
54, in which only the carboxyl group was protected as a 2–(trimethylsilyl)ethyl (TMSE) ester, showed activity at the micromolar level against pancreatic ductal adenocarcinoma PT45 and hepatocellular carcinoma HEPG2 [
44].
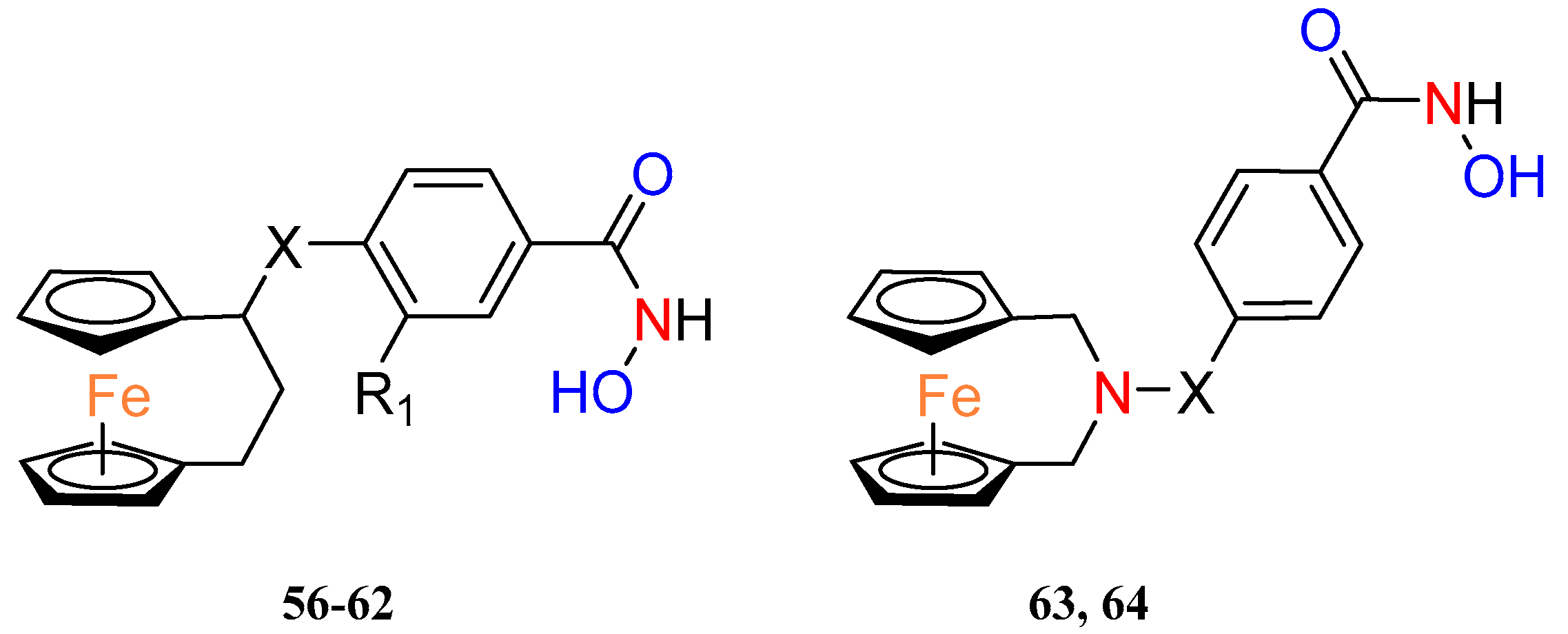
Recently, Yan et al. combined [3]ferrocenophanes and 2–aza–[3]ferrocenophanes
56–64 with benzohydroxamic acid, via a piperazine or piperidine linker, to obtain a new series of ferrocenyl hydroxamic acids (
Table 5). Two
ansa–ferrocene hybrids have shown inhibition of histone deacetylase 6 (HDAC6) at nanomolar concentrations, and this enzyme has been considered a possible target for anticancer and antineurodegenerative therapies [
45]. Selected effective inhibitors of HDAC6 were further tested on other HDAC subtypes to identify compound
60 as the most selective to HDCA6 with selectivity ratios, to others from the HDCA family, ranging from 13 to more than 261. Both HDAC6 inhibitors
60 and
63 revealed moderate antiproliferative activity against a panel of cancer cell lines including a prostate cancer cell line (22RV1), an immunoglobulin λ myeloma cell line (MM1.S), a monocyte leukemia cell line (MV4–11), a mantle cell lymphoma cell line (JEKO–1) and a breast cancer cell line (4T1). Compound
63 also induced apoptosis in 4T1 cancer cells in a dose–dependent manner. Western blot studies showed altered levels of proteins involved in the apoptosis pathway under the influence of compound
63, e.g., PARP or caspase–3. The possible synergism of the pro–apoptotic effects of the compounds was also pointed out by the fact that HDAC inhibitors have the potential to induce apoptosis spontaneously and ferrocene complexes can induce apoptosis through ROS. Thus, this was confirmed by showing an increase in total ROS in 4T1 cancer cells after incubation with
63.
