Abstract
An unusual series of germylenes and stannylenes stabilized by new tetradentate bis(amidine) ligands RNC(R′)N-linker-NC(R′)NR with a rigid naphthalene backbone has been prepared by protonolysis reaction of Lappert’s metallylenes [M(HMDS)2] (M = Ge or Sn). Germylenes and stannylenes were fully characterized by NMR spectroscopy and X-ray diffraction analysis. DFT calculations have been performed to clarify the structural and electronic properties associated with tetradentate bis(amidine) ligands. Stannylene L1Sn shows reactivity through oxidation, oxidative addition, and transmetalation reactions, affording the corresponding gallium and aluminum derivatives.
1. Introduction
Amidines have been employed as ligands for numerous metallic and semi-metallic elements [1], and one of their best features is the ability to adjust both steric and electronic properties, leading to many substitution patterns of the CN2 framework [2,3,4]. In recent years, the trend has shifted to develop bulkier systems to stabilize a broader range of metal centers or to control the complex geometry via rigid coordination, as seen with bridged bis(amidine) ligands.
Amidinate ligands have shown a remarkable ability to stabilize low-valent main group elements, such as tetrylenes, forming planar NCN-Ge(II) and NCN-Sn(II) four-membered rings [5]. The first bis(amidinato)germylene, reported by Richeson et al. in 1997, exhibited one chelating and one dangling amidinate ligand, with the three-coordinate germanium center adopting a trigonal–pyramidal geometry (Figure 1) [6]. Using the same synthetic method, Karsch et al. reported in 1998 a bis(amidinato)germylene with both amidinate ligands acting as chelates due to the less hindered substituents on the nitrogen atoms [7]. On the other hand, the first mono- and bis-(amidinato)stannylenes were reported by Tolman et al. in 2002, using a N-silylated benzamidinate ligand (Figure 1) [8].
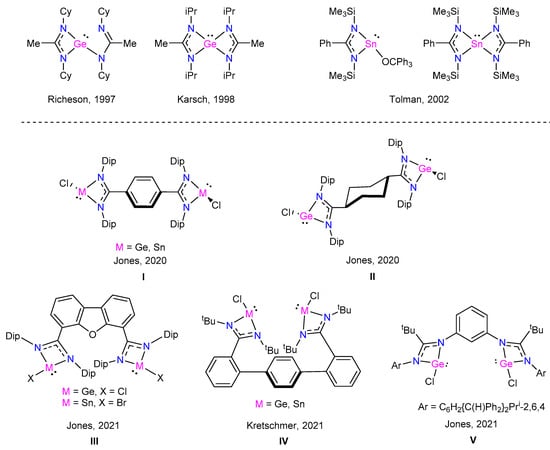
Figure 1.
Examples of germylenes and stannylenes with bis(amidine) ligands [6,7,8,9,10,11].
More recently, the first tetrylenes with 1,4-phenylene (I) and 1,4-cyclohexylene (II) bridged bis(amidine) ligands were reported by Jones et al. in 2020 [9]. In 2021, the same group reported a bis-germylene and a bis-stannylene with a dibenzofurandiyl-linked Dipp-substituted bis(amidine) ligand (III) [10]. In the same year, Kretschmer et al. reported bis-germylenes and bis-stannylenes using a terphenyl-linked bis(amidine) ligand (IV) [11]. It is important to note that all these examples correspond to amidine fragments connected through the carbon atom, and only one bis-germylene with a bis(amidine) ligand nitrogen connected via a 1,3-phenylene bridge has been synthesized by Jones et al. (V) [10]. The main characteristic of bridged bis(amidine) ligands exhibited is that the amidine functionalities act as independent ligands, allowing the convenient preparation of stable bis-metallylenes. Moreover, there is no report of a bis(amidine) system acting as a tetradentate ligand with both amidine moieties coordinated to the same metal center. We therefore considered the hypothesis that such a bis(amidine) system could influence the stabilization and the reactivity of the corresponding tetrylenes.
Herein, we report the synthesis and structural characterization of novel stannylene and germylene compounds stabilized by naphthalene-linked bis(amidine) ligands, with the amidine functionalities connected to the naphthalene bridge through the nitrogen atoms. In addition, the reactivity of synthesized stannylenes was explored in oxidation, oxidative addition, and transmetalation reactions.
2. Results and Discussion
2.1. Synthesis of Bis(amidine) Ligands with a Rigid Naphthalene Backbone
Bis(amidine) ligands L1H2 and L2H2, with a rigid planar 1,8-diaminonaphthalene linker, were prepared following the reported procedures [12,13], while the synthesis of L3H2 was carried out through the reaction between one equivalent of N,N′-(naphthalene-1,8-diyl)bis(2,2-dimethylpropanimidoyl chloride) and two equivalents of p-tolylamine in dry toluene under reflux for 4 h in the presence of Et3N (Scheme 1). L3H2 was isolated as yellow crystals in 63% yield and characterized by NMR spectroscopy. The 1H NMR spectrum shows two NH signals at 9.43 and 8.50 ppm, similar to those previously reported bis(amidine) ligands [13,14]. In addition, CH3 resonances of p-tolyl and tbutyl fragments appear as singlets at 2.05 and 2.01 ppm and at 1.38 and 1.23 ppm, respectively. The 13C NMR spectrum also exhibits two different C=N groups at 161.3 and 160.7 ppm, confirming the non-symmetrical character of L3H2 ligand in solution due to NH…N hydrogen bonds.
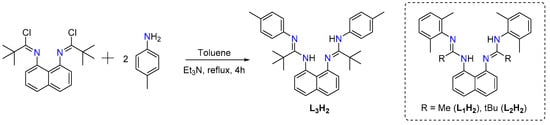
Scheme 1.
Synthesis of ligand L3H2.
The presence of an intramolecular hydrogen bond between the N-H group and the nitrogen atom of the amidine moiety [ N1-(H1)…N(3) 1.95(5) Å] is confirmed in the solid-state structure of ligand L3H2 (Figure 2), in line with similar observations for ligands L1H2 and L2H2 [12,13].
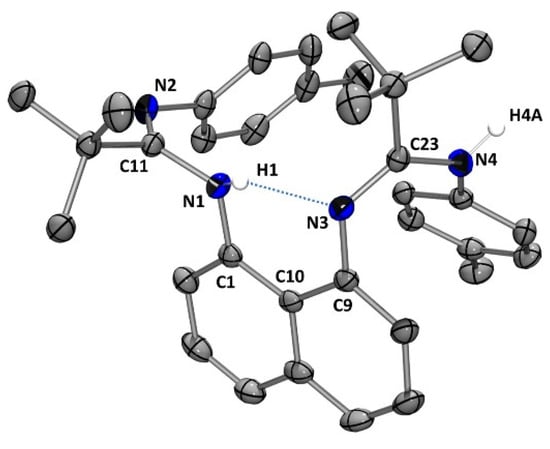
Figure 2.
Molecular structure of L3H2. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms (except H1 and H4A) have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: N(1)-C(11) 1.421(5); N(2)-C(11) 1.272(5); N(3)-C(23) 1.286(5); N(4)-C(23) 1.373(5); N1-(H1)…N(3) 1.95(5); N(1)-C(11)-N(2) 122.5(3); N(3)-C(23)-N(4) 126.1(3).
2.2. Synthesis of Stannylenes L1–3Sn
The synthesis of stannylene L1–3Sn was carried out using protonolysis reactions due to the simplicity of the procedure [15,16,17]. Therefore, the corresponding bis(amidine) ligand L1–3H2 reacted with one equivalent of Sn(HMDS)2 in dry THF at 60 °C for 3 h (Scheme 2). Stannylene L1Sn was isolated as colorless crystals from a pentane solution at −30 °C in 77% yield. The formation of L1Sn was confirmed by 1H, 13C, and 119Sn NMR spectroscopy and mass spectrometry. The 1H NMR spectrum shows a singlet at 1.97 ppm with an integration of 6 H, corresponding to the two amidine methyl groups, and a singlet at 2.18 ppm with an integration of 12 H, corresponding to the methyl groups of 2,6-dimethylphenyl fragments. This observed NMR pattern agrees with a symmetrical structure around the tin center. The 13C NMR spectrum shows a characteristic signal for the NCN fragment at 169.0 ppm, similar to those (amidinato)stannylenes previously reported [8,15,16,18]. In the 119Sn NMR spectrum, a resonance at −276.7 ppm confirms the tetracoordinated nature of the tin atom, comparable to that of reported homoleptic four-coordinated bis(amidinate) tin(II) [15]. L3H2 also reacts with Sn(HMDS)2 in the same manner to give the corresponding tetracoordinated stannylene L3Sn, as shown by the NMR analysis. Indeed, the 1H NMR spectrum shows a singlet at 1.30 ppm that integrates for 18 H, corresponding to the amidine tbutyl groups, and a singlet at 2.29 ppm that integrates for 6 H, corresponding to the p-tolyl methyl groups. The 13C NMR spectrum shows a characteristic signal for the NCN fragment at 177.5 ppm. The symmetrical tetracoordination of the tin center is also confirmed by a resonance at −254.9 ppm in the 119Sn NMR spectrum. In contrast, the use of the bulky bis(amidinate) ligand L2H2 leads to the formation of a dimeric structure (L2Sn)2.
