
Theoretical Investigation into Polymorphic Transformation between β-HMX and δ-HMX by Finite Temperature String

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Peaks in Pair Distribution Function of β-HMX and δ-HMX
2.2. Convergence of FTS and K-Means Clustering
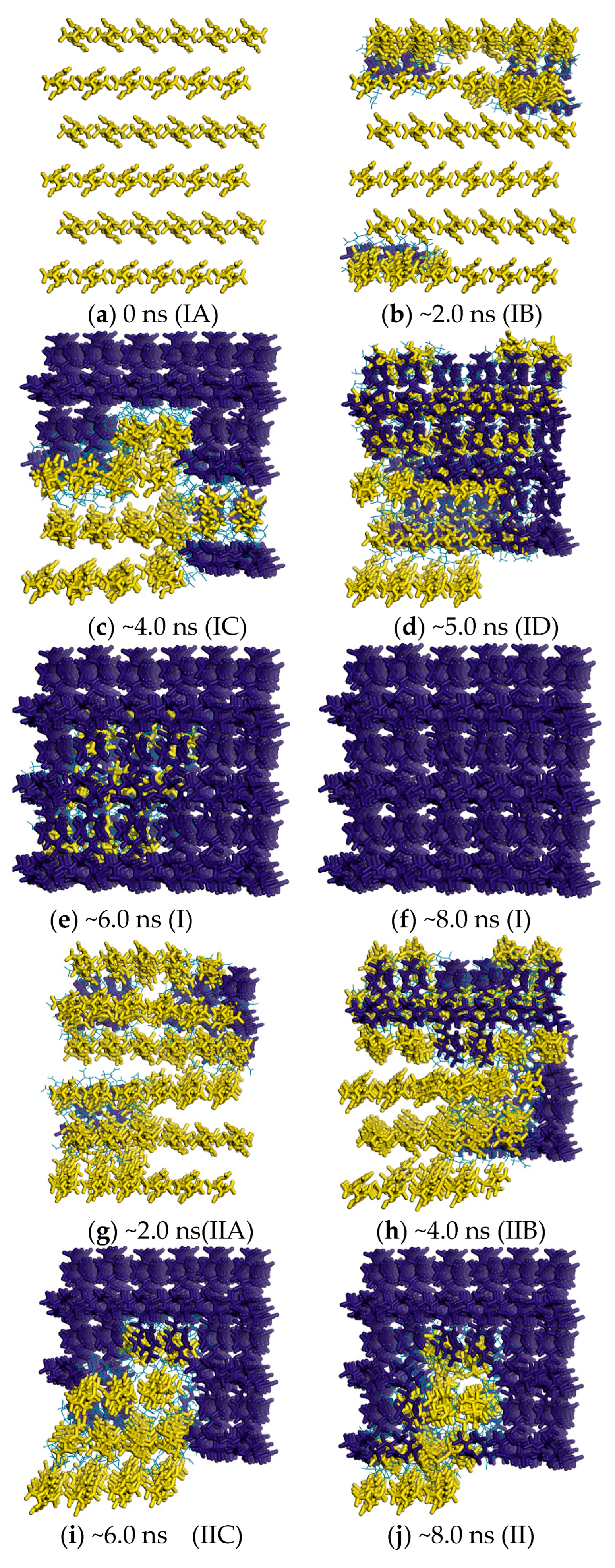
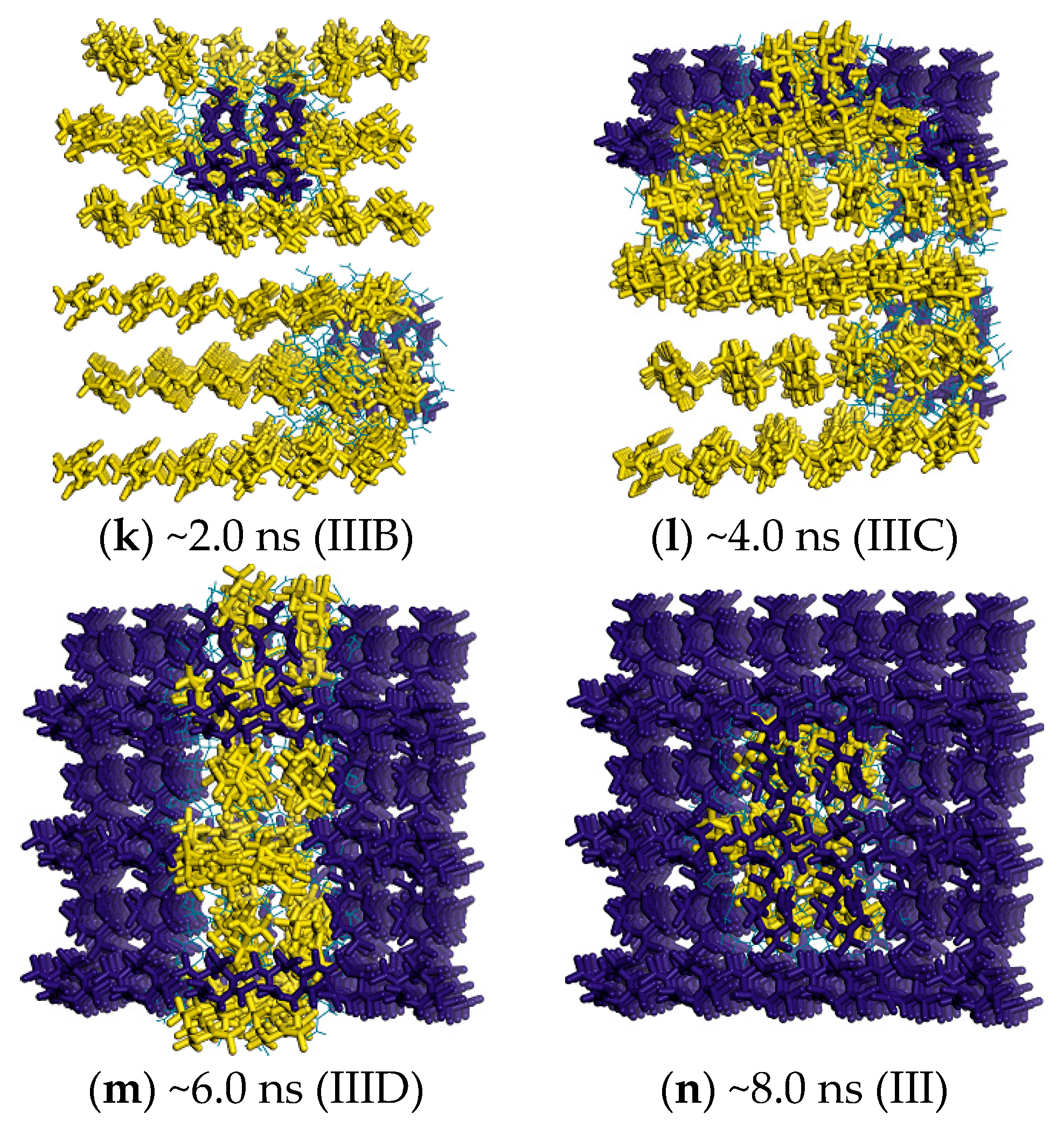
2.3. Minimum Free-Energy Path
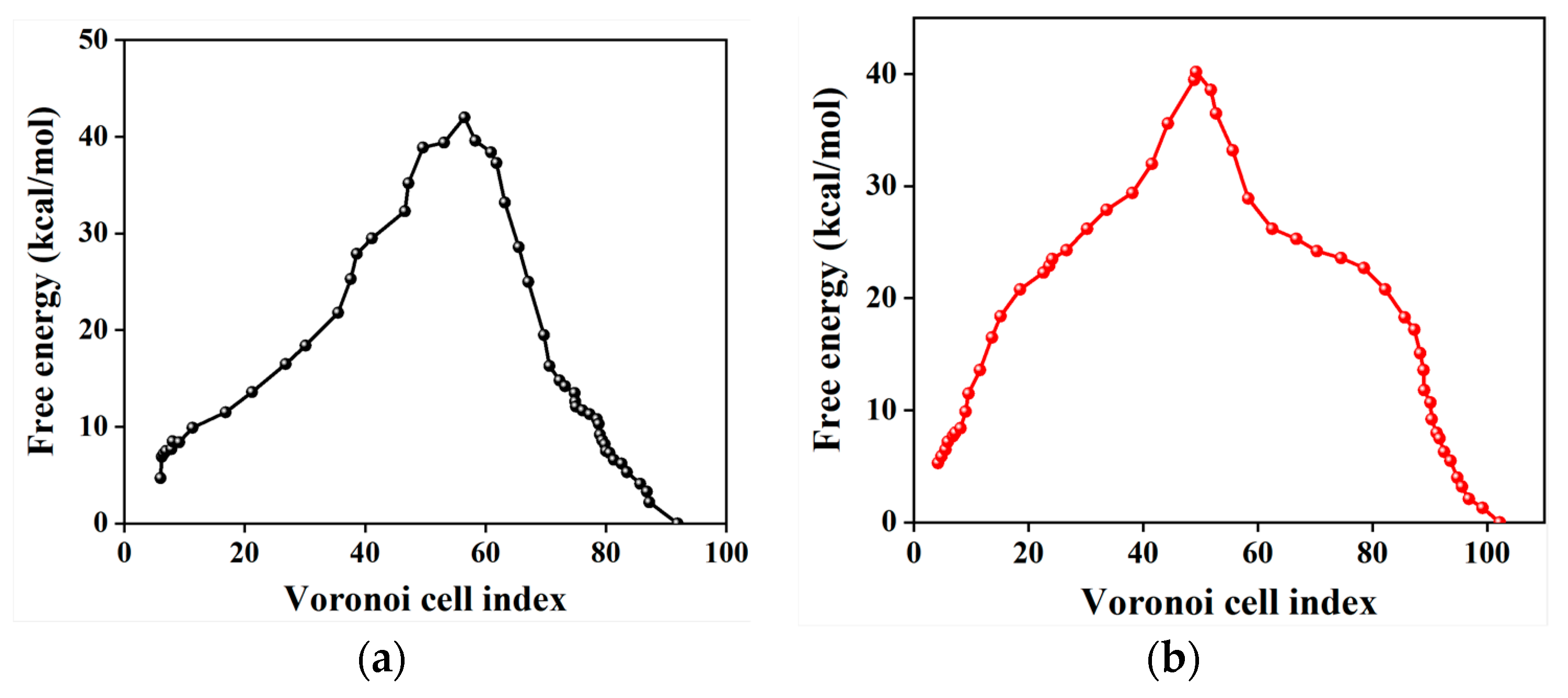
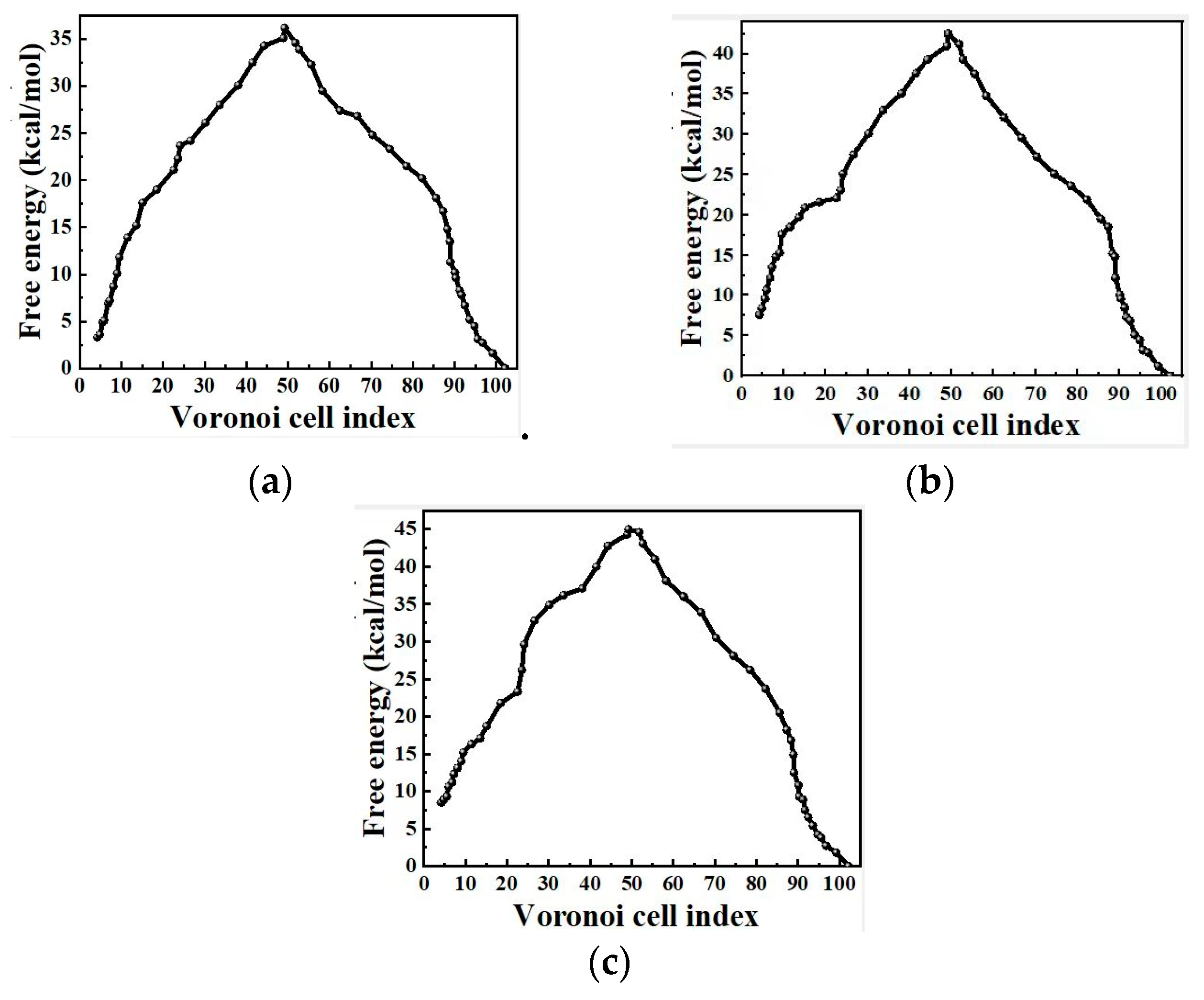
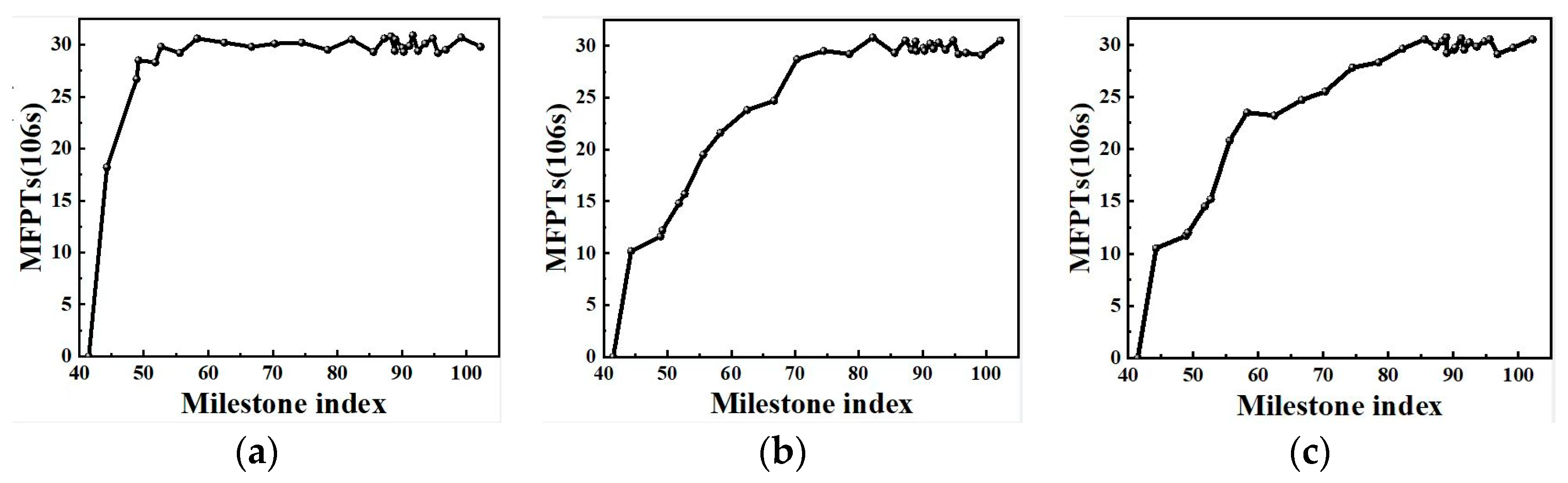
2.4. Free Energy from Markovian Milestoning with Voronoi Tessellations
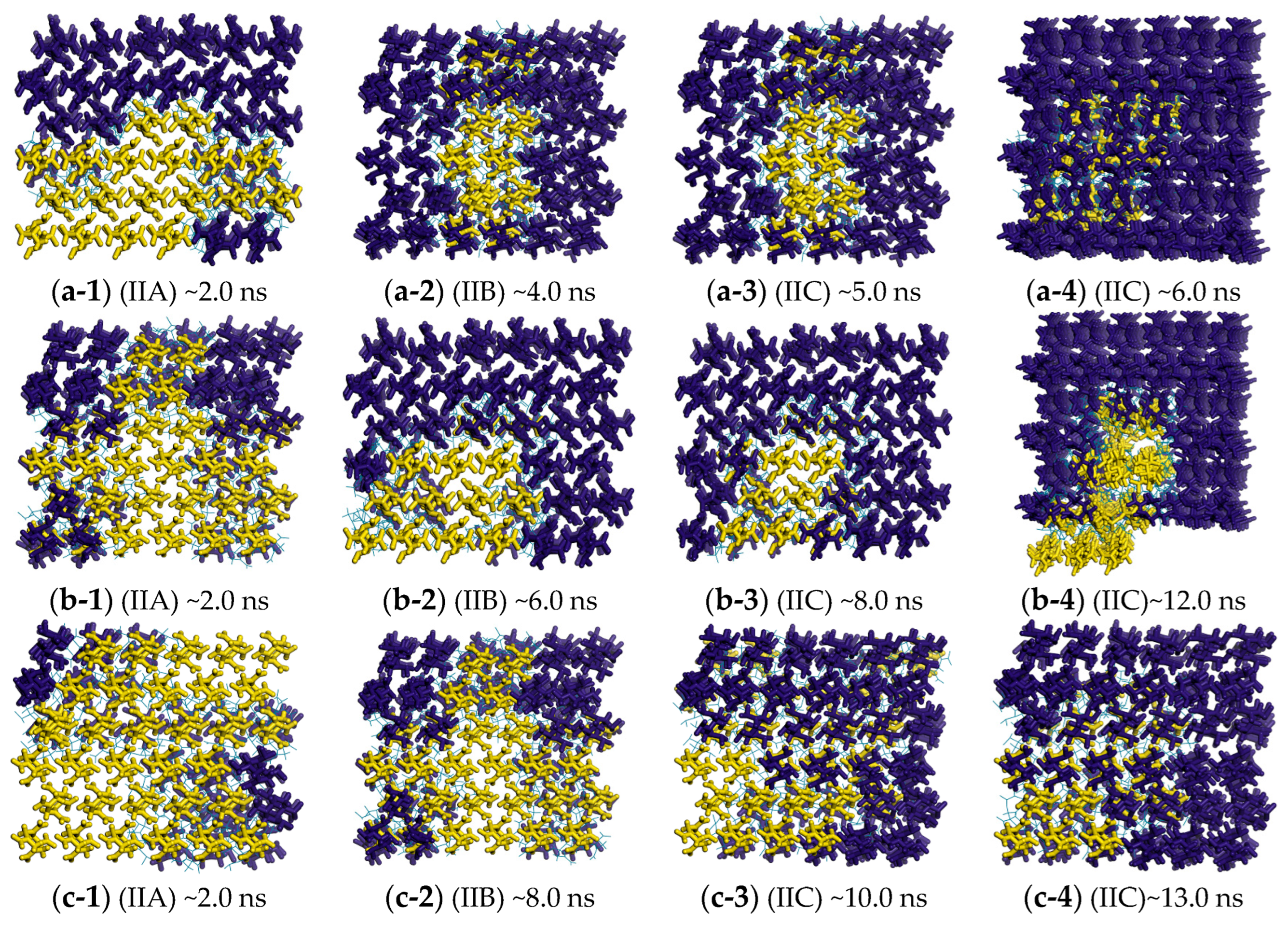
2.5. Temperature Effect on Polymorphic Transformation
2.5.1. Peaks in Pair Distribution Function
2.5.2. Polymorphic Transformation from β-HMX to δ-HMX
2.6. Prediction of Impact Sensitivity for HMX Polymorph
3. Theory
3.1. Order Parameters
3.2. String Method
3.3. Determining MFEP by FTS
3.3.1. Initial Trajectory
3.3.2. K-Means Clustering
3.3.3. Determining MFEP by SMCV
3.4. Markovian Milestoning with Voronoi Tessellations
3.4.1. Construction of Markovian Milestoning with Voronoi Tessellations
3.4.2. Accumulating Statistics of the Number Ni,j, , and
3.4.3. Calculating the Probabilities πi and Free Energy Fi
3.4.4. Calculating Mean First Passage Times (MFPTs)
4. MD Simulation Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tai, B.T.; Andrew, J.S.; David, A.K. Efficient calculation of α- and β-nitrogen free energies and coexistence conditions via overlap sampling with targeted perturbation. J. Chem. Phys. 2011, 135, 044125. [Google Scholar]
- Cardew, P.T.; Davey, R.J.; Ruddick, A.J. Kinetics of polymorphic solid-state transformations. J. Chem. Soc. Faraday Trans. 1984, 80, 659–668. [Google Scholar] [CrossRef]
- Davey, R.J.; Maginn, S.J.; Andrews, S.J.; Buckley, A.M.; Cottier, D.; Dempsay, P.; Rout, J.E.; Stanley, D.R.; Taylor, A. Stabilization of a metastable crystalline phase by twinning. Nature 1993, 366, 248–250. [Google Scholar] [CrossRef]
- Morris, K.R.; Griesser, U.J.; Eckhardt, C.J.; Stowell, J.G. Theoretical approaches to physical transformations of active pharmaceutical ingredients during manufacturing processes. Adv. Drug Delivery Rev. 2001, 48, 91–114. [Google Scholar] [CrossRef]
- Zhang, G.G.Z.; Gu, C.; Zell, M.T.; Burkhardt, R.T.; Munson, E.J.; Grant, D.J.W. Crystallization and Transitions of Sulfamerazine Polymorphs. J. Pharm. Sci. 2002, 91, 1089–1100. [Google Scholar] [CrossRef]
- Kim, Y.S.; Paskow, H.C.; Rousseau, R.W. Propagation of solid-state transformations by dehydration and stabilization of pseudopolymorphic crystals of sodium naproxen. Cryst. Growth Des. 2005, 5, 1623–1632. [Google Scholar] [CrossRef]
- Herbstein, F.H. On the mechanism of some first-order enantiotropic solid-state phase transitions: From Simon through Ubbelohde to Mnyukh. Acta Crystallogr. 2006, 62, 341–383. [Google Scholar] [CrossRef] [PubMed]
- Sadovnikov, S.I.; Gusev, A.I.; Chukin, A.V.; Rempel, A.A. High-temperature X-ray diffraction and thermal expansion of nanocrystalline and coarse-crystalline acanthite α-Ag2S and argentite β-Ag2S. Phys. Chem. Chem. Phys. 2016, 18, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.D.; Vega, C. Melting point and phase diagram of methanol as obtained from computer simulations of the OPLS model. J. Chem. Phys. 2010, 132, 094505. [Google Scholar] [CrossRef]
- Wei, X.F.; Xu, J.J.; Li, H.Z.; Long, X.P.; Zhang, C.Y. Comparative study of experiments and calculations on the polymorphisms of 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20) precipitated by solvent/antisolvent method. J. Phys. Chem. C 2016, 120, 5042–5051. [Google Scholar] [CrossRef]
- Irie, M.; Kobatake, S.; Horichi, M. Reversible surface morphology changes of a photochromic diarylethene single crystal by photoirradiation. Science 2001, 291, 1769–1772. [Google Scholar] [CrossRef]