Buchowicz and coworkers designed and synthesized new uracil–triazole–[4]ferrocenophane hybrids with antitumor activity against breast and lung cancers. The antitumor potential of
ansa–ferrocene (±)–
65 was evaluated on three cell lines: the hormone–dependent breast cancer cell line MCF–7, the triple–negative breast cancer line MDA–MB–231 and A549 lung cancer cells (
Table 6). Compound (±)–
65 acted at the cisplatin level against all cancer lines studied and showed better activity than its nonbridged analogue. The compound also showed significantly lower toxicity against normal MRC–5 cells than cisplatin, particularly in the case of lung cancer A549, where SI for (±)–
65 was 4.2 vs. 1.8 for cisplatin [
46].
The same research group published a synthesis and biological evaluation of (allylaminomethyl)
ansa–ferrocene derivatives (±)–
66 and (±)–
67, which were then compared with their ferrocene analogues [
47]. The anticancer effects of (±)–
66 and (±)–
67 were also evaluated against hormone–dependent MCF–7 breast cancer and A–549 lung cancer cells. Furthermore, the compounds were studied using prostate adenocarcinoma PC–3 and mouse noncancerous fibroblasts line Balb 3T3. The tested cell lines showed twice as much sensitivity to
ansa analogues as their ferrocene counterparts (the same order of magnitude). Both compounds demonstrated the highest response against MC–7 cells. Compound (±)–