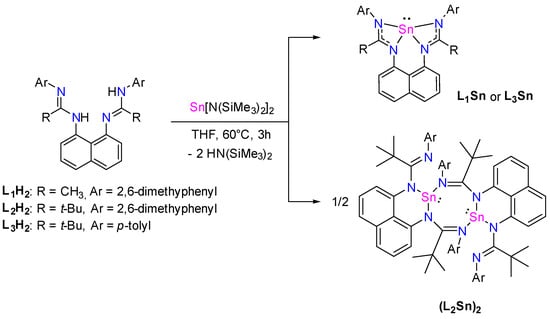
Scheme 2.
Synthesis of stannylene L1–3Sn.
Stannylene L1Sn was characterized in the solid state by single-crystal X-ray diffraction analysis (Figure 3). The molecular structure indicates a four-coordinate tin center with a distorted square-based pyramidal geometry. A few stannylenes with unbridged bis(amidine) ligands display a similar geometry [16]. The acute N1–Sn–N2 and N3–Sn–N4 angles of 57.38° and 57.74° are comparable to values previously recorded for Sn(II) amidinate complexes [15,18].
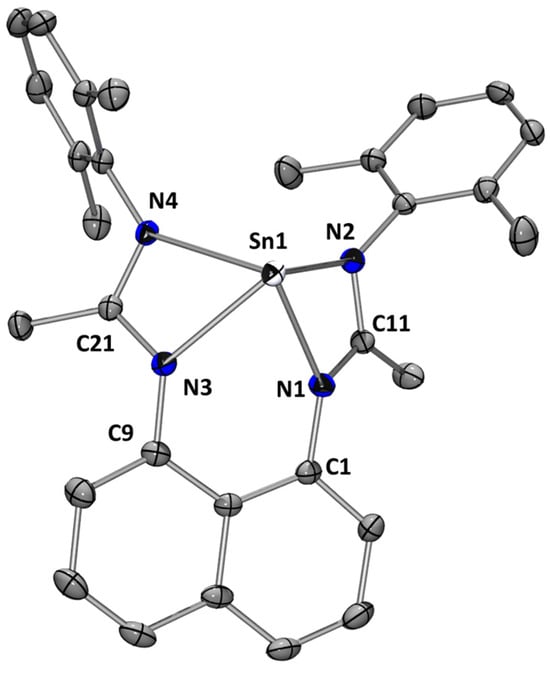
Figure 3.
Molecular structure of L1Sn. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Sn(1)-N(1) 2.2435(17); Sn(1)-N(2) 2.3327(17); Sn(1)-N(3) 2.2232(17); Sn(1)-N(4) 2.3181(16); N(1)-C(11) 1.326(3); N(2)-C(11) 1.328(3); N(3)-C(21) 1.328(3); N(4)-C(21) 1.329(3); N(3)-Sn(1)-N(1) 72.57(6); N(3)-Sn(1)-N(4) 57.74(6); N(1)-Sn(1)-N(4) 107.35(6); N(3)-Sn(1)-N(2) 112.07(6); N(1)-Sn(1)-N(2) 57.38(6); N(4)-Sn(1)-N(2) 95.29(6); N(1)-C(11)-N(2) 111.85(17); N(3)-C(21)-N(4) 111.37(17).
The coordinates of the experimental solid-state structure are consistent with the calculated values in DFT (see Supplementary Materials for more information). Indeed, the modeling calculates values between 2.27 and 2.42 Å for the Sn–N bonds, probably due to steric repulsion, and angles at the tin atom of 56°. Moreover, the energy of molecular orbitals of this species (computed using DFT) shows an electrophilic character (the first vacant orbital on the Sn atom is computed at −0.60 eV) and a potential nucleophilicity, as the HOMO centered on the non-bonding pair of tin is rather accessible (−5.42 eV) (Figure 4).
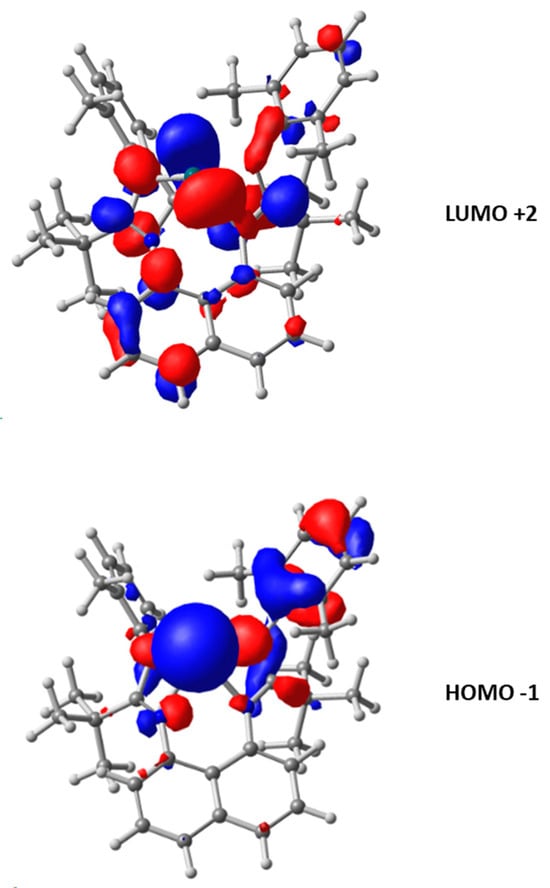
Figure 4.
LUMO +2 (up) and HOMO −1 (down) for L1Sn.
The modeling demonstrates that this compound can be obtained from the bis(amidine) ligand through the addition of Sn(HMDS)2. Indeed, starting from the resulting complex, a prototropic shift can occur, leading to the facile formation of the corresponding RSnH (TS = 21.63 kcal/mol) (see Supplementary Materials for more information). Subsequently, the elimination of HMDS results in the obtaining of L1Sn, exhibiting a higher but consistent kinetic rate with the applied experimental conditions (60 °C/3 h).
Because of the poor solubility of stannylene dimer (L2Sn)2 in common organic solvents, the NMR characterization cannot be obtained once crystallized. Nevertheless, the X-ray diffraction analysis shows an eight-membered cyclic structure with a Sn…Sn distance of 3.7311(5) Å (Figure 5). Each Sn center has a trigonal pyramidal geometry with a sum of angles around the Sn atom of 272°. Interestingly, the tin atoms are coordinated to both nitrogen atoms directly linked to the naphthalene core and, simultaneously, to one of the nitrogen atoms from another amidinate fragment. Unfortunately, providing theoretical arguments for the formation of dimer (L2Sn)2 is challenging. Indeed, calculations reveal an energetically favorable reaction pathway (ΔG = −27.74 kcal/mol) to obtain the corresponding monomer. We can postulate that the presence of the 2,6-dimethylphenyl and tbutyl groups in the ligand introduces significant steric hindrance, inhibiting the arrangement of a tetracoordinated tin center and thereby influencing the preferential formation of the dimer.
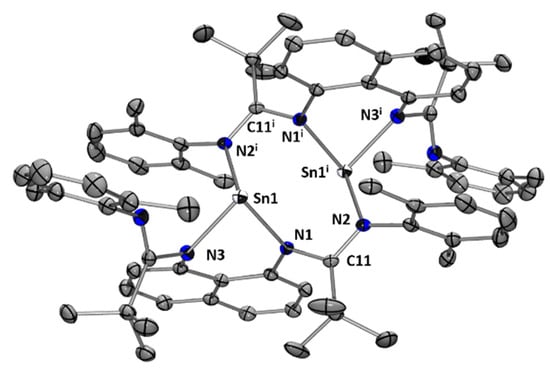
Figure 5.
