
Multitasking Pharmacophores Support Cabotegravir-Based Long-Acting HIV Pre-Exposure Prophylaxis (PrEP)

 ,
,  ,
,  and
and
Abstract
1. Introduction
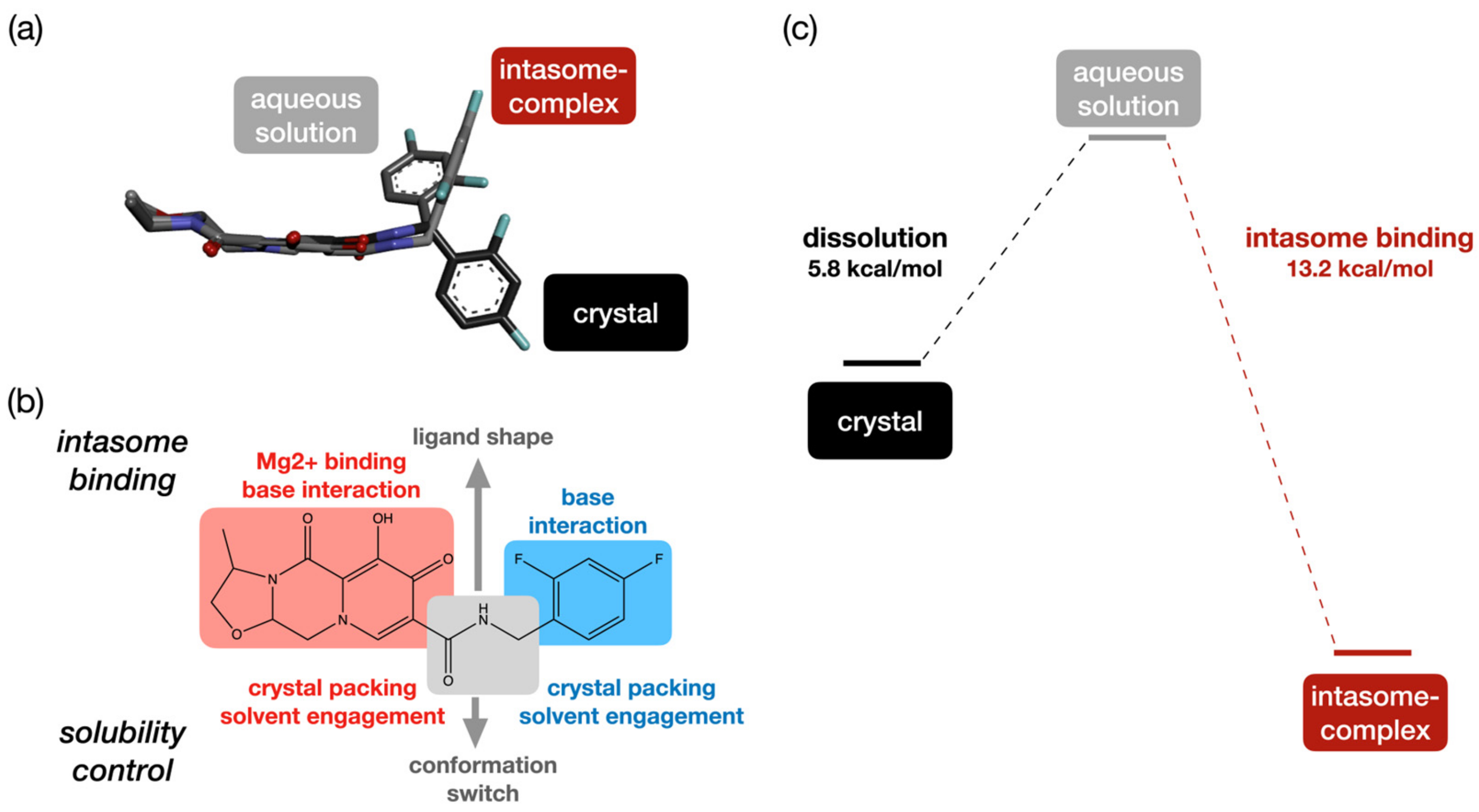
2. Results and Discussion
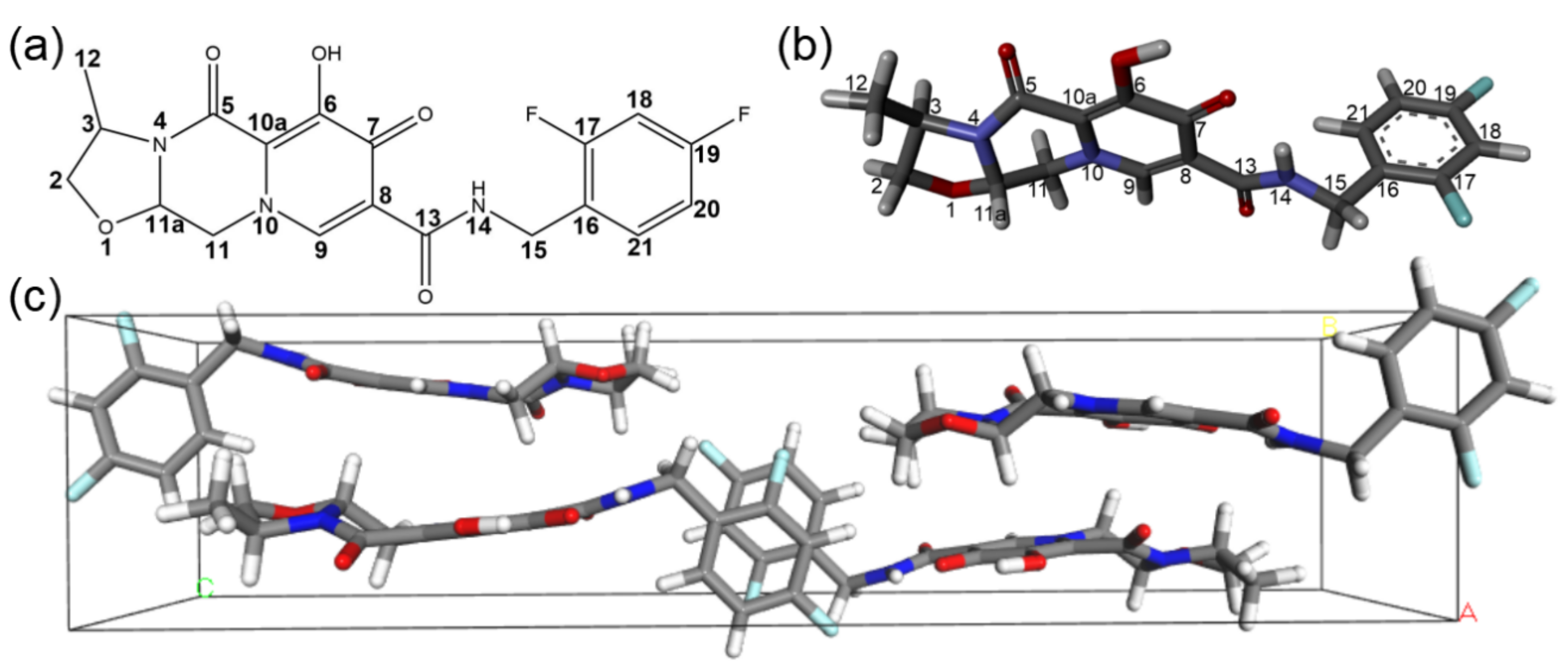
2.1. The Crystal Structure of Cabotegravir
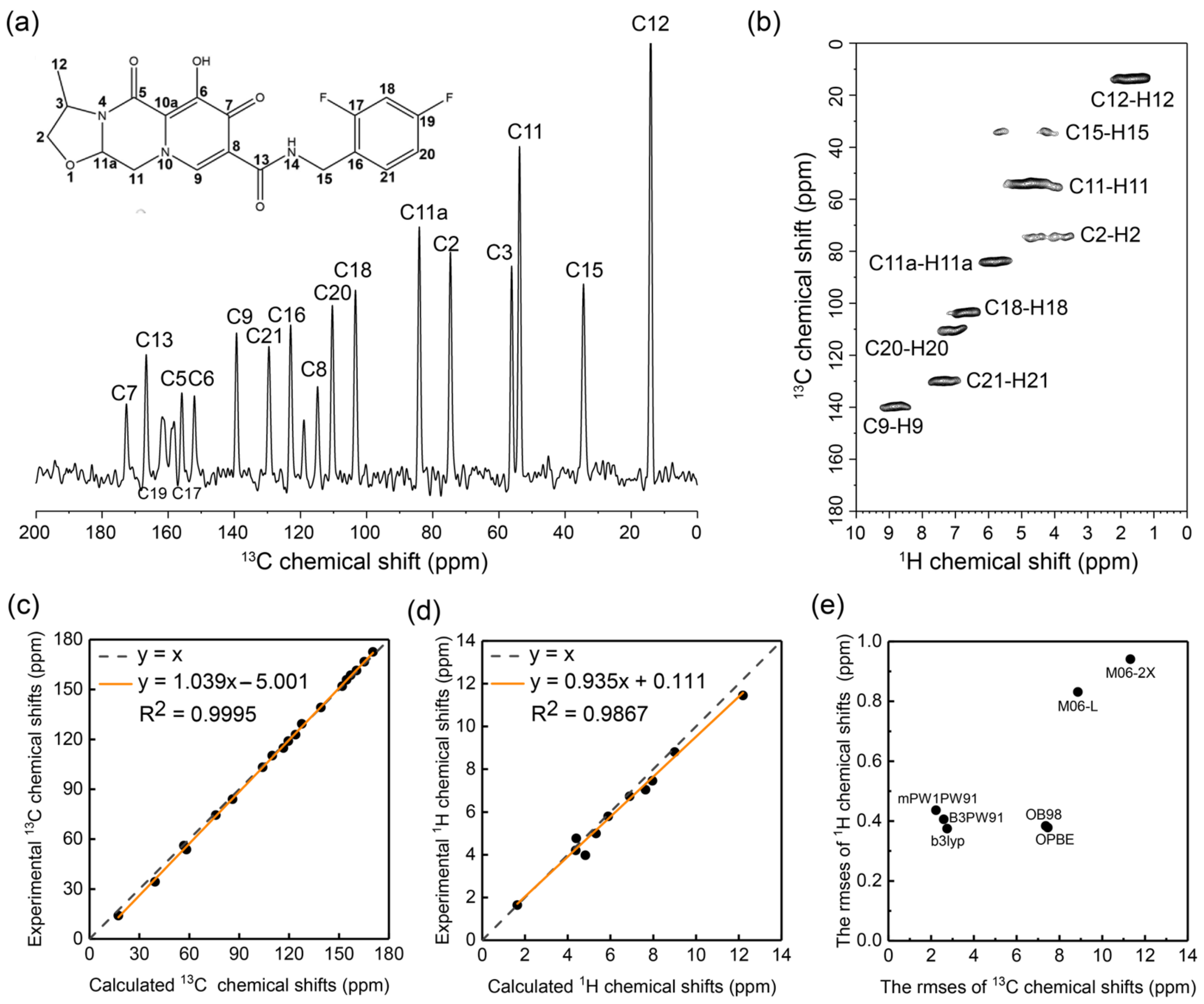
2.2. Optimizing the Chemical Shift Prediction for Cabotegravir
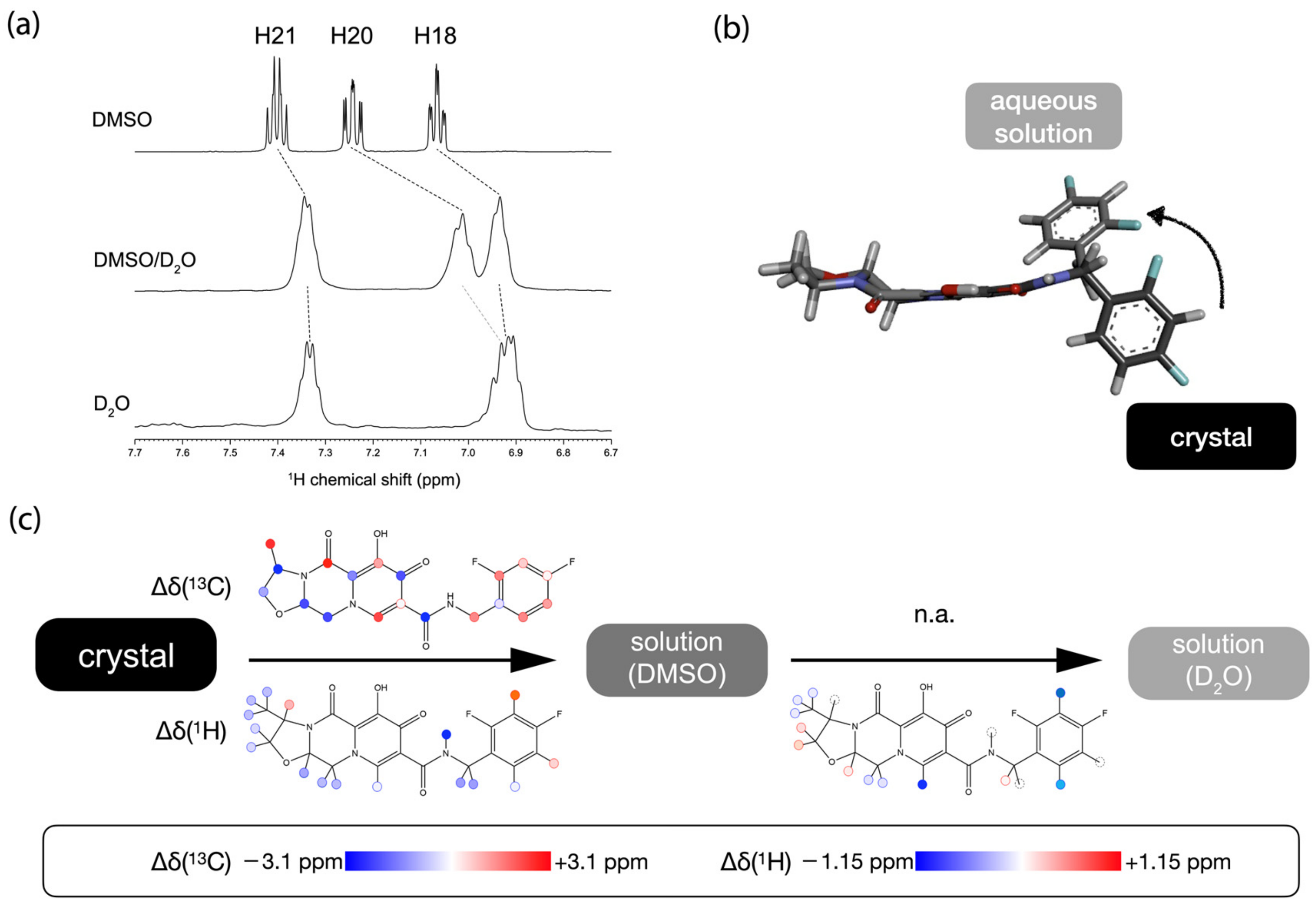
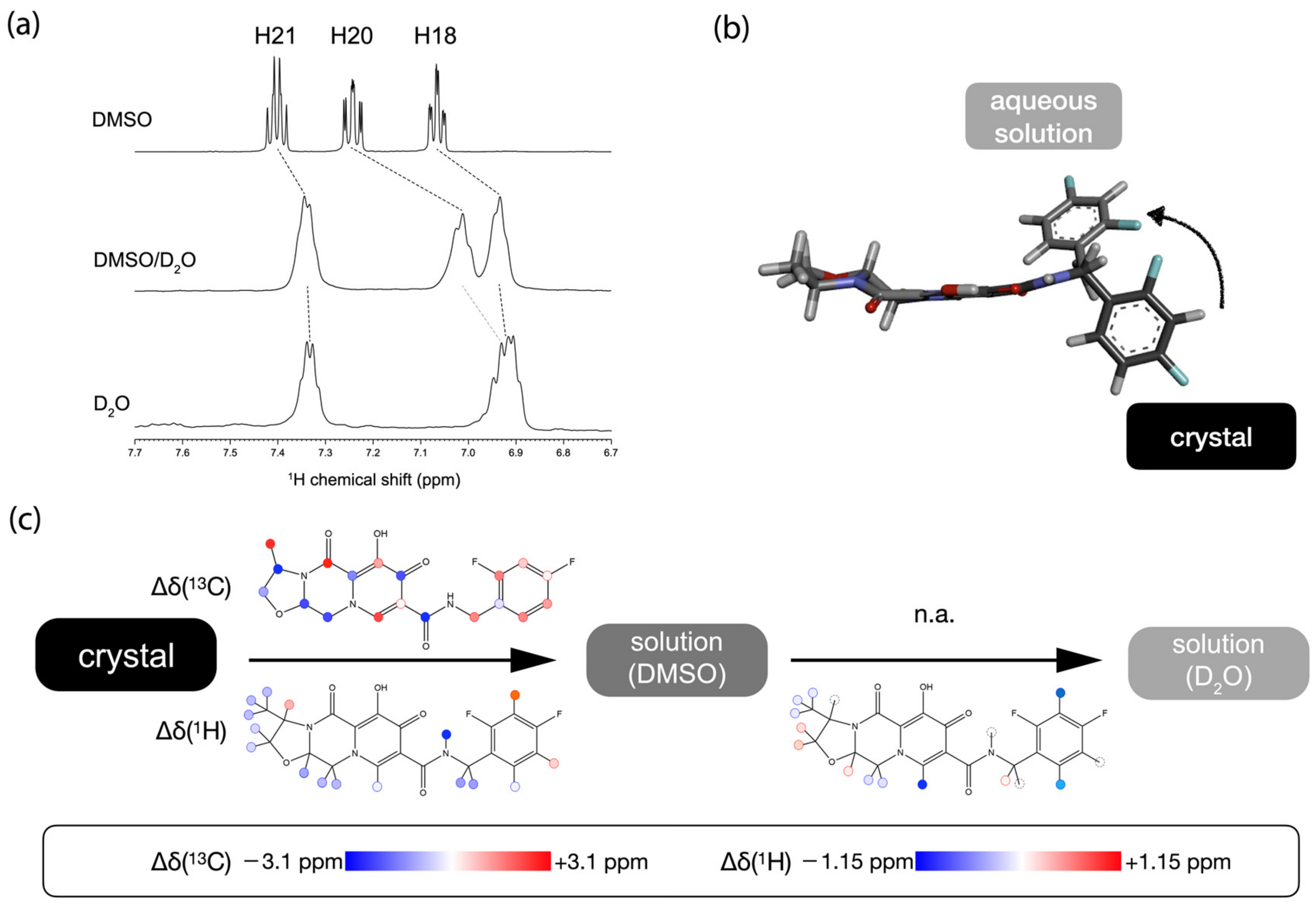
2.3. The Conformational Changes of Cabotegravir upon Dissolution
2.4. Multi-Tasking Pharmacophores in Cabotegravir
3. Methods
3.1. The Crystallization and XRD Structural Determination of Cabotegravir
3.2. SSNMR Spectroscopy
3.3. Solution NMR Spectroscopy
3.4. The AF-QM/MM Method for Chemical Shift Calculations
3.5. The Structure Modeling of Cabotegravir in Different Environments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rambaut, A.; Posada, D.; Crandall, K.A.; Holmes, E.C. The causes and consequences of HIV evolution. Nat. Rev. Genet. 2004, 5, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Barré-Sinoussi, F.; Ross, A.L.; Delfraissy, J.