67 was, therefore, administered to examine the effect on the cell cycle progression in this cancer line. The studies showed cycle arrest in the S–phase of DNA replication following administration of increasing concentrations of (±)–
67. In addition, a sub–G1 phase was observed in MCF–7 cells treated with 40 µM and 60 µM of compound (±)–
67, indicating the apoptotic nature of an action of (±)–
67.
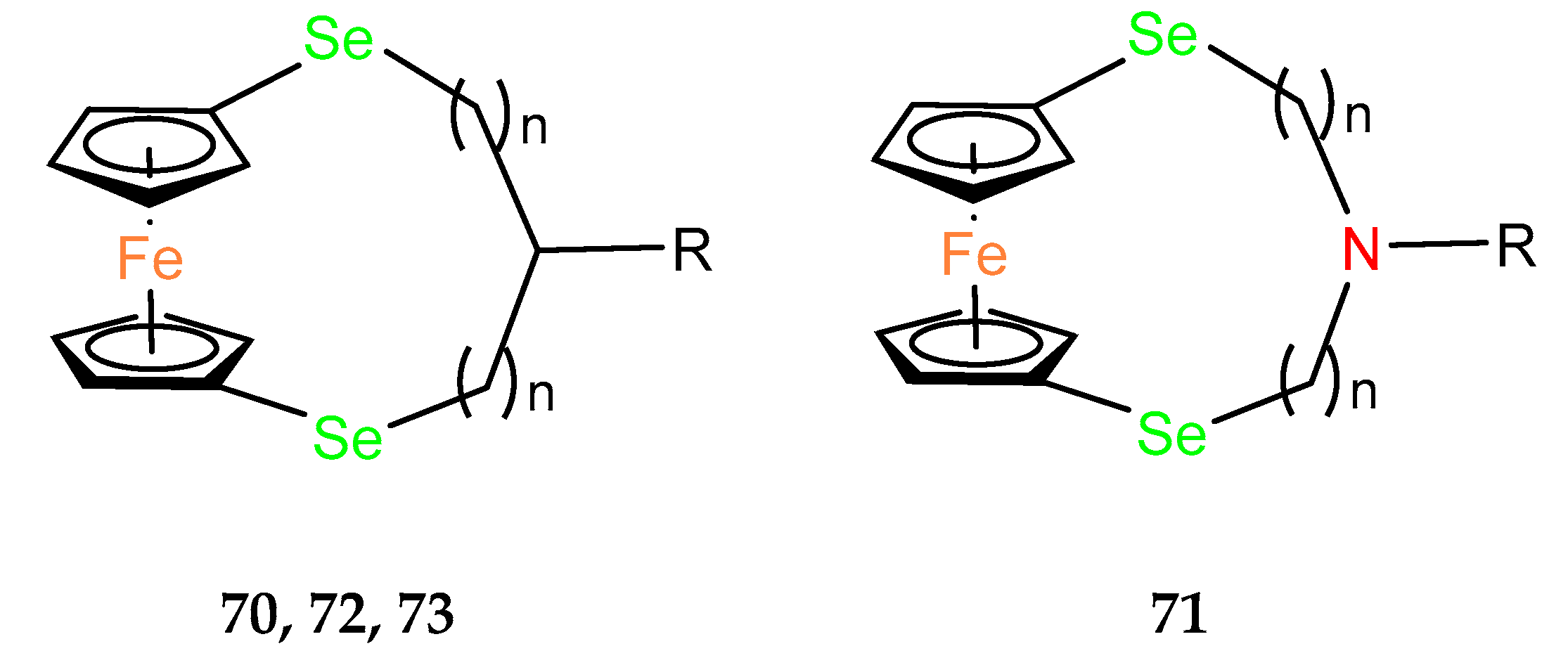
2.3. Ferrocenophanes with Bridges Containing Heteroatoms
The biological properties of ferrocenophanes have been tested and demonstrated in several compounds. In addition to the most extensively studied carbon–bridged compounds, the other examples of
ansa–ferrocenes contain nitrogen (e.g.,
63,
64), selenium or phosphorus. We note that research on the selenium–bridged compounds is a significant contribution to the broader studies on the bioactivity of different organoselenium derivatives related to their antioxidant and anticancer properties [
48].
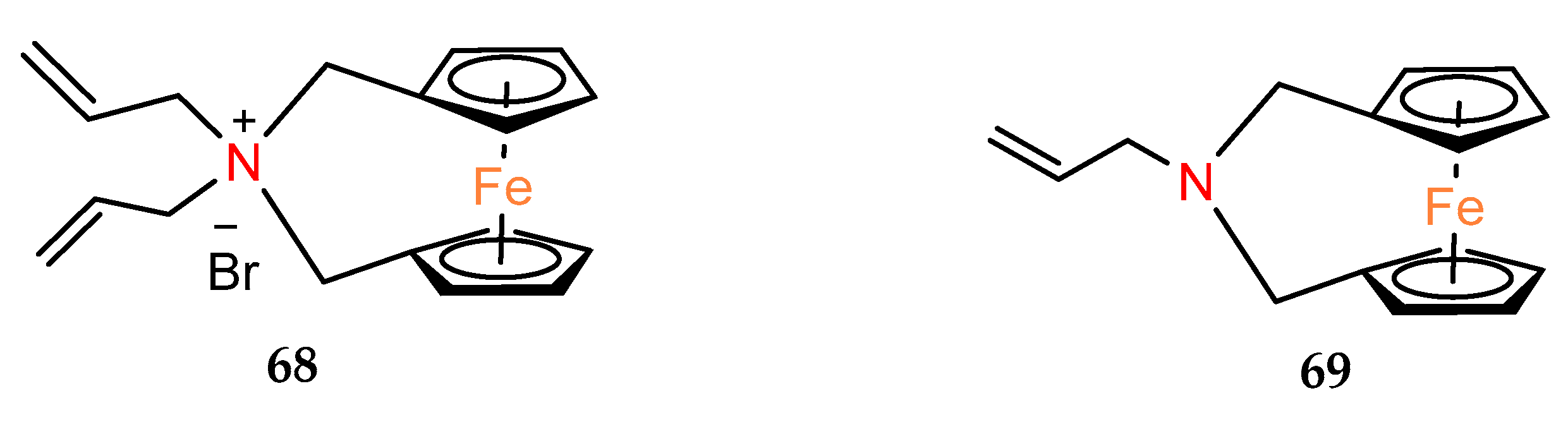
In studies on allyl derivatives of ferrocene [
47], the cytotoxicity of
ansa–ferrocenes with nitrogen in the bridge was also checked. Compounds
68 and
69 (
Figure 4) showed weak activity (EC
50 > 100 µM) for each of the tested cell lines.