Molecular structure of (L2Sn)2. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Sn(1)-N(1) 2.243(2); Sn(1)-N(2i) 2.327(2); Sn(1)-N(3) 2.144(3); C(11)-N(1) 1.378(4); C(11)-N(2) 1.309(4); N(1)-Sn(1)-N(3) 78.80(9); N(3)-Sn(1)-N(2i) 97.73(9); N(1)-Sn(1)-N(2i) 95.21(9); C(11)-N(1)-Sn(1) 112.43(18); N(1)-C(11)-N(2) 114.2(3).
Stannylene L3Sn and dimer (L2Sn)2 can also be prepared through the reaction of deprotonated bis(amidine) ligand (L2K2 or L3K2) with one equivalent of SnCl2. The same products, (L2Sn)2 and L3Sn, were also obtained using two equivalents of SnCl2, instead of the expected bis-stannylenes L2Sn2 and L3Sn2 (Scheme 3).
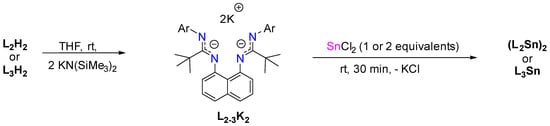
Scheme 3.
Synthetic routes to obtain L2-3Sn from deprotonated bis(amidine) ligands.
Other attempts to synthesize bis-stannylenes L2Sn2 and L3Sn2, by reacting the corresponding ligand (L2H2 or L3H2) with two equivalents of Sn(HMDS)2 (via protonolysis reactions), have also failed (Scheme 4). In both cases, at room temperature, the formation of an intermediate LH-SnN(TMS)2 was first detected by 1H NMR spectroscopy. The subsequent heating at 60 °C for 1h affords the corresponding stannylenes (L2Sn)2 or L3Sn (44% and 61% yield, respectively), accompanied by the formation of H-HMDS and a black solid of Sn0 as by-products.
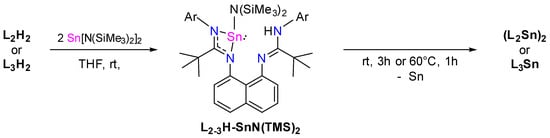
Scheme 4.
Synthetic routes to obtain L2-3Sn by protonolysis.
2.3. Synthesis of Germylenes Stabilized by Bis(amidine) Ligands
Following the same synthetic strategy, we have considered the reaction of bis(amidinato) ligands L1–3H2 with Ge(HMDS)2. In the case of L1H2, only dimer (L1Ge)2 was obtained as yellow crystals in 42% yield (Scheme 5). Because of poor solubility, it was only possible to characterize (L1Ge)2 by an X-ray diffraction analysis. The eight-membered cyclic molecular structure of (L1Ge)2 exhibits two trigonal pyramidal Ge centers with a Ge–Ge distance of 3.4984(3) Å (Figure 6).
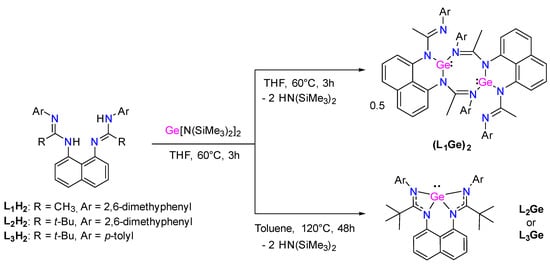
Scheme 5.
Synthesis of germylenes L1–3Ge.
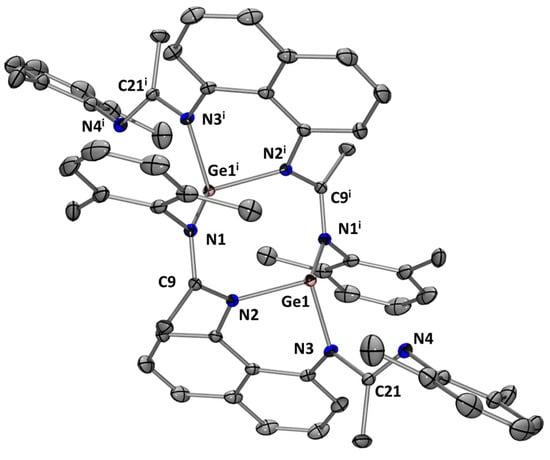
Figure 6.
Molecular structure of (L1Ge)2. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Ge(1)-N(1i) 2.0961(11); Ge(1)-N(2) 2.0048(10); Ge(1)-N(3) 1.9332(10); C(9)-N(1) 1.3074(16); C(9)-N(2) 1.3740(16); N(3)-Ge(1)-N(2) 87.48(4); N(3)-Ge(1)-N(1i) 95.69(4); N(2)-Ge(1)-N(1i) 96.79(4); N(1)-C(9)-N(2) 116.31(11); C(9)-N(2)-Ge(1) 112.38(8).
The reaction of L2H2 with Ge(HMDS)2 afforded germylene L2Ge after heating at 110 °C for 48 h to complete the reaction (Scheme 5). Germylene L2Ge was isolated as yellow crystals from a pentane solution at −30 °C in 52% yield. The 1H NMR spectrum shows a singlet at 1.60 ppm with an integration of 18 H, corresponding to the tbutyl groups, and a singlet at 2.18 ppm with an integration of 12 H, corresponding to the methyl groups of the 2,6-dimethylphenyl substituent. In addition, the 13C NMR spectrum shows the characteristic signal for the NCN fragment at 170.9 ppm. These data are in agreement with a symmetrical tetracoordinated germanium center in solution. However, in the solid state, the X-ray diffraction analysis of L2Ge shows a tri-coordinated germanium atom with a distorted trigonal pyramidal geometry. Here, the coordination is through the two nitrogen atoms of the naphthalene bridge and one nitrogen atom of amidine moiety, leaving the remaining nitrogen atom dangling (Figure 7). The N–C distances [1.275(5)–1.417(5) Å] indicate an electron delocalization within the NCN fragments, and due to the bulky nature of the tbutyl substituents, they are in opposite directions to each other, distorting the naphthalene ring.
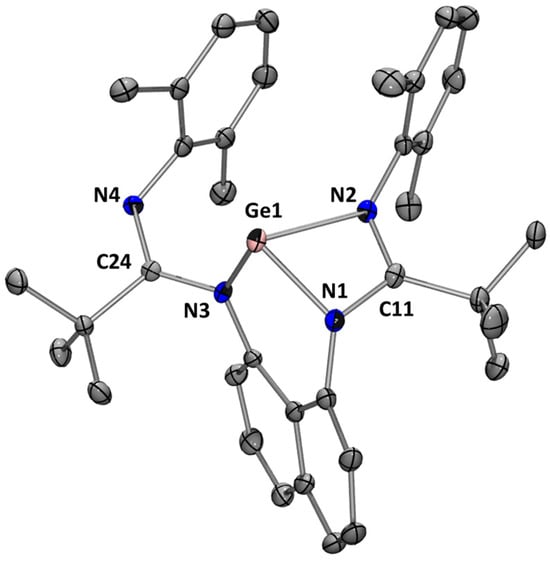
Figure 7.
Molecular structure of L2Ge. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Ge(1)-N(1) 1.969(3); Ge(1)-N(2) 2.073(3); Ge(1)-N(3) 1.912(3); C(11)-N(2) 1.308(5); C(11)-N(1) 1.374(5); C(24)-N(4) 1.275(5); C(24)-N(3) 1.417(5); N(1)-Ge(1)-N(2) 64.89(13); N(1)-Ge(1)-N(3) 90.95(13); N(2)-Ge(1)-N(3) 102.00(13); N(1)-C(11)-N(2) 108.0(3); N(3)-C(24)-N(4) 124.0(3).
2.4. Reactivity of Stannylene L1Sn
Several tests of reactivity were carried out with stannylene L1Sn, which is easy to obtain in good yields. We first considered the activation of small molecules such as ethylene, NH3, and CO2, but without any success because of no reactions, despite the multiple conditions evaluated (temperature, time, and pressure).