-F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Archin, N.; Cannon, P.; Collins, S.; Jones, R.B.; de Jong, M.A.; Lambotte, O.; Lamplough, R.; Ndungu, T.; Sugarman, J.; et al. Research priorities for an HIV cure: International AIDS society global scientific strategy 2021. Nat. Med. 2021, 27, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Fonner, V.A.; Dalglish, S.L.; Kennedy, C.E.; Baggaley, R.; O’reilly, K.R.; Koechlin, F.M.; Rodolph, M.; Hodges-Mameletzis, I.; Grant, R.M. Effectiveness and safety of oral HIV preexposure prophylaxis for all populations. AIDS 2016, 30, 1973. [Google Scholar] [CrossRef] [PubMed]
- Unemo, M.; Bradshaw, C.S.; Hocking, J.S.; de Vries, H.J.; Francis, S.C.; Mabey, D.; Marrazzo, J.M.; Sonder, G.J.; Schwebke, J.R.; Hoornenborg, E.; et al. Sexually transmitted infections: Challenges ahead. Lancet Infect. Dis. 2017, 17, e235–e279. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Meanwell, N.A.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Riddell, J.; Amico, K.R.; Mayer, K.H. HIV preexposure prophylaxis: A review. JAMA 2018, 319, 1261–1268. [Google Scholar] [CrossRef]
- Irungu, E.M.; Baeten, J.M. PrEP rollout in Africa: Status and opportunity. Nat. Med. 2020, 26, 655–664. [Google Scholar] [CrossRef]