In studies on the cytotoxicity of organoselenium dopamine conjugates, the
ansa–ferrocene derivative
70 was found to be the most active in the cytotoxicity assays using AGS (gastric adenocarcinoma), A2780 (ovarian carcinoma), A549 (non–small–cell lung carcinoma), BxPC–3 (pancreatic cancer), HepG2 (hepatocellular carcinoma) and MGC–803 (human gastric cancer) cell lines (
Table 7) [
49]. HepG2 appeared to be the most sensitive line, with an IC
50 value of 2.2 ± 0.5 μM. Therefore, the cell cycle distribution in HepG2 was analyzed after administration of 10 or 20 µM of the compound. G1 arrest was observed by indicating a dose–dependent increase in the population of cells in this phase. Subsequently, the population of cells in the S–phase decreased significantly. A dose–dependent presence of cells in the G2/M phase was also observed. Because cell cycle arrest plays a role in apoptosis, the dose–dependent ability of the tested compound to induce apoptosis or necrosis in HepG2 cells was further confirmed by flow cytometry in the presence of annexin stain. The late–stage apoptosis rates of HepG2 cells treated with 10 and 20 µM increased in a dose–dependent manner to 13.09% and 33.62%, respectively. The necrotic rate of HepG2 cells also increased. As a result, compound
70 was effective in directly killing cells or inhibiting the cells growth. Biochemical tests also showed an increase in the pro–apoptotic enzymes caspase 3 and caspase 9. The expression of the pro–apoptotic protein Bax also increased, while a decrease in the anti–apoptotic protein Bcl–2 was observed. Immunoblotting studies also showed an increase in the expression level of the p53 suppressor protein after administration of compound
70. Encouraging results from in vitro tests led the authors to in vivo experiments, where compound
70 was shown to inhibit tumor growth in nude mice bearing HepG2 tumor xenografts. In addition,
70 inhibited both tubule formation and endothelial cell (HUVEC) migration in the anti–vascular activity assay.
Based on the promising results for the seleno–organic derivative of dopamine
70, a similar dopamine, aza–
ansa–ferrocene
71, was obtained among a novel group of compounds. The mechanistic study revealed that the cytotoxicity of these ferrocenyl seleno–dopamine derivatives was mainly related to the Fenton–like reaction under physiological conditions [
50].
Core–shell conversion nanoparticles (UCNPs) were functionalized via surface coordination chemistry protocols using
ansa–ferrocene derivatives
70 and
71 to give conjugates Fc–UCNPs [
51]. A drug carrier form (Fc–UCNP) was also obtained by encapsulating Fc–UCNPs in liposomes. It was assumed that the obtained conjugates would be able to release OH radicals in a Fenton–photo reaction activated by NIR radiation to induce cancer cell death. In vitro studies on AGS and MGC–803 cancer lines evidenced the radiation effect in compound
72 (
Table 7). None of the two
ansa–ferrocene derivatives,
72 nor
73, was selected for further in vitro studies, which demonstrated that the UCNP–Lipo model conjugate, in a xenograft model using AGS cancer cells in BALB/c nude mice, underwent preferential accumulation in a tumor site followed by its enhanced uptake to cancer cells.