However, when exposed to N2O (five bars) in THF solution, L1Sn slowly reacts (3 h at 70 °C) to give a dimeric amidinate-stannoxane 1a, which was isolated as colorless crystals in 46% yield (Scheme 6). Compound 1a is moderately soluble in THF, and the 1H NMR signals tend to be broadened. Therefore, the coupling of signals in the aromatic region cannot be clearly seen, but the integration is as expected, with two broad singlets at 2.07 and 1.85 ppm that integrate for 12 H, corresponding to the methyl groups of the 2,6-dimethylphenyl fragment. In addition, a resonance at 1.76 ppm, integrating for 12 H and corresponding to the methyl groups of the amidine moieties, was observed. The characteristic signal of the carbon amidinate fragments in the 13C NMR spectrum resonates at 168.7 ppm. In addition, a 119Sn chemical shift at −494.5 ppm was observed, consistent with reported stannoxane dimers [19]. The crystal structure of 1a shows a dinuclear species with a central planar Sn2O2 ring with two hexacoordinated tin atoms in a distorted pseudo-octahedral geometry (Figure 8). The O–Sn distances (~2.000 Å), Sn–Sn contact (2.9779(2) Å), and O–Sn–O angles (84.01°) are in the range of other dimeric tin amidinate-stannoxanes reported [19].
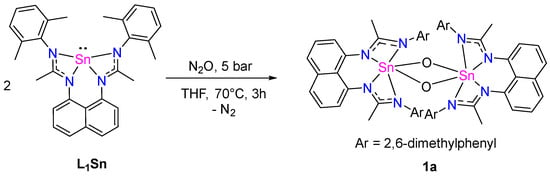
Scheme 6.
Synthesis of 1a.
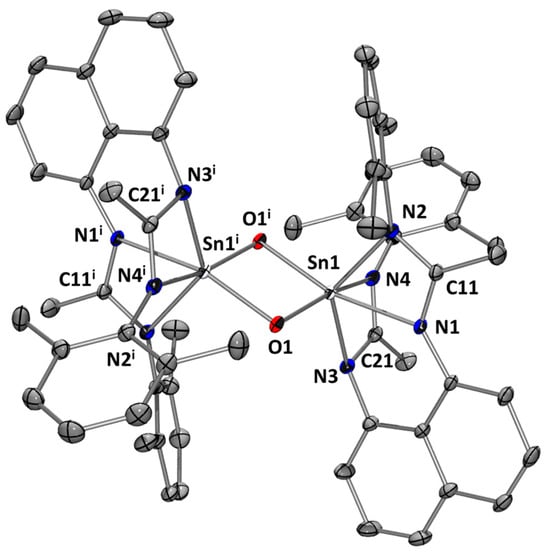
Figure 8.
Molecular structure of 1a. Thermal ellipsoids are represented with a 30% probability. Hydrogens and solvent molecules have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Sn(1)-N(1) 2.2236(10); Sn(1)-N(2) 2.1644(11); Sn(1)-N(3) 2.1423(10); Sn(1)-N(4) 2.2742(10); Sn(1)-O(1) 2.0079(9); Sn(1)-O(1i) 1.9996(8); Sn(1)-Sn(1i) 2.97787(19); C(11)-N(1) 1.3285(16); C(11)-N(2) 1.3415(16); C(21)-N(4) 1.3113(17); C(21)-N(3) 1.3439(17); O(1)-Sn(1)-O(1i) 84.01(4); Sn(1)-O(1)-Sn(1i) 95.98(4); N(1)-C(11)-N(2) 110.50(11); N(3)-C(21)-N(4) 112.95(11).
Usually, stannylenes readily react with chalcogenides [6,20,21,22,23,24,25], and therefore we have investigated the reaction of L1Sn with elemental sulfur (Scheme 7). After the reaction mixture had been stirred at room temperature for 4 h, 1H, and 119Sn NMR spectroscopy showed a mixture of new signals and starting stannylene. In the 119Sn NMR spectrum, next to the resonance of L1Sn, a new signal appears at −371 ppm, which evolves to a resonance singlet at −648.0 ppm after 24 h, corresponding to dimer 1b. After purification, 1b was isolated as pale-yellow crystals from a THF solution at −30 °C in 51% yield. The dimeric structure of 1b has been determined by an X-ray diffraction analysis, revealing a central planar Sn2S2 ring with the two tin centers in a distorted pseudo-octahedral geometry (Figure 9). The S–Sn distances are ~2.400 Å and Sn–Sn contact is 3.3847(10) Å [25], longer than that observed for the di-oxygen analog dimer 1a due to the size of the sulfur atom.
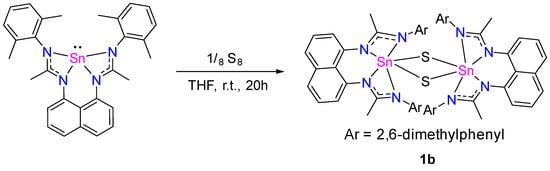
Scheme 7.
Synthetic route to obtain 1b.
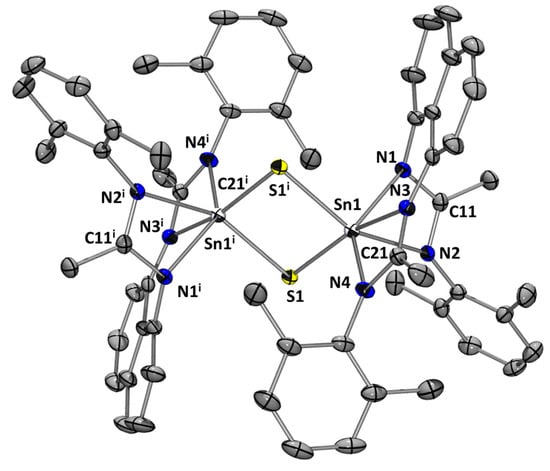
Figure 9.
Molecular structure of 1b. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms and solvent molecules have been omitted for clarity. The asymmetric unit contains two independent but very similar molecules; data for only one molecule are discussed. Selected bond distances [Å] and bond angles [deg]: Sn(1)-N(1) 2.130(4); Sn(1)-N(2) 2.374(3); Sn(1)-N(3) 2.240(3); Sn(1)-N(4) 2.198(4); Sn(1)-S(1) 2.4172(14); Sn(1)-S(1i) 2.4585(13); C(11)-N(1) 1.345(6); C(11)-N(2) 1.315(5); C(21)-N(3) 1.332(5); C(21)-N(4) 1.328(5); Sn(1)-S(1)-Sn(1i) 87.92(4); S(1)-Sn(1)-S(1i) 92.08(4); N(1)-C(11)-N(2) 112.8(4); N(3)-C(21)-N(4) 110.9(4).
In addition, compound 1b was characterized by NMR spectroscopy. The 1H NMR spectrum exhibits broadened signals. In the 13C NMR spectrum, the characteristic signal of the NCN fragment appears at 167.4 ppm. In the 119Sn NMR spectrum, a very high field chemical shift at −648.0 ppm is observed, a resonance signal that is more high-field shifted than those of previously reported Sn2S2-bridged dimeric complexes [26,27].
L1Sn reacts with p-tolyldisulfide in THF at 70 °C overnight via an oxidative addition reaction with a tin insertion into the S–S bond (Scheme 8) to give a hexacoordinate Sn(IV) species 2a [22]. This molecule presents a characteristic 119Sn chemical shift at −456.7 ppm, consistent with a hexacoordinated tin center [24]. In addition, the X-ray diffraction analysis of 2a reveals a monomeric Sn(IV) species in a distorted octahedral coordination sphere that included the four nitrogen atoms of the two chelating amidinates and the two p-tolyl sulfur fragments (Figure 10). The equivalence of C–N bond lengths within NCN frameworks and their magnitudes (1.301 to 1.361 Å) indicate that the π electrons within the ligands are delocalized.
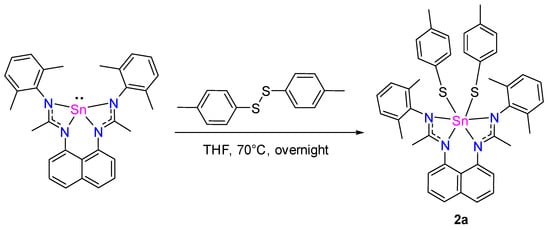
Scheme 8.
Synthesis of 2a.