- Andrews, C.D.; Spreen, W.R.; Mohri, H.; Moss, L.; Ford, S.; Gettie, A.; Russell-Lodrigue, K.; Bohm, R.P.; Cheng-Mayer, C.; Hong, Z.; et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science 2014, 343, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.D.; Yueh, Y.L.; Spreen, W.R.; Bernard, L.S.; Boente-Carrera, M.; Rodriguez, K.; Gettie, A.; Russell-Lodrigue, K.; Blanchard, J.; Ford, S.; et al. A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci. Transl. Med. 2015, 7, 270ra4. [Google Scholar] [CrossRef] [PubMed]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; et al. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J. Med. Chem. 2013, 56, 5901–5916. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef]
- Kulkarni, T.A.; Bade, A.N.; Sillman, B.; Shetty, B.L.D.; Wojtkiewicz, M.S.; Gautam, N.; Hilaire, J.R.; Sravanam, S.; Szlachetka, A.; Lamberty, B.G.; et al. A year-long extended release nanoformulated cabotegravir prodrug. Nat. Mater. 2020, 19, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Krovi, S.A.; Johnson, L.M.; Luecke, E.; Achilles, S.L. Advances in long-acting injectables, implants, and vaginal rings for contraception and HIV prevention. Adv. Drug Deliv. Rev. 2021, 176, 113849. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Luo, H.; Lu, Q.; Liu, Z.; Liu, J.; Zhang, J.; Wei, Z.; Li, J. Quantum Mechanical-Based Stability Evaluation of Crystal Structures for HIV-Targeted Drug Cabotegravir. Molecules 2021, 26, 