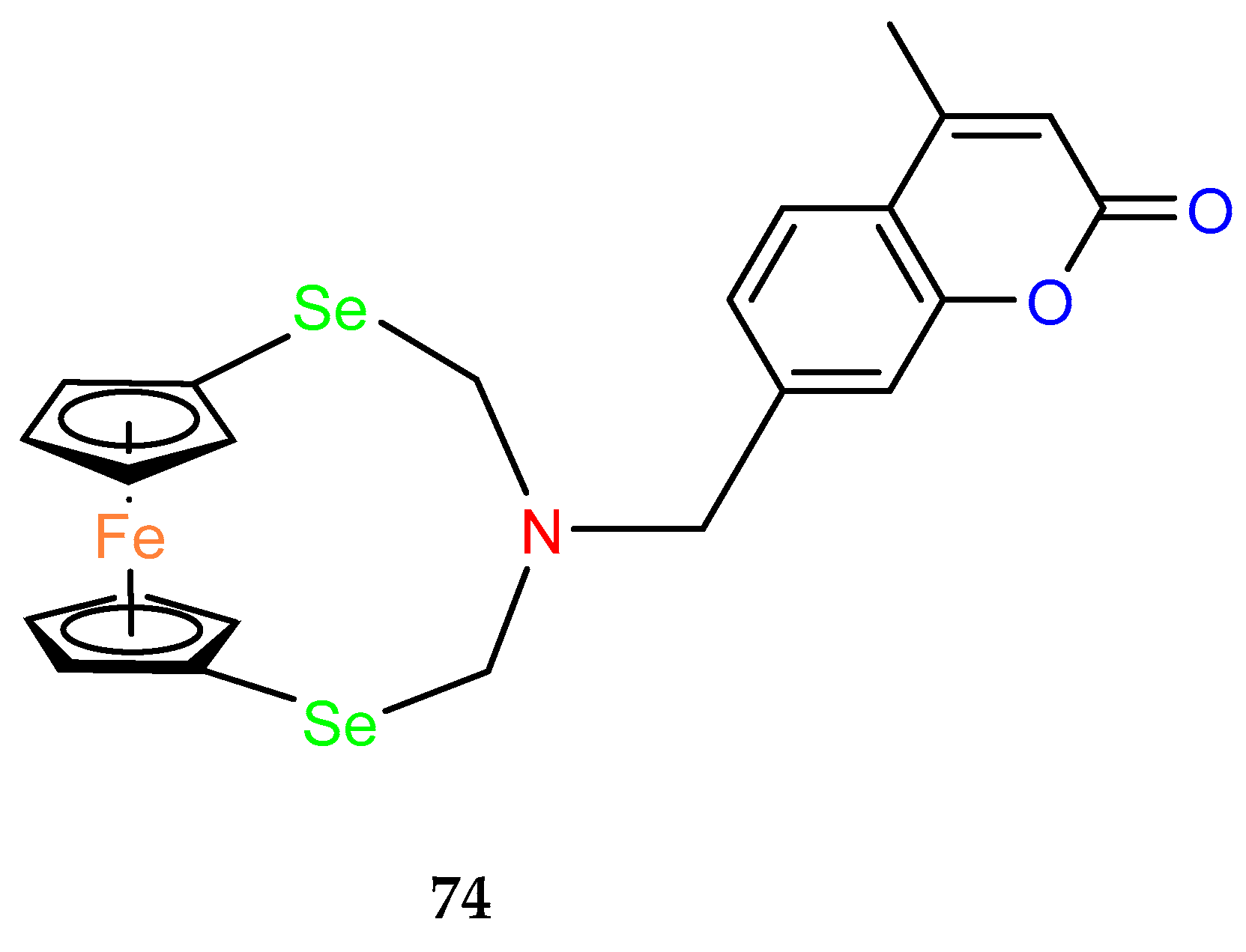
To investigate the utility of
ansa–ferrocene derivatives as compounds with phototriggering properties, a noncovalent complex containing an Eu
3+ cation and a ClO
4– anion in the structure of a ferrocenophane derivative with coumarin fluorophore
74 was prepared (
Figure 5). In in vitro cell line studies, the compound, which exhibits photoluminescence upon UV irradiation ≥ 365 nm, showed phototriggered anticancer activity against HepG2 liver cancer cells, as confirmed by confocal imaging [
52].