- Vippagunta, S.R.; Brittain, H.G.; Grant, D.J.W. Crystalline solids. Adv. Drug Delivery Rev. 2001, 48, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Tonauer, C.M.; Bauer, M.; Loerting, T. The impact of temperature and unwanted impurities on slow compression of ice. Phys. Chem. Chem. Phys. 2022, 24, 35–41. [Google Scholar] [CrossRef]
- Yi, P.; Falk, M.L.; Weihs, T.P. Suppression of homogeneous crystal nucleation of the NiAl intermetallic by a composition gradient: A molecular dynamics study. J. Chem. Phys. 2017, 146, 184501. [Google Scholar] [CrossRef]
- Bernshtein, J. Polymorphism in Molecular Crystals; Clarendon Press: Oxford, UK, 2002. [Google Scholar]
- Sikder, A.K.; Sikder, N. A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications. J. Hazard. Mater. 2004, 112, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sivabalan, R.; Gore, G.M.; Nair, U.R.; Saikia, A.; Venugopalan, S.; Gandhe, B.R. Study on ultrasound assisted precipitation of CL-20 and its effect on morphology and sensitivity. J. Hazard. Mater. 2007, 139, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Vrcelj, R.M.; Gallagher, H.G.; Sherwood, J.N. Polymorphism in 2,4,6-Trinitrotoluene Crystallized from Solution. J. Am. Chem. Soc. 2001, 123, 2291–2295. [Google Scholar] [CrossRef]
- Parrish, D.A.; Deschamps, J.R.; Gilardi, R.D.; Butcher, R.J. Polymorphs of Picryl Bromide. Cryst. Growth Des. 2008, 8, 57–62. [Google Scholar] [CrossRef]
- Cady, H.H.; Smith, L.C. Studies on the Polymorphs of HMX; Los Alamos Scientific Laboratory: Los Alamos, NM, USA, 1962. [Google Scholar]
- Smilowitz, L.B.; Henson, B.F.; Asay, B.W. Interfacial and volumetric kinetics of the β→δ phase transition in the energetic nitramine octahydro-l,3,5,7-tetranitro-l,3,5,7-tetrazocine based on the virtual melting mechanism. J. Chem. Phys. 2006, 124, 026101-1–026101-4. [Google Scholar]
- Smilowitz, L.B.; Henson, B.F.; Asay, B.W. Kinetics of the β→δ phase transition in PBX9501. Shock. Campression Condens. Matter. 2001, 620, 1077–1080. [Google Scholar]
- Brill, T.B.; Karpowicz, R.J. Solid phase transition kinetics The role of intermolecular forces in the condensed-phase decamposition of octahydro-l,3,5,7-tetranitro-l,3,5,7-tetrazocine. J. Phys. Chem. 1982, 86, 4260–4265. [Google Scholar] [CrossRef]
- Weese, R.K. Kinetics of β→δ Solid-Solid Phase Transition of HMX. UCRL-LR-143960; Lawrence Livermore National Lab. (LLNL): Livermore, CA, USA, 2000. [Google Scholar]
- Smilowitz, L.B.; Henson, B.F.; Asay, B.W.; Dickson, P. The β→δ phase transition in the energetic nitramine octahydmo-l,3,5,7-tetranitmo-l,3,5,7-tetrazocine. Kinetics. J. Chem. Phys. 2002, 117, 3789–3798. [Google Scholar] [CrossRef]
- Bao, J.N.; Fan, H.B.; Xue, X.J.; Xie, Q.; Pan, P.J. Temperature-dependent crystalline structure and phase transition of poly(butylene adipate) end-functionalized by multiple hydrogen-bonding groups. Phys. Chem. Chem. Phys. 2018, 20, 26479–26488. [Google Scholar] [CrossRef]
- Handle, P.H.; Loerting, T. Temperature-induced amorphisation of hexagonal ice. Phys. Chem. Chem. Phys. 2015, 17, 5403–5412. [Google Scholar] [CrossRef] [PubMed]
- Diana, H.; Pilar, C.; Rico, G.; Benjamin, W.; Rajadurai, C.; Svetlana, K.; Mario, R.; Roberto-Carlos, S.; Klaus, K.; Doris, G. Polymorphism and metal-induced structural transformation in 5,5′-bis(4-pyridyl)(2,2′-bispyrimidine) adlayers on Au(111). Phys. Chem. Chem. Phys. 2018, 20, 15960–15969. [Google Scholar]
- Venugopal, K.; Deepak, D.; Suresh, D.; Sunil, V. Transformation of photophysical properties from solution to solid state in alkoxy-cyano-diphenylacetylene molecules. Phys. Chem. Chem. Phys. 2015, 17, 18768–18779. [Google Scholar]
- Beckham, G.T.; Peters, B.; Trout, B.L. Evidence for a size dependent nucleation mechanism in solid state polymorph transformations. J. Phys. Chem. B 2008, 112, 7460–7466. [Google Scholar] [CrossRef]