7178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Mroue, K.H.; Ramamoorthy, A. Proton-based ultrafast magic angle spinning solid-state NMR spectroscopy. Acc. Chem. Res. 2017, 50, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Joedicke, L.; Mao, J.; Kuenze, G.; Reinhart, C.; Kalavacherla, T.; Jonker, H.R.; Richter, C.; Schwalbe, H.; Meiler, J.; Preu, J.; et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat. Chem. Biol. 2018, 14, 284–290. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Duong, N.T. Practical guides for 1H detected solid-state NMR under fast MAS for small molecules. J. Magn. Reson. Open 2022, 10, 100062. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Hou, G.; Agarwal, V.; Su, Y.; Ramamoorthy, A. Ultrafast magic angle spinning solid-state NMR spectroscopy: Advances in methodology and applications. Chem. Rev. 2022, 123, 918–988. [Google Scholar] [CrossRef] [PubMed]
- Bravetti, F.; Bordignon, S.; Alig, E.; Eisenbeil, D.; Fink, L.; Nervi, C.; Gobetto, R.; Schmidt, M.U.; Chierotti, M.R. Solid-State NMR-Driven Crystal Structure Prediction of Molecular Crystals: The Case of Mebendazole. Chem. Eur. J. 2022, 28, e202103589. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Su, Y. Solid-state NMR of organic molecules: Characterising solid-state form. Solid State Nucl. Magn. Reson. 2023, 126, 101876. [Google Scholar]