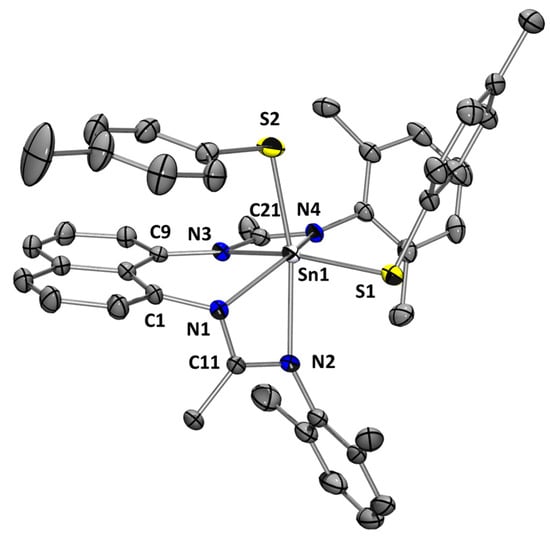
Figure 10.
Molecular structure of 2a. Thermal ellipsoids are represented with a 30% probability. Hydrogen atoms have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Sn(1)-N(1) 2.132(3); Sn(1)-N(2) 2.411(3); Sn(1)-N(3) 2.230(3); Sn(1)-N(4) 2.180(3); Sn(1)-S(1) 2.4297(15); Sn(1)-S(2) 2.4483(16); C(11)-N(1) 1.361(4); C(11)-N(2) 1.301(4); C(21)-N(3) 1.334(4); C(21)-N(4) 1.337(4); N(1)-Sn(1)-N(4) 132.96(11); N(1)-Sn(1)-N(3) 78.01(11); N(3)-Sn(1)-N(4) 59.72(10); N(1)-Sn(1)-N(2) 58.27(11); N(2)-Sn(1)-N(4) 97.20(12); N(2)-Sn(1)-N(3) 86.71(11); N(1)-Sn(1)-S(1) 111.48(9); N(4)-Sn(1)-S(1) 102.83(8); N(3)-Sn(1)-S(1) 158.14(7); N(2)-Sn(1)-S(1) 82.44(9); N(1)-Sn(1)-S(2) 105.90(9); N(4)-Sn(1)-S(2) 97.26(9); N(3)-Sn(1)-S(2) 94.06(9); N(2)-Sn(1)-S(2) 163.67(8); S(1)-Sn(1)-S(2) 101.67(7); N(2)-C(11)-N(1) 113.5(3); N(3)-C(21)-N(4) 110.6(3).
3,5-di-tert-butyl-ortho-quinone also reacts with L1Sn to afford the corresponding [4 + 1]-cycloadduct 2b (Scheme 9). The 1H NMR spectrum reveals the characteristic signals of the quinone group in the aromatic region, doublets at 6.57 and 6.39 ppm (JHH = 2.4 Hz), and two singlets at 1.06 and 1.16 ppm, corresponding to the CH3 groups of tbutyl in the aliphatic area. Also, the signals corresponding to the amidine CH3 groups’ moieties integrating for 6 H as a singlet at 2.00 ppm and to the CH3 groups of 2,6-dimethylphenyl moieties integrating for 12 H as a singlet at 2.08 ppm, respectively, are observed. The 119Sn NMR spectrum shows a singlet at −512.2 ppm, in agreement with the cycloadduct structure. The mass analysis (DCI/CH4) spectrum exhibits a peak at 786.29, which corresponds to [M]+ of compound 2b, evidencing its formation.
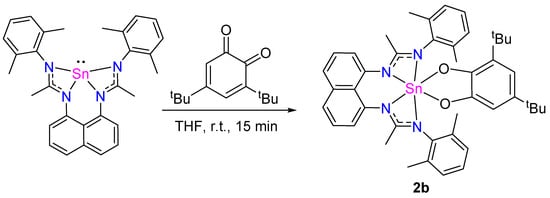
Scheme 9.
Synthesis of 2b.
As reported by So et al., an amidinatogermylene can act as a good ligand to form a germylene→ECl2-type adduct with GeCl2 and SnCl2 [28]. In contrast, L1Sn reacts with GeCl2(dioxane) in THF at room temperature via a transmetalation reaction to give a germylene dimer (L1Ge)2 (Scheme 10). The 119Sn NMR spectrum shows a resonance at −214.0 ppm, in agreement with the formation of SnCl2 [29].
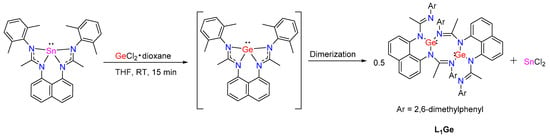
Scheme 10.
Synthesis of L1Ge by transmetalation reaction.
Transmetalation reactions were also observed with AlCl3 and GaCl3, affording the corresponding Al(III) 3a and Ga(III)-chlorides 3b, and in both cases, forming one equivalent of SnCl2 (Scheme 11). The modeling confirms an exergonic reaction (−18.92 kcal/mol) during this “metal” exchange process. L1Sn reacts with AlCl3 quickly in THF at room temperature for 30 min to give 3a, which was isolated as a white solid in 83% yield. The 1H NMR spectrum shows, in the aromatic region, the expected resonances and integrals, and in the aliphatic area, two singlets at 2.33 and 1.90 ppm, integrating for 12 H, corresponding to the methyl groups of 2,6-dimethylphenyl fragments and a singlet at 2.13 ppm (6 H) related to the -CH3 of the amidine groups. In the 13C NMR spectrum, the characteristic NCN signal is observed at 176.4 ppm. The symmetry of the 1H and 13C NMR spectra are in agreement with a pentacoordinated aluminum complex. Using the same experimental conditions, L1Sn also reacts with GaCl3 to give Ga(III)-chloride 3b, which was isolated as colorless crystals from a pentane solution at −30 °C in 88% yield. Gallium chloride 3b was fully characterized by NMR spectroscopy (very similar to that of 3a) and by X-ray diffraction analysis. The molecular structure of 3b (Figure 11) shows a pentacoordinated gallium center with a distorted square-based pyramidal geometry. The Ga–Cl bond distance of 2.183 Å is in the range expected for such compounds [30].
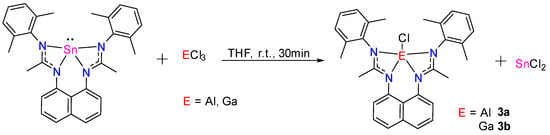
Scheme 11.
Synthesis of 3a and 3b via transmetalation reactions.
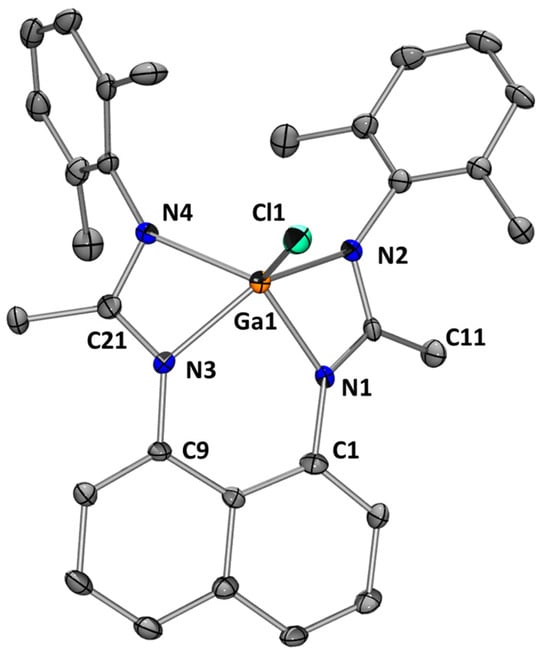
Figure 11.
Molecular structure of 3b. Thermal ellipsoids are represented with a 30% probability. Hydrogens have been omitted for clarity. Selected bond distances [Å] and bond angles [deg]: Ga(1)-N(1) 1.984(4); Ga(1)-N(2) 2.017(4); Ga(1)-N(3) 1.981(4); Ga(1)-N(4) 2.051(4); Ga(1)-Cl(1) 2.1828(14); N(1)-Ga(1)-N(3) 85.32(15); C(11)-N(1) 1.320(5); C(11)-N(2) 1.345(6); C(21)-N(3) 1.327(6); C(21)-N(4) 1.342(6); N(2)-Ga(1)-N(3) 139.06(16); N(1)-Ga(1)-N(2) 65.43(16); N(3)-Ga(1)-N(4) 65.69(15); N(1)-Ga(1)-N(4) 130.88(15); N(2)-Ga(1)-N(4) 112.06(16); N(3)-Ga(1)-Cl(1) 110.32(12); N(1)-Ga(1)-Cl(1) 115.30(11); N(2)-Ga(1)-Cl(1) 107.98(12); N(4)-Ga(1)-Cl(1) 111.79(12); N(1)-C(11)-N(2) 108.5(4); N(3)-C(21)-N(4) 110.1(4).