- Mnyukh, Y.V. Polymorphic transitions in crystals: Nucleation. J. Cryst. Growth 1976, 32, 371–377. [Google Scholar] [CrossRef]
- Tuble, S.C.; Anwar, J.; Gale, J.D. An Approach to developing a force field for molecular simulation of martensitic phase transitions between phases with subtle differences in energy and structure. J. Am. Chem. Soc. 2004, 126, 396–405. [Google Scholar] [CrossRef]
- Beckham, G.T.; Peters, B.; Starbuck, C.; Variankaval, N.; Trout, B.L. Surface-mediated nucleation in the solid-state polymorph transformation of terephthalic acid. J. Am. Chem. Soc. 2007, 129, 4714–4723. [Google Scholar] [CrossRef]
- Bayés-García, L.; Calvet, T.; Cuevas-Diarte, M.À.; Ueno, S.; Sato, S. Crystallization and transformation of polymorphic forms of trioleoyl glycerol and 1,2-dioleoyl-3-rac-linoleoyl glycerol. J. Phys. Chem. B 2013, 117, 9170–9181. [Google Scholar] [CrossRef] [PubMed]
- Santiso, E.E.; Trout, B.L. A general method for molecular modeling of nucleation from the melt. J. Chem. Phys. 2015, 143, 174109. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J. Colloidal crystals model real world. Nature 1995, 378, 231. [Google Scholar] [CrossRef]
- Price, S.L. Computed crystal energy landscapes for understanding and predicting organic crystal structures and polymorphism. Acc. Chem. Res. 2009, 42, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. From crystal structure prediction to polymorph prediction: Interpreting the crystal energy landscape. Phys. Chem. Chem. Phys. 2008, 10, 1996–2009. [Google Scholar] [CrossRef]
- Dickson, A.; Warmflash, A.; Dinner, A.R. Nonequilibrium umbrella sampling in spaces of many order parameters. J. Chem. Phys. 2009, 130, 074104. [Google Scholar] [CrossRef]
- Faradjian, A.K.; Elber, R. Computing time scales from reaction coordinates by milestoning. J. Chem. Phys. 2004, 120, 10880–10889. [Google Scholar] [CrossRef]
- Santiso, E.E.; Trout, B.L. A general set of order parameters for molecular crystals. J. Chem. Phys. 2011, 134, 064109. [Google Scholar] [CrossRef]
- Carter, E.; Ciccotti, G.; Hynes, J.; Kapral, R. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 1989, 156, 472–477. [Google Scholar] [CrossRef]
- Anwar, J.; Zahn, D. Uncovering molecular processes in crystal nucleation and growth by using molecular simulation. Angew. Chem. Int. Ed. 2011, 50, 1996–2013. [Google Scholar] [CrossRef]
- Dan, S.; Mondal, B.; Saha, S.K.; Mondal, S.; Ranganathan, R.; Kumar, M.; Mazumdar, C. Similar and dissimilar properties of polymorphic phases of NdIr3. J. Phys. Chem. C 2022, 126, 16514–16521. [Google Scholar] [CrossRef]
- Bellucci, M.A.; Trout, B.L. Bézier curve string method for the study of rare events in complex chemical systems. J. Chem. Phys. 2014, 141, 074110. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, V.; Karplus, M. Investigations of α-helix↔β-sheet transition pathways in a miniprotein using the finite-temperature string method. J. Chem. Phys. 2014, 140, 175103. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.S.; Ladbury, J.E.; Pitt, W.R.; Williams, M.A. Extent of enthalpy–entropy compensation in protein–ligand interactions. Protein Sci. 2011, 20, 1607–1618. [Google Scholar] [CrossRef]
- Berendsen, H.J. A Glimpse of the Holy Grail? Science 1998, 282, 642–643. [Google Scholar] [CrossRef]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126, 054103. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Park, S.; Khalili-Araghi, F.; Tajkhorshid, E.; Schulten, K. Free energy calculation from steered molecular dynamics simulations using Jarzynski’s equality. J. Chem. Phys. 2003, 119, 3559–3566. [Google Scholar] [CrossRef]
- Weinan, E.; Ren, W.; Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 66, 052301. [Google Scholar]
- Kulshrestha, A.; Punnathanam, S.N.; Ayappa, K.G. Finite temperature string method with umbrella sampling using path collective variables: Application to secondary structure change in a protein. Soft Matter. 2022, 18, 7593–7603. [Google Scholar] [CrossRef]