- Cordova, M.; Emsley, L. Chemical Shift-Dependent Interaction Maps in Molecular Solids. J. Am. Chem. Soc. 2023, 127, 11094–11102. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Jin, X.; Wan, Z.; He, X. Automated fragmentation quantum mechanical calculation of 13C and 1H chemical shifts in molecular crystals. J. Chem. Phys. 2021, 154, 064502. [Google Scholar] [CrossRef] [PubMed]
- Belov, K.; Dyshin, A.; Krestyaninov, M.; Efimov, S.; Khodov, I.; Kiselev, M.J. Conformational preferences of tolfenamic acid in DMSO-CO2 solvent system by 2D NOESY. Mol. Liq. 2022, 367, 120481. [Google Scholar] [CrossRef]
- Khodov, I.; Belov, K.; Krestyaninov, M.; Sobornova, V.; Dyshin, A.; Kiselev, M.J. Does DMSO Affect the Conformational Changes of Drug Molecules in Supercritical CO2 Media? Mol. Liq. 2023, 384, 122230. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Shaka, A.; Keeler, J.; Frenkiel, T.; Freeman, R.J. An improved sequence for broadband decoupling: WALTZ-16. Magn. Reson. 1983, 52, 335–338. [Google Scholar] [CrossRef]
- Shaka, A.; Barker, P.B.; Freeman, R.J. Computer-optimized decoupling scheme for wideband applications and low-level operation. Magn. Reson. 1985, 64, 547–552. [Google Scholar] [CrossRef]