3. Materials and Methods
3.1. General Comments
All manipulations were performed under an inert argon or nitrogen atmosphere using standard Schlenk-line and glovebox techniques. Dry, oxygen-free solvents were employed. Reagents were obtained from commercial suppliers unless otherwise stated. N-(2,6-dimethylphenyl)acetamide [31], N-(2,6-dimethylphenyl)acetimidoyl chloride [29], N,N′-(naphthalene-1,8-diyl)bis(2,2-dimethylpropanamide) [13], N,N′(naphthalene-1,8-diyl)bis(2,2-dimethylpropanimidoyl chloride) [13], L1 [12], and L2 [13] were synthesized following reported procedures. The Lappert’s germanium(II) and tin(II) derivatives were prepared according to the literature procedures [32]. The 1D and 2D NMR spectra were recorded with the following spectrometers for 1H, 13C, and 119Sn: Bruker Avance II 300 MHz, Avance III HD 400 MHz, and Avance I and II 500 MHz spectrometers. The chemical shift has been counted positively versus the low field and expressed in part per million (ppm). The mass spectrometric analysis was performed using three techniques: direct chemical ionization (DCI-CH4) methods recorded on a GCT Premier Waters mass spectrometer (Headquarters, MN, USA); electrospray ionization (ESI, Los Angeles, CA, USA) recorded on a Waters Xevo G2 Q-TOF mass spectrometer; and a Maldi micro-MX micro–mass in a pyrene matrix (ratio of product/matrix 1/100). Melting points were measured with capillary Electrothermal Stuart SMP40 apparatus, and samples were prepared in the glovebox before the analysis. FT-IR spectra were measured on a ThermoNicolet 6700 (Waltham, MA, USA) Nexus and recovered in a solid state (KBr). Single-crystal X-ray data were collected at a low temperature (193(2)K) on a Bruker APEX II Quazar (Billerica, MA, USA) diffractometer equipped with a 30W air-cooled microfocus source [(L2Sn)2, L2Ge, 2a and 3b] or on a Bruker D8 VENTURE diffractometer equipped with a PHOTON III detector [L3H2, L1Sn, (L1Ge)2, 1a and 1b] using MoKα radiation (λ = 0.71037 Å). The structures were solved by the intrinsic phasing method (SHELXT) [33] and refined by the full-matrix least-squares method on F2 [34]. All non-H atoms were refined with anisotropic displacement parameters and all the hydrogen atoms were refined isotropically at calculated positions using a riding model. For 2a, some solvent molecules were highly disordered and difficult to model correctly. Therefore, the SQUEEZE function of PLATON [35] was used to eliminate the contribution of the electron density of those solvent molecules from the intensity data. Calculations were performed with the Gaussian 16 suite of programs [36] using the density functional method B3LYP with dispersion (D3) [37,38,39,40]. Tin atoms were treated with a Stuttgart–Dresden pseudopotential in combination with its adapted basis set [41]. All other atoms have been described with a 6–31G(d,p) basis set. Geometry optimizations were carried out without any symmetry restrictions. Frequency calculations were undertaken to confirm the nature of the stationary points, yielding one imaginary frequency for transition states (TS) and all of them were positive for minima. The connectivity of the transition states and their adjacent minima was confirmed by intrinsic reaction coordinate (IRC) calculations [42,43].
3.2. Synthesis
Synthesis of L3H2. p-toluidine (591 mg, 5.6 mmol) was added to a solution of N,N′-(naphthalene-1,8-diyl)bis(2,2-dimethylpropanimidoyl chloride) (1 g, 2.8 mmol) in dry toluene (50 mL). Then, Et3N (559 mg, 5.6 mmol) was added to the mixture. The reaction was stirred for 4 h under reflux. All volatiles were removed under reduced pressure. The solid residue was taken up in 40 mL of Et2O and washed with 25 mL of a saturated solution of Na2CO3. Then, the organic layer was washed with water (3 × 30 mL) and dried over Na2SO4. After that, the solvent was removed, and the crude product was recrystallized with CH2Cl2/pentane (1:2). Yellow crystals were obtained (63% yield). Melting point: 116–122 °C. 1H NMR (DMSO-d6, 500 MHz): δ 9.43 (s, 1H, NH); 8.50 (s, 1H, NH); 7.26 (d, JHH = 6.5 Hz, 1H, aryl); 6.94 (d, JHH = 7.9 Hz, 1H, aryl); 6.88–6.83 (m, 3H, aryl); 6.80 (m, 3H, aryl); 6.66 (d, JHH = 8.1 Hz, 2H, aryl); 6.58–6.51 (m, 2H, aryl); 6.32–6.20 (m, 2H, aryl); 2.05 (s, 3H, CH3); 2.01 (s, 3H, CH3); 1.38 (s, 9H, C(CH3)3); 1.23 (s, 9H, C(CH3)3). 13C{1H} NMR (DMSO-d6, 125 MHz): δ 161.3 (C=N); 160.7 (C=N); 147.2 (aryl, C); 146.5 (aryl, C); 138.6 (aryl, C); 137.2 (aryl, C); 135.6 (aryl, C); 131.2 (aryl, C); 130.4 (aryl, C); 128.7 (aryl, CH); 128.0 (aryl, CH); 127.7 (aryl, CH); 124.8 (aryl, CH); 121.4 (aryl, CH); 120.9 (aryl, CH); 119.8 (aryl, CH); 116.5 (aryl, C); 114.8 (aryl, CH); 28.8 (C(CH3)3); 28.3 (C(CH3)3); 20.3 (CH3). IR (KBr, cm−1): 3373 (˅NH); 1623 (˅C=N). ESI m/z: 504.33 ([M]+).
3.2.1. General Synthetic Procedure of Metallylenes
THF (5 mL) was added to a mixture of one equiv. of MCl2 (M = Sn or Ge) and two equiv. of K[N(SiMe3)2]2 in a 20 mL vial inside the glovebox. The mixture was stirred for 30 min at room temperature. After this time, a white precipitate was obtained. The mixture was filtered, and the solution was added to one equiv. of the corresponding L1–3H2, stirring at 60 °C for 3 h. Then, all the volatiles were removed and the residual solid was washed with pentane (3 × 5 mL).
L1Sn. Colorless crystals were obtained by recrystallization from pentane at −30 °C (77% yield). Melting point: 202 °C (decomposition). 1H NMR (THF-d8, 500 MHz): δ 7.42 (dd, JHH = 8.2, 1.1 Hz, 2H, C10H6); 7.31–7.27 (m, 2H, C10H6); 7.07 (dd, JHH = 7.5, 1.1 Hz, 2H, C10H6); 6.96 (d, JHH = 7.4 Hz, 4H, C6H3); 6.83 (t, JHH = 7.5 Hz, 2H, C6H3); 2.18 (s, 12H, CH3); 1.97 (s, 6H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 169.0 (NCN); 145.3 (C6H3ipso); 144.5 (C10H6ipso); 138.6 (C10H6ipso); 134.4 (C6H3ipso); 128.8 (C6H3); 126.4 (C10H6); 124.8 (C6H3); 123.4 (C10H6); 119.8 (C10H6); 20.0 (CH3); 17.9 (CH3). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −276.7. ESI m/z: 565.15 ([M]+); 449.27 ([M − Sn]+).
(L2Sn)2. Yellow crystals (44% yield). Melting point: 243 °C (decomposition). MS (Maldi-TOF) m/z: 649.3 ([M/2, monomer]+). Meaningful solution state spectroscopic data for the compound could not be obtained for the compound as it shows negligible solubility in normal non-coordinating deuterated solvents once crystallized.