- Weinan, E.; Ren, W.; Vanden-Eijnden, E. Finite temperature string method for the study of rare events. J. Phys. Chem. B 2005, 109, 6688–6693. [Google Scholar]
- Vanden-Eijnden, E.; Venturoli, M. Revisiting the finite temperature string method for the calculation of reaction tubes and free energies. J. Chem. Phys. 2009, 130, 194103. [Google Scholar] [CrossRef] [PubMed]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged elastic band method for finding minimum energy paths of transitions. Class. Quantum Dyn. Condens. Phase Simul. 1998, 385–404. [Google Scholar] [CrossRef]
- Maragliano, L.; Fischer, A.; Vanden-Eijnden, E.; Ciccotti, G. Learning Markovian dynamics with spectral maps. J. Chem. Phys. 2006, 125, 024106. [Google Scholar] [CrossRef]
- Ren, W.; Vanden-Eijnden, E.; Maragakis, P.; Weinan, E. Nonlinear discovery of slow molecular modes using state-free reversible VAMPnets. J. Chem. Phys. 2005, 123, 134109. [Google Scholar] [CrossRef] [PubMed]
- Zinovjev, K.; Tuñón, I. Adaptive finite temperature string method in collective variables. J. Phys. Chem. A 2017, 121, 9764–9772. [Google Scholar] [CrossRef]
- Dickson, B.M.; Huang, H.; Post, C.B. Unrestrained computation of free energy along a path. J. Phys. Chem. B 2012, 116, 11046–11055. [Google Scholar] [CrossRef]
- Díaz, L.G.; Ensing, B. Path finding on high-dimensional free energy landscapes. Phys. Rev. Lett. 2012, 109, 020601. [Google Scholar] [CrossRef]
- Maragliano, L.; Roux, B.; Vanden-Eijnden, E. Comparison between mean forces and swarms-of-trajectories string methods. J. Chem. Theory Comput. 2014, 10, 524–533. [Google Scholar] [CrossRef]
- Song, H.D.; Zhu, F.Q. Morphology evolution of polymer blends under intense shear during high speed thin-wall injection molding. J. Phys. Chem. B 2017, 121, 3376–3386. [Google Scholar] [CrossRef]
- Cao, L.R.; Lv, C.; Yang, W. Hidden conformation events in DNA base extrusions: A generalized-ensemble path optimization and equilibrium simulation study. J. Chem. Theory Comput. 2013, 9, 3756–3768. [Google Scholar] [CrossRef] [PubMed]
- Badaoui, M.; Buigues, P.J.; Berta, D.; Mandana, G.M.; Gu, H.; Földes, T.; Dickson, C.J.; Hornak, V.; Kato, M.; Molteni, C.; et al. Combined free-energy calculation and machine learning methods for understanding ligand unbinding kinetics. J. Chem. Theory Comput. 2022, 18, 2543–2555. [Google Scholar] [CrossRef] [PubMed]
- He, X.X.; Shen, Y.; Hung, F.R.; Santiso, E.E. Molecular simulation of homogeneous nucleation of crystals of an ionic liquid from the melt. J. Chem. Phys. 2015, 143, 124506. [Google Scholar] [CrossRef]
- Salvalaglio, M.; Vetter, T.; Giberti, F.; Mazzotti, M.; Parrinello, M. Uncovering molecular details of urea crystal growth in the presence of additives. J. Am. Chem. Soc. 2012, 134, 17221–17233. [Google Scholar] [CrossRef]
- Shah, M.; Santiso, E.E.; Trout, B.L. Computer simulations of homogeneous nucleation of benzene from the melt. J. Phys. Chem. B 2011, 115, 10400–10412. [Google Scholar] [CrossRef]
- Gobbo, G.; Bellucci, M.A.; Tribello, G.A.; Ciccotti, G.; Trout, B.L. Nucleation of molecular crystals driven by relative information entropy. J. Chem. Theory Comput. 2018, 14, 959–972. [Google Scholar] [CrossRef]
- Ren, F.D.; Liu, Y.Z.; Ding, K.W.; Chang, L.L.; Cao, D.L.; Liu, S.B. Finite temperature string by K-means clustering sampling with order parameter as collective variables for molecular crystal: Application to polymorphic transformation between β-CL-20 and ε-CL-20. Phys. Chem. Chem. Phys. 2024, 26, 3500–3515. [Google Scholar] [CrossRef]
- Ren, F.D.; Wang, X.L.; Zhang, Q.; Wang, X.J.; Chang, L.L.; Zhang, Z.T. Experimental and theoretical investigation of external electric-field-induced crystallization of TKX-50 from solution by finite-temperature string with order parameters as collective variables for ionic crystals. Molecules 2024, 29, 1159. [Google Scholar] [CrossRef] [PubMed]