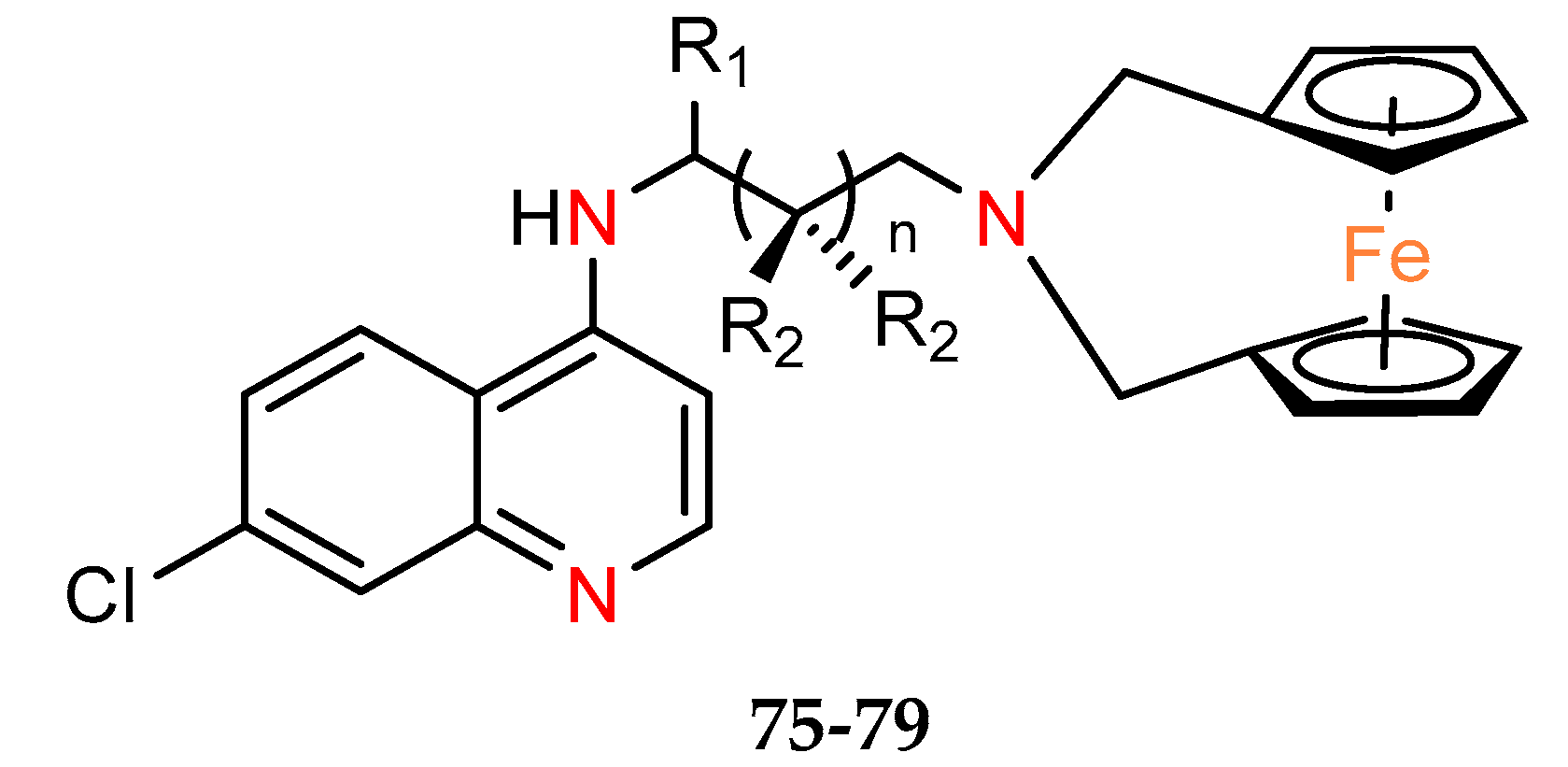
Five ferrocene chloroquine derivatives with the terminal nitrogen of the chloroquine derivative bridging the two cyclopentadienyl rings of ferrocene (
75–79) were synthesized by Salas et al. [
53]. They have been characterized in terms of antiplasmodial activity, as well as cytotoxicity on breast epithelial cells cell line MCF–10A, commonly used as a model for normal human breast cells [
54] and melanoma cancer line MDA–MB–435S (but not a breast cancer cell line [
55] as mentioned in the cited paper [
45]) (
Table 8). All tested compounds were found to lower the viability of the cancer cell culture but with low selectivity toward normal cells. Of the series studied, compound
79 appears to be the most active and with the worst selectivity index versus normal line SI = 0.5.
Ferrocenophane with a bridge of two phosphorus atoms coordinated to gold(I) was tested in vitro on various cancer cell lines in the 1990s [
56]. Compound
80 showed the best activity against the SW1116 cell line (IC
50 = 48.9 µg mL
–l). The cytotoxicity, although improved by the presence of gold, was not better than the cytotoxicity of the well–known drug cisplatin (
Table 9).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}