L3Sn. Yellow solid (61% yield). Melting point: 189 °C (decomposition). 1H NMR (THF-d8, 400 MHz): δ 7.38 (d, JHH = 7.5 Hz, 2H, C10H6); 7.24–7.19 (m, 2H, C10H6); 7.09 (d, JHH = 7.1 Hz, 2H, C10H6); 7.05 (d, JHH = 8.0 Hz, 4H, C6H4); 6.97 (d, JHH = 8.2 Hz, 4H, C6H4); 2.29 (s, 6H, CH3); 1.30 (s, 18H, C(CH3)3). 13C{1H} NMR (THF-d8, 125 MHz): δ 177.5 (NCN); 146.7 (C6H4ipso); 145.7 (C10H6ipso); 138.4 (C10H6ipso); 133.0 (C6H4ipso); 130.4 (C6H4); 126.3 (C10H6); 124.9 (C6H4); 123.4 (C10H6); 121.3 (C10H6); 43.6 (C(CH3)3); 31.8 (C(CH3)3); 21.0 (CH3). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −254.9. MS (Maldi-TOF) m/z: 621.2 ([M]+); 503.4 ([M–Sn]+).
(L1Ge)2. Yellow crystals (42% yield). Melting point: 259 °C (decomposition). MS (Maldi-TOF) m/z: 519.14 ([M/2, monomer]+). Meaningful solution state spectroscopic data for the compound could not be obtained for the compound as it shows negligible solubility in normal non-coordinating deuterated solvents once crystallized.
L2Ge. Toluene was used instead of THF as solvent. Yellow crystals were obtained by recrystallization from pentane at −30 °C. (52% yield). Melting point: 199 °C (decomposition). 1H NMR (C6D6, 300 MHz): δ 6.98–6.91 (m, 2H, C10H6); 6.89–6.81 (m, 2H, C10H6); 6.62 (d, JHH = 7.7 Hz, 4H, C6H3); 6.47–6.40 (m, 2H, C6H3); 6.33 (d, JHH = 6.3 Hz, 2H. C10H6); 2.18 (s, 12H, CH3); 1.60 (s, 18H, C(CH3)3). 13C{1H} NMR (C6D6, 125 MHz): δ 170.9 (NCN); 151.8 (C6H3ipso); 148.8 (C10H6ipso); 139.7 (C10H6ipso); 138.5 (C6H3ipso); 128.3 (C6H3); 126.3 (C10H6); 120.4 (C6H3); 114.9 (C10H6); 108.8 (C10H6); 41.9 (C(CH3)3); 30.6 (C(CH3)3); 19.7 (CH3).
Synthesis of 1a. In a J. Young valve NMR tube, a THF (0.4 mL) solution of L1Sn (30 mg, 0.053 mmol) was exposed to 5 bar of N2O. The reaction was monitored by NMR and proceeded quantitatively after 3 h at 70 °C. Colorless crystals were observed and separated from the solution by filtration. Then, the crystals were washed with pentane (3 × 2 mL). Colorless crystals were obtained (46% yield). Melting point: 204 °C. 1H NMR (THF-d8, 500 MHz): δ 7.42 (d, JHH = 6.4 Hz, 4H, C10H6); 7.26 (t, JHH = 7.8 Hz; 4H, C10H6); 7.13 (d, JHH = 6.8 Hz, 4H, C10H6), 7.01 (s, 4H, C6H3); 6.92–6.83 (m, 8H, C6H3); 2.07 (s, 12H, CH3); 1.85 (s, 12H, CH3); 1.76 (s, 12H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 168.7 (NCN); 143.6 (C6H3ipso); 142.7 (C10H6ipso); 138.5 (C10H6ipso); 137.0 (C10H6ipso); 135.3 (C6H3ipso); 128.7 (C6H3); 128.3 (C6H3); 126.3 (C10H6); 125.7 (C6H3); 124.8 (C10H6); 120.6 (C10H6); 19.8 (CH3); 19.0 (CH3); 15.8 (CH3). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −494.5. MS (Maldi-TOF) m/z: 1161.26 ([M]+); 583.13 ([M/2, monomer]+).
3.2.2. General Reactivity Evaluation Procedure
THF was added to a mixture of one equiv. of L1Sn and one equiv. of S8, p-tolyldisulfide, 3,5-di-tert-butyl-o-benzoquinone, AlCl3, or GaCl3, accordingly. The mixture was stirred for the corresponding time and temperature. Then, the solvent was removed and the residual solid was washed with pentane (3 × 0.5 mL).
1b. Stirred overnight at room temperature. Pale-yellow crystals were obtained by recrystallization from THF at −30 °C (51% yield). Melting point: 229 °C (decomposition). 1H NMR (THF-d8, 300 MHz): δ 7.40 (d, JHH = 7.5 Hz, 4H, C10H6); 7.22 (t, JHH = 7.8 Hz, 4H, C10H6); 7.11–7.02 (m, 4H, C10H6); 6.93 (bs, 12H, C6H3); 2.11 (br s, 24H, CH3); 1.75 (s, 12H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 167.4 (NCN); 142.7 (C10H6ipso); 138.2 (C10H6ipso); 135.9 (C6H3ipso); 128.8 (C6H3); 126.4 (C10H6); 125.7 (C6H3); 124.8 (C10H6); 121.5 (C10H6); 19.7 (CH3); 16.4 (CH3)). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −648.0. MS (Maldi-TOF) m/z: 1193.10 ([M]+); 597.04 ([M/2, monomer]+).
2a. Stirred overnight at 70 °C. Pale-brown crystals (51% yield). Melting point: 199 °C. 1H NMR (THF-d8, 500 MHz): δ 7.40 (dd, JHH = 8.1, 1.1 Hz, 2H, C10H6); 7.25–7.21 (m, 2H, C10H6); 7.17 (dd, JHH = 7.5, 1.2 Hz, 2H, C10H6); 7.05–6.96 (m, 6H, C6H3); 6.63 (d, JHH = 8.1 Hz, 4H, C6H4); 6.44 (d, JHH = 7.9 Hz, 4H, C6H4); 2.19 (s, 12H, CH3); 2.09 (s, Sn satellite: JHSn = 7.3 Hz, 6H, CH3); 1.79 (s, Sn satellite: JHSn = 2.4 Hz, 6H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 168.8 (NCN); 143.8 (Sn satellite: JCSn = 5.2 Hz, C10H6ipso); 143.3 (Sn satellite: JCSn = 7.2 Hz, C6H3); 138.4 (Sn satellite: JCSn = 3.3 Hz, C10H6ipso); 136.1 (Sn satellite: JCSn = 12.8 Hz, C6H4); 135.6 (Sn satellite: JCSn = 3.1 Hz, C6H3); 135.5 (C6H4ipso); 131.4 (C6H4ipso); 129.2 (C6H3); 129.0 (Sn satellite: JCSn = 6.6 Hz, C6H4); 127.6 (C10H6ipso); 126.4 (C10H6); 126.4 (C6H3); 125.2 (C10H6); 121.1 (Sn satellite: JCSn = 11.2 Hz, C10H6); 21.1 (CH3); 19.9 (CH3); 16.4 (Sn satellite: JCSn = 21.40 Hz, CH3). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −456.7. MS (Maldi-TOF) m/z: 687.13 ([M − S(tolyl)]+); 565.05 ([M − 2 S(tolyl)]+).
2b. Stirred for 15 min at room temperature. White solid (96% yield). Melting point: 240 °C (decomposition). 1H NMR (THF-d8, 500 MHz): δ 7.65–7.62 (m, 2H, C10H6); 7.44–7.39 (m, 4H, C10H6); 6.98–6.92 (m, 6H, C6H3); 6.57 (d, JHH = 2.4 Hz, 1H, C6H2); 6.39 (d, JHH = 2.4 Hz, 1H, C6H2); 2.08 (s, 12H, CH3); 2.00 (s, Sn satellite: JHSn = 3.5 Hz, 6H, CH3); 1.16 (s, 9H, C(CH3)3); 1.06 (s, 9H, C(CH3)3). 13C{1H} NMR (THF-d8, 125 MHz): δ 171.6 (NCN); 151.3 (Sn satellite: JCSn = 5.1 Hz, C6H2ipso); 147.1 (Sn satellite: JCSn = 3.3 Hz, C6H2ipso); 142.0 (Sn satellite: JCSn = 3.9 Hz, C10H6ipso); 140.9 (Sn satellite: JCSn = 7.5 Hz, C6H3ipso); 138.7 (C6H2ipso); 138.5 (C10H6ipso); 136.1 (C6H3ipso); 134.1 (C6H2ipso); 129.1 (C6H3); 127.1 (C6H3); 126.7 (C10H6); 126.1 (C10H6ipso); 126.0 (C10H6); 121.2 (Sn satellite: JCSn = 11.6 Hz, C10H6); 111.5 (C6H2); 110.3 (Sn satellite: JCSn = 40.2 Hz, C6H2); 35.3 (C(CH3)3); 34.9 (C(CH3)3); 32.4 (C(CH3)3); 30.2 (C(CH3)3); 19.1 (CH3); 15.5 (Sn satellite: JCSn = 33.7 Hz, CH3). 119Sn{1H} NMR (THF-d8, 186 MHz): δ −512.2. ESI m/z: 786.29 ([M]+).