- Adelman, J.L.; Grabe, M. Simulating rare events using a weighted ensemble-based string method. J. Chem. Phys. 2013, 138, 044105. [Google Scholar] [CrossRef]
- Mac, Q.J. Some Methods for Classification and Analysis of Multivariate Observations; Berkeley Symposium on Mathematical Statistics and Probability: Berkeley, CA, USA, 1967. [Google Scholar]
- Nawaz, M.; Mehmood, Z.; Nazir, T.; Naqvi, R.A.; Rehman, A.; Iqbal, M.; Saba, T. Skin cancer detection from dermoscopic images using deep learning and fuzzy k-means clustering. Microsc. Res. Techniq. 2022, 85, 339–351. [Google Scholar] [CrossRef]
- Abo-Elnaga, Y.; Nasr, S. K-means cluster interactive algorithm-based evolutionary approach for solving bilevel multi-objective programming problems. Alex. Eng. J. 2022, 61, 811–827. [Google Scholar] [CrossRef]
- Rong, H.; Ramirez-Serrano, A.; Guan, L.; Gao, Y. Image object extraction based on semantic detection and Improved K-Means algorithm. IEEE Access 2020, 8, 171129–171139. [Google Scholar] [CrossRef]
- Ovchinnikov, V.; Karplus, M.; Vanden-Eijnden, E. A traveling-salesman based automated path searching method for functional conformational changes of biological macromolecules. J. Chem. Phys. 2011, 134, 085103. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E.; Roux, B. Free energy and kinetics of conformational transitions from voronoi tessellated milestoning with restraining potentials. J. Chem. Theory Comput. 2009, 5, 2589–2594. [Google Scholar] [CrossRef]
- Vanden-Eijnden, E.; Venturoli, M. Supervised learning and the finite-temperature string method for computing committor functions and reaction rates. J. Chem. Phys. 2009, 130, 194101. [Google Scholar] [CrossRef]
- Xue, C.; Sun, J.; Song, G.B.; Kang, B.; Xia, Y.X. Review on B→δ phase transition of HMX. Chin. J. Energ. Mater. 2008, 6, 753–756. [Google Scholar]
- Abreu, J.; Rico-Juan, J. A new iterative algorithm for computing a quality approximate median of strings based on edit operations. Pattern Recognit. Lett. 2014, 36, 74–80. [Google Scholar] [CrossRef]
- Henikoff, J.G.; Henikoff, S. Blocks database and its applications. Method. Enzymol. 1996, 266, 88–105. [Google Scholar]
- Mirabal, P.; Abreu, J.; Seco, D. Assessing the best edit in perturbation-based iterative refinement algorithms to compute the median string. Pattern Recognit. Lett. 2019, 120, 104–111. [Google Scholar] [CrossRef]
- Maragliano, L.; Vanden-Eijnden, E. On-the-fly string method for minimum free energy paths calculation. Chem. Phys. Lett. 2007, 446, 182–190. [Google Scholar] [CrossRef]
- Norris, J.R.; Markov, C. Cambridge Series in Statistical and Porbabilistic Mathematics; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Rong, C.; Zhao, D.; Zhou, T.; Liu, S.; Yu, D.; Liu, S. Homogeneous molecular systems are positively cooperative, but charged molecular systems are negatively cooperative. J. Phys. Chem. Lett. 2019, 10, 1716–1721. [Google Scholar] [CrossRef]
- Ren, F.D.; Shi, W.J.; Cao, D.L.; Li, Y.X.; Liu, L.L.; Gao, L. A theoretical investigation into the cooperativity effect on the TNT melting point under external electric field. J. Mol. Model. 2021, 27, 4. [Google Scholar] [CrossRef] [PubMed]
- Elishav, O.; Podgaetsky, R.; Meikler, O.; Hirshberg, B. Collective variables for conformational polymorphism in molecular crystals. J. Phys. Chem. Lett. 2023, 14, 971–976. [Google Scholar] [CrossRef]
- Giberti, F.; Salvalaglio, M.; Mazzotti, M.; Parrinello, M. Insight into the nucleation of urea crystals from the melt. Chem. Eng. Sci. 2015, 121, 51–59. [Google Scholar] [CrossRef]
- Tribello, G.A.; Giberti, F.; Sosso, G.C.; Salvalaglio, M.; Parrinello, M. Analyzing and driving cluster formation in atomistic simulations. J. Chem. Theory Comput. 2017, 13, 1317–1327. [Google Scholar] [CrossRef]