- Tang, S.; Case, D.A. Vibrational averaging of chemical shift anisotropies in model peptides. J. Biomol. NMR 2007, 38, 255–262. [Google Scholar] [CrossRef]
- Swails, J.; Zhu, T.; He, X.; Case, D.A. AFNMR: Automated fragmentation quantum mechanical calculation of NMR chemical shifts for biomolecules. J. Biomol. NMR 2015, 63, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhu, T.; Zhang, J.Z.; He, X. A systematic study on RNA NMR chemical shift calculation based on the automated fragmentation QM/MM approach. RSC Adv. 2016, 6, 108590–108602. [Google Scholar] [CrossRef]
- Jin, X.; Zhu, T.; Zhang, J.Z.; He, X. Automated fragmentation QM/MM calculation of NMR chemical shifts for protein-ligand complexes. Front. Chem. 2018, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ding, X.; Sun, C.; Watts, A.; He, X.; Zhao, X. Dynamic Coupling of Tyrosine 185 with the Bacteriorhodopsin Photocycle, as Revealed by Chemical Shift Assisted AF-QM/MM Calculations and Molecular Dynamic Simulations. Int. J. Mol. Sci. 2021, 22, 13587. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Frisch, M.E.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakat-suji, H.; et al. Gaussian 16, Rev. C. 01. 2016. Available online: https://gaussian.com/gaussian16/ (accessed on 11 September 2023).