- Gimondi, I.; Salvalaglio, M. CO2 packing polymorphism under pressure: Mechanism and thermodynamics of the I-III polymorphic transition. J. Chem. Phys. 2017, 147, 114502. [Google Scholar] [CrossRef]
- Francia, N.F.; Price, L.S.; Nyman, J.; Price, S.L.; Salvalaglio, M. Systematic finite-temperature reduction of crystal energy landscapes. Cryst. Growth. Des. 2020, 20, 6847–6862. [Google Scholar] [CrossRef]
- Samanta, A.; Chen, W.E.M.; Yu, T.; Tuckerman, M. Sampling saddle points on a free energy surface. J. Chem. Phys. 2014, 140, 164109. [Google Scholar] [CrossRef]
- Cuendet, M.A.; Tuckerman, M.E. Free energy reconstruction from metadynamics or adiabatic free energy dynamics simulations. J. Chem. Theory Comput. 2014, 10, 2975–2986. [Google Scholar] [CrossRef]
- Yu, T.Q.; Tuckerman, M.E. Temperature-accelerated method for exploring polymorphism in molecular crystals based on free Energy. Phys. Rev. Lett. 2011, 107, 015701. [Google Scholar] [CrossRef]
- Piaggi, P.M.; Parrinello, M. Entropy based fingerprint for local crystalline order. J. Chem. Phys. 2017, 147, 114112. [Google Scholar] [CrossRef]
- Piaggi, P.M.; Valsson, O.; Parrinello, M. Enhancing entropy and enthalpy fluctuations to drive crystallization in atomistic simulations. Phys. Rev. Lett. 2017, 119, 015701. [Google Scholar] [CrossRef] [PubMed]
- Neha; Mondal, S.; Kumari, N.; Karmakar, T. Collective variables for crystallization simulations-from early developments to recent advances. ACS Omega 2022, 8, 127–146. [Google Scholar]
- Turnbull, D.; Fisher, J.C. Rate of Nucleation in Condensed Systems. J. Chem. Phys. 1949, 17, 71–73. [Google Scholar] [CrossRef]
- Rice, B.M.; Sahu, S.; Owens, F.J. Density functional calculations of bond dissociation energies for NO2 scission in some nitroaromatic molecules. J. Mol. Struct. Theochem. 2002, 583, 69–72. [Google Scholar] [CrossRef]
- Aina, A.A.; Misquitta, A.J.; Phipps, M.J.S.; Price, S.L. Charge Distributions of Nitro Groups within Organic Explosive Crystals: Effects on Sensitivity and Modeling. ACS Omega 2019, 4, 8614–8625. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Impact sensitivity and crystal lattice compressibility/free space. J. Mol. Model. 2014, 20, 2233. [Google Scholar] [CrossRef]
- Michalchuk, A.A.L.; Trestman, M.; Rudić, S.; Portius, P.; Fincham, P.T.; Pulham, C.R.; Morrison, C.A. Predicting the reactivity of energetic materials: An ab initio multi-phonon approach. J. Mater. Chem. A 2019, 7, 19539–19553. [Google Scholar] [CrossRef]
- Atceken, N.; Hemingway, J.; Bull, C.L.; Liu, X.J.; Michalchuk, A.A.L.; Konar, S.; Morrison, C.A.; Pulham, C.R. High-pressure structural studies and pressure-induced sensitisation of 3,4,5-trinitro-1H-pyrazole. Phys. Chem. Chem. Phys. 2023, 25, 31646–31654. [Google Scholar] [CrossRef]
- Christopher, I.L.; Pulham, C.R.; Michalchuk, A.A.L.; Morrison, C.A. Is the impact sensitivity of RDX polymorph dependent? J. Chem. Phys. 2023, 158, 124115. [Google Scholar] [CrossRef]
- Michalchuk, A.A.L.; Hemingway, J.; Morrison, C.A. Predicting the impact sensitivities of energetic materials through zone-center phonon up-pumping. J. Chem. Phys. 2021, 154, 064105. [Google Scholar] [CrossRef] [PubMed]
- Michalchuk, A.A.L.; Rudić, S.; Pulham, C.R.; Morrison, C.A. Predicting the impact sensitivity of a polymorphic high explosive: The curious case of FOX-7. Chem. Commun. 2021, 57, 11213–11216. [Google Scholar] [CrossRef]
- Hunter, S.; Sutinen, T.; Parker, S.F.; Morrison, C.A.; Williamson, D.M.; Thompson, S.; Gould, P.J.; Pulham, C.R. Experimental and DFT-D Studies of the Molecular Organic Energetic Material RDX. J. Phys. Chem. C 2013, 117, 80628. [Google Scholar] [CrossRef]
- Jolliffe, I. Principal Component Analysis, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Available online: http://www.ks.uiuc.edu/Research/namd/ (accessed on 8 October 2024).
- MacKerell, A.; Wiorkiewicz-Kuczera, J.; Karplus, M. An all-atom empirical energy function for the simulation of nucleic acids. J. Am. Chem. Soc. 1995, 117, 11946–11975. [Google Scholar] [CrossRef]
- MacKerell, A.; Bashford, D.; Bellott, M.; Dunbrack, R.; Evanseck, J.; Field, M.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Phillips, J.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.; Kale, L.; Schulten, K. Sire: An interoperability engine for prototyping algorithms and exchanging information between molecular simulation programs. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald: An N⋅log(N) method for ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
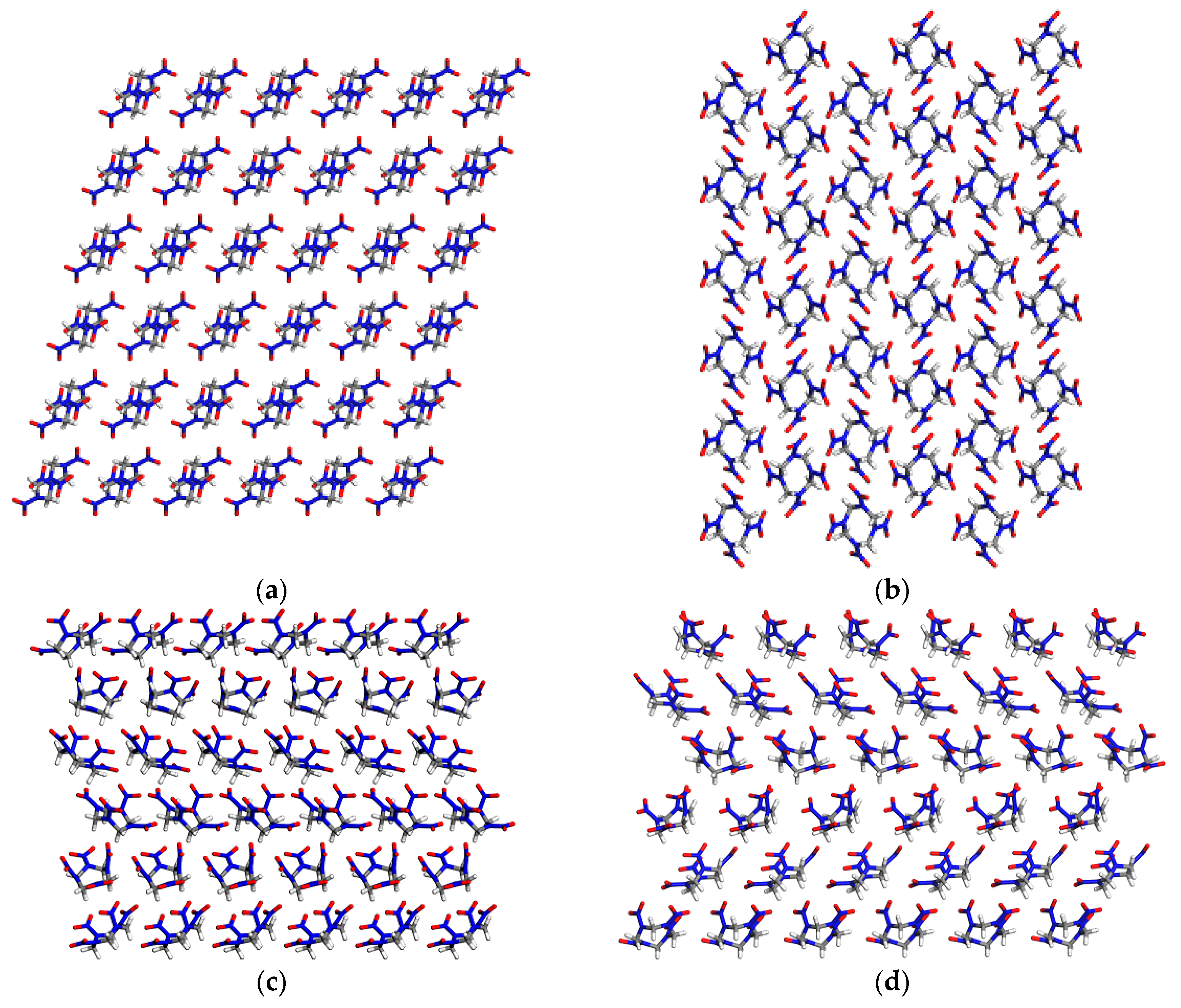
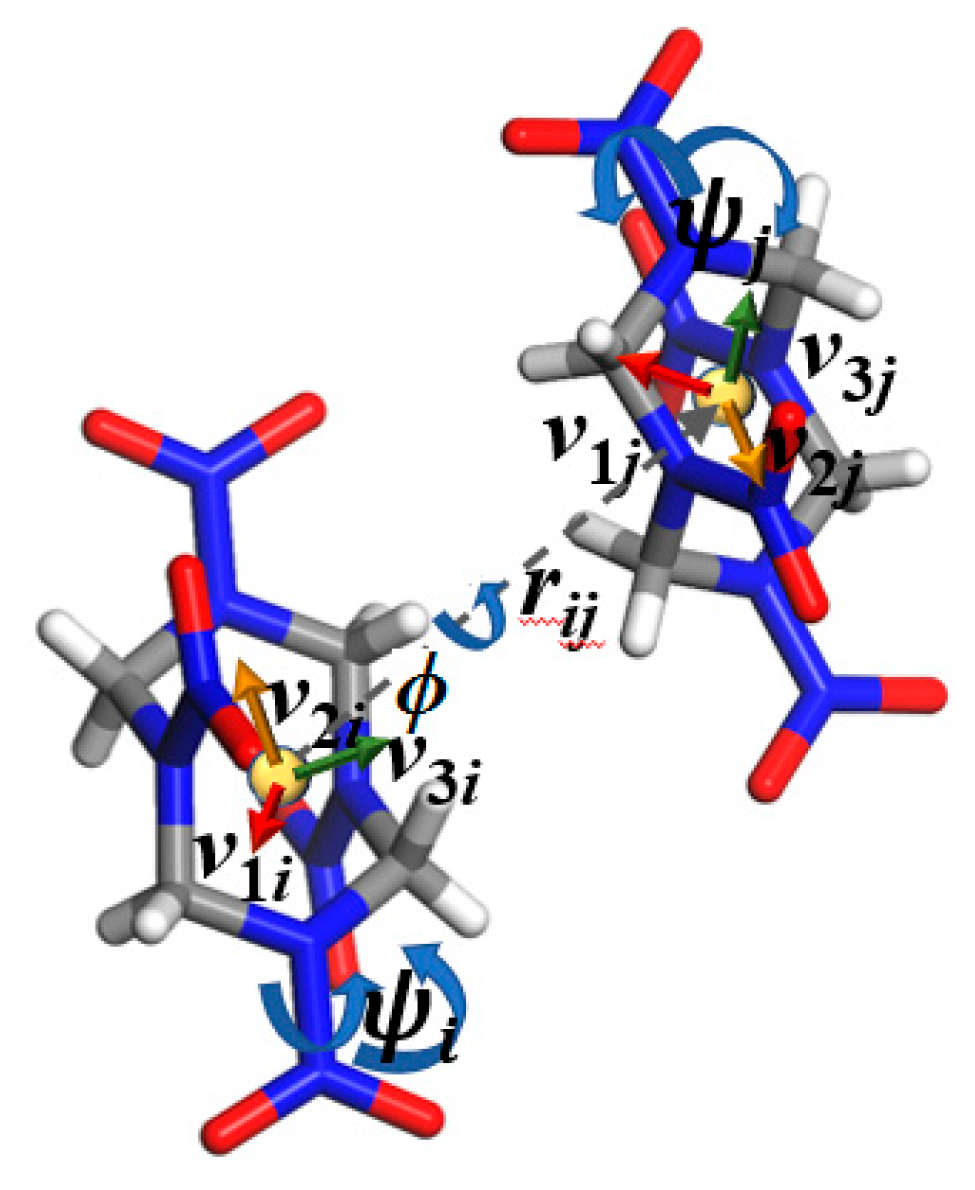
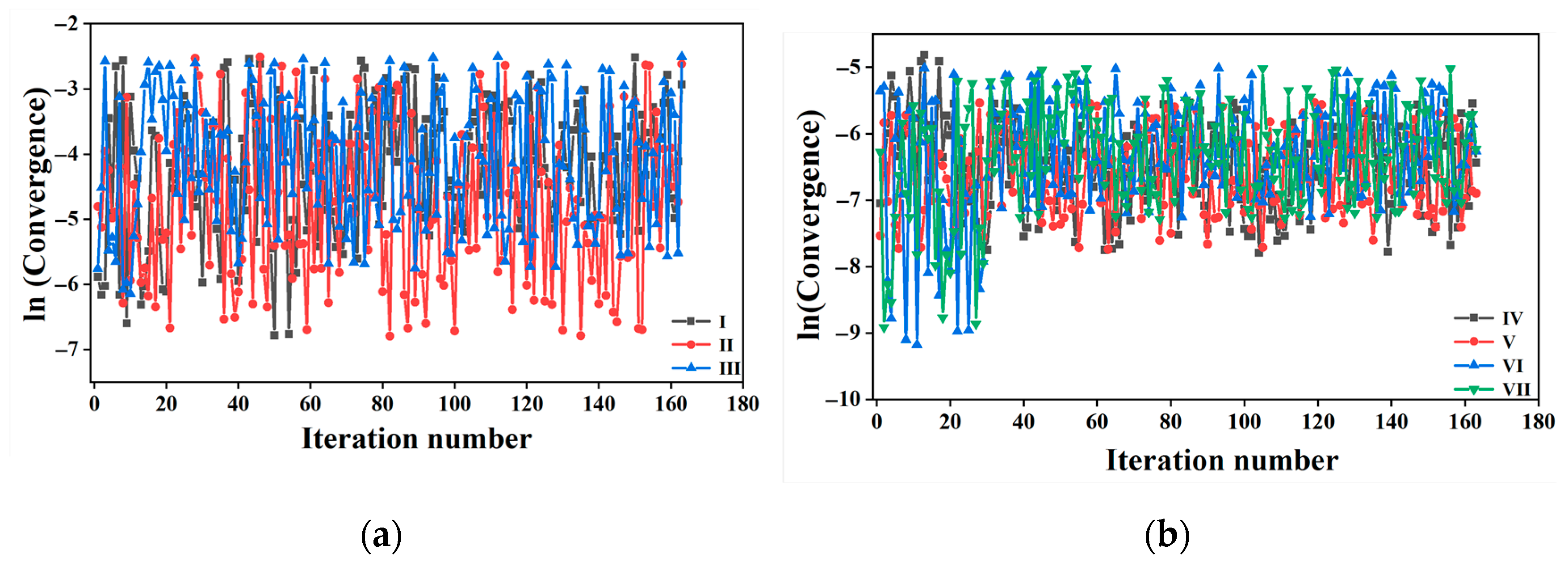
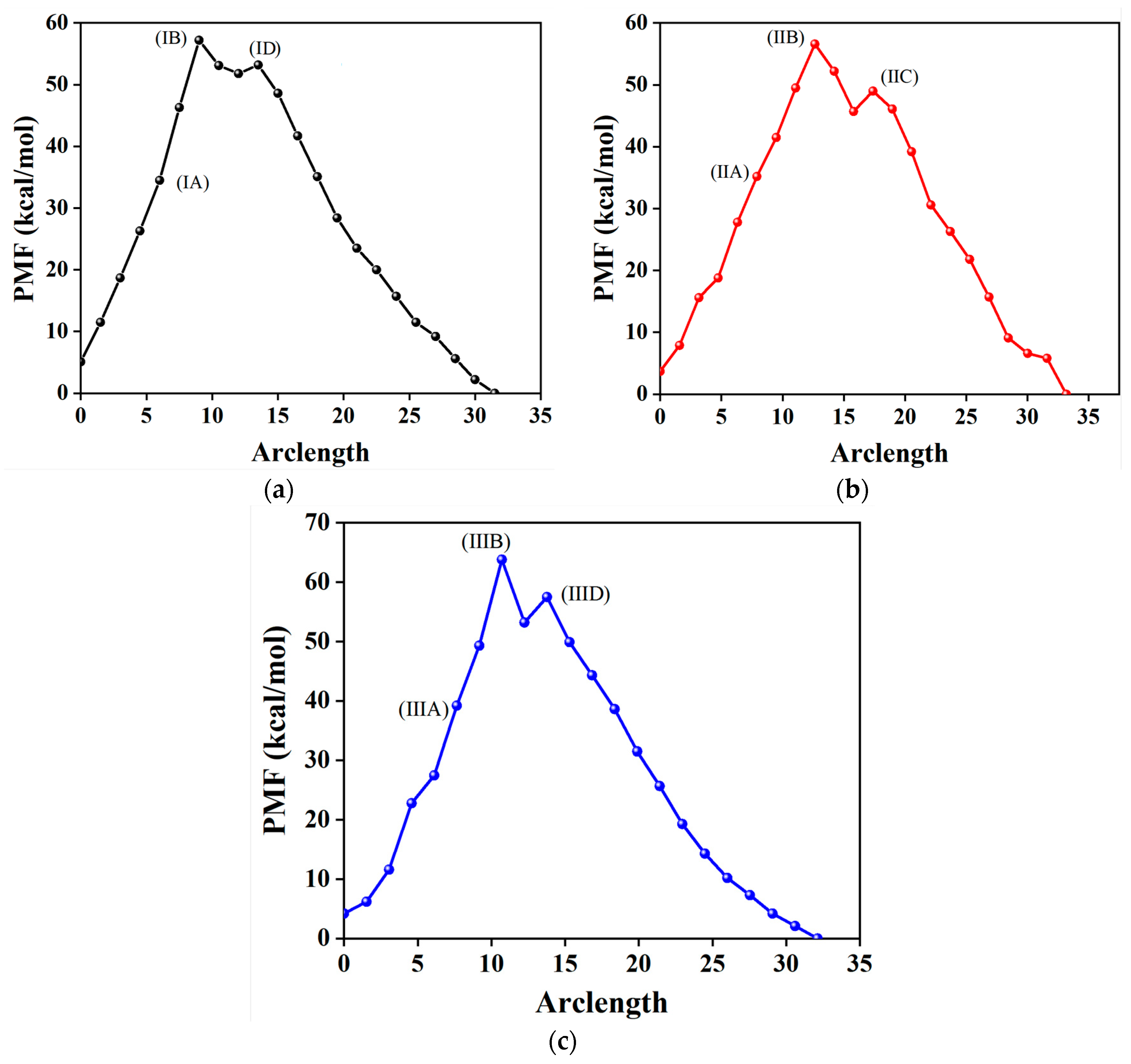

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; Xin, Z.; Fu, Y.; Duan, H. Theoretical Investigation into Polymorphic Transformation between β-HMX and δ-HMX by Finite Temperature String. Molecules 2024, 29, 4819. https://doi.org/10.3390/molecules29204819
Jia X, Xin Z, Fu Y, Duan H. Theoretical Investigation into Polymorphic Transformation between β-HMX and δ-HMX by Finite Temperature String. Molecules. 2024; 29(20):4819. https://doi.org/10.3390/molecules29204819
Chicago/Turabian StyleJia, Xiumei, Zhendong Xin, Yizheng Fu, and Hongji Duan. 2024. "Theoretical Investigation into Polymorphic Transformation between β-HMX and δ-HMX by Finite Temperature String" Molecules 29, no. 20: 4819. https://doi.org/10.3390/molecules29204819
APA StyleJia, X., Xin, Z., Fu, Y., & Duan, H. (2024). Theoretical Investigation into Polymorphic Transformation between β-HMX and δ-HMX by Finite Temperature String. Molecules, 29(20), 4819. https://doi.org/10.3390/molecules29204819