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | Cabotegravir |
|---|---|
| Empirical formula | C19H17F2N3O5 |
| Formula weight | 405.35 g/mol |
| Temperature | 182(2) K |
| Wavelength | 1.54178 Å |
| Crystal system, space group | Orthorhombic, P212121 |
| Unit cell dimensions | a = 7.3002(5) Å alpha = 90 deg. |
| b = 7.3002(1) Å beta = 90 deg. | |
| c = 32.343(2) Å gamma = 90 deg. | |
| Volume | 1723.63(16) Å3 |
| Z, Calculated density | 4, 1.562 Mg/m3 |
| Absorption coefficient | 1.103 mm−1 |
| F(000) | 840 |
| Crystal size | 0.260 × 0.140 × 0.080 mm |
| Theta range for data collection | 5.471 to 68.809 deg. |
| Limiting indices | −8 h 8, −8 k 8, −31 l 39 |
| Reflections collected/unique | 17,089/3165 [R(int) = 0.0349] |
| Completeness to theta = 67.679 | 99.4% |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.7531 and 0.6369 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3165/0/262 |
| Goodness-of-fit on F2 | 1.059 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0274, wR2 = 0.0722 |
| R indices (all data) | R1 = 0.0279, wR2 = 0.0730 |
| Absolute structure parameter | 0.01(4) |
| Extinction coefficient | n/a |
| Largest diff. peak and hole | 0.157 and −0.172 e·A−3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Z.; Shi, M.; Gong, Y.; Lucci, M.; Li, J.; Zhou, J.; Yang, X.-L.; Lelli, M.; He, X.; Mao, J. Multitasking Pharmacophores Support Cabotegravir-Based Long-Acting HIV Pre-Exposure Prophylaxis (PrEP). Molecules 2024, 29, 376. https://doi.org/10.3390/molecules29020376
Wan Z, Shi M, Gong Y, Lucci M, Li J, Zhou J, Yang X-L, Lelli M, He X, Mao J. Multitasking Pharmacophores Support Cabotegravir-Based Long-Acting HIV Pre-Exposure Prophylaxis (PrEP). Molecules. 2024; 29(2):376. https://doi.org/10.3390/molecules29020376
Chicago/Turabian StyleWan, Zheng, Man Shi, Yanqing Gong, Massimo Lucci, Jinjin Li, Jiahai Zhou, Xiao-Liang Yang, Moreno Lelli, Xiao He, and Jiafei Mao. 2024. "Multitasking Pharmacophores Support Cabotegravir-Based Long-Acting HIV Pre-Exposure Prophylaxis (PrEP)" Molecules 29, no. 2: 376. https://doi.org/10.3390/molecules29020376
APA StyleWan, Z., Shi, M., Gong, Y., Lucci, M., Li, J., Zhou, J., Yang, X.-L., Lelli, M., He, X., & Mao, J. (2024). Multitasking Pharmacophores Support Cabotegravir-Based Long-Acting HIV Pre-Exposure Prophylaxis (PrEP). Molecules, 29(2), 376. https://doi.org/10.3390/molecules29020376