3a. Stirred for 30 min at room temperature. White solid (85% yield). Melting point: 124 °C. 1H NMR (THF-d8, 500 MHz): δ 7.46 (dd, JHH = 8.3, 1.1 Hz, 2H, C10H6); 7.35–7.31 (m, 2H, C10H6); 7.24 (dd, JHH = 7.5, 1.2 Hz, 2H, C10H6); 7.02–6.90 (m, 6H, C6H3); 2.33 (s, 6H, CH3); 2.13 (s, 6H, CH3); 1.90 (s, 6H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 176.4 (NCN); 141.9 (C10H6ipso); 141.8 (C10H6ipso); 140.6 (C6H3ipso); 138.4 (C10H6ipso); 135.8 (C6H3ipso); 135.1 (C6H3ipso); 129.4 (C6H3); 128.8 (C6H3); 126.6 (C10H6); 126.2 (C6H3); 124.1 (C10H6); 123.9 (C10H6ipso); 117.8 (C10H6); 19.9 (CH3); 19.3 (CH3); 15.6 (CH3). 27Al{1H} NMR (THF-d8, 130 MHz): δ 69.4.
3b. Stirred for 30 min at room temperature. Colorless crystals were obtained from recrystallization with pentane at −30 °C (88% yield). Melting point: 209 °C (decomposition). 1H NMR (THF-d8, 500 MHz): δ 7.46 (dd, JHH = 8.3, 1.1 Hz, 2H, C10H6); 7.34–7.30 (m, 2H, C10H6); 7.20 (dd, JHH = 7.5, 1.2 Hz, 2H, C10H6); 7.03–6.90 (m, 6H, C6H3); 2.34 (s, 6H, CH3); 2.12 (s, 6H, CH3); 1.98 (s, 6H, CH3). 13C{1H} NMR (THF-d8, 125 MHz): δ 172.3 (NCN); 142.9 (C10H6ipso); 141.9 (C6H3ipso); 138.6 (C10H6ipso); 136.1 (C6H3ipso); 135.5 (C6H3ipso); 128.8 (C6H3); 126.4 (C10H6); 126.4 (C6H3); 124.3 (C10H6); 123.1 (C10H6ipso); 118.3 (C10H6); 19.8 (CH3); 15.1 (CH3). MS (Maldi-TOF) m/z: 551.06 ([M]+); 517.04 ([M − Cl]+); 449.10 ([M − GaCl]+).
3.3. X-ray Data
CCDC-2303009 (L3H2), CCDC-2266345 (L1Sn), CCDC-2266346 [(L2Sn)2], CCDC-2266347 [(L1Ge)2], CCDC-2266348 (L2Ge), CCDC-2266349 (1a), CCDC-2266350 (1b), CCDC-2266351 (2a), and CCDC-2266352 (3b) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
4. Conclusions
Three tetradentate bis(amidine) ligands RNC(R′)N-linker-NC(R′)NR with a rigid naphthalene linker L1–3 were successfully used for the stabilization of metallylenes. The corresponding stannylenes and germylenes (LSn and LGe) have been prepared by protonolysis reaction of Lappert’s metallylenes [M(HMDS)2] (M = Ge or Sn) and were fully characterized by NMR spectroscopy and X-ray diffraction analysis. Structures in the solid state show either a monomer or a dimer depending on the different substituents of the amidine moiety, demonstrating that the nature of the bis(amidine) system can influence the stabilization and the reactivity of the corresponding tetrylenes. DFT calculations have been performed in order to define the electronic properties associated with tetradentate ligands. The reactivity of stannylene L1Sn was explored in oxidation, oxidative addition, and transmetalation reactions to form in particular the corresponding gallium and aluminum derivatives.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29020325/s1, NMR spectra, crystal structure refinements, and computational investigations (PDF).
Author Contributions
Conceptualization, R.S.R. and D.M.; investigation, A.A.; X-ray structural studies: S.M.-L.; DFT calculations, J.-M.S.; writing—original draft preparation, A.A., R.S.R. and D.M.; writing—review and editing, all authors; supervision, E.M., A.R.C., A.B., T.K., R.S.R. and D.M. project administration, R.S.R. and D.M.; funding acquisition, T.K. and R.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Université de Toulouse (UPS), ECOS-Sud Chili (No. C19E04), and FONDECYT (project No. 1200748; 1230537). A. A. acknowledges funding from Ph.D. ANID 2019 fellowship No. 21190209.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Aly, A.A.; Brâse, S.; Gomaa, M.A.-M. Amidines: Their synthesis, reactivity, and applications in heterocycle synthesis. Arkivoc 2018, vi, 85–138. [Google Scholar] [CrossRef]
- Aly, A.A.; El-Din, A.M.N. Functionality of Amidines and Amidrazones. Arkivoc 2008, i, 153–194. [Google Scholar] [CrossRef]
- Quek, J.Y.; Davis, T.P.; Lowe, A.B. Amidine Functionality as a Stimulus-Responsive Building Block. Chem. Soc. Rev. 2013, 42, 7326–7334. [Google Scholar] [CrossRef] [PubMed]
- Coles, M.P. Application of Neutral Amidines and Guanidines in Coordination Chemistry. Dalton Trans. 2006, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Rodriguez, L.; Cabeza, J.-A.; Garcia-Alvarez, P.; Polo, D. The transition-metal chemistry of amidinatosilylenes, -germylenes and -stannylenes. Coord. Chem. Rev. 2015, 300, 1–28. [Google Scholar] [CrossRef]
- Foley, S.R.; Bensimon, C.; Richeson, D.S. Facile Formation of Rare Terminal Chalcogenido Germanium Complexes with Alkylamidinates as Supporting Ligands. J. Am. Chem. Soc. 1997, 119, 10359–10363. [Google Scholar] [CrossRef]
- Karsch, H.H.; Schlüter, P.A.; Reisky, M. Bis(Amidinate) Complexes of Silicon and Germanium. Eur. J. Inorg. Chem. 1998, 433–436. [Google Scholar] [CrossRef]
- Aubrecht, K.B.; Hillmyer, M.A.; Tolman, W.B. Polymerization of Lactide by Monomeric Sn(II) Alkoxide Complexes. Macromolecules 2002, 35, 644–650. [Google Scholar] [CrossRef]
- Garg, P.; Dange, D.; Jones, C. S- and p-Block Dinuclear Metal(Loid) Complexes Bearing 1,4-Phenylene and 1,4-Cyclohexylene Bridged Bis(Amidinate) Ligands. Eur. J. Inorg. Chem. 2020, 2020, 4037–4044. [Google Scholar] [CrossRef]
- Garg, P.; Dange, D.; Jones, C. Bulky Arene-Bridged Bis(Amide) and Bis(Amidinate) Complexes of Germanium(II) and Tin(II). Dalton Trans. 2021, 50, 9118–9122. [Google Scholar] [CrossRef]
- Dehmel, M.; Wünsche, M.A.; Görls, H.; Kretschmer, R. Dinuclear Chlorotetrylenes of Silicon, Germanium, and Tin Based on a Backbone-Bridged Bis(Amidine). Eur. J. Inorg. Chem. 2021, 2021, 4806–4811. [Google Scholar] [CrossRef]
- Saltarini, S.; Villegas-Escobar, N.; Martínez, J.; Daniliuc, C.G.; Matute, R.A.; Gade, L.H.; Rojas, R.S. Toward a Neutral Single-Component Amidinate Iodide Aluminum Catalyst for the CO2 Fixation into Cyclic Carbonates. Inorg. Chem. 2021, 60, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Yakovenko, M.V.; Cherkasov, A.V.; Fukin, G.K.; Cui, D.; Trifonov, A.A. Lanthanide Complexes Coordinated by a Dianionic Bis(Amidinate) Ligand with a Rigid Naphthalene Linker. Eur. J. Inorg. Chem. 2010, 2010, 3290–3298. [Google Scholar] [CrossRef]
- Osorio Meléndez, D.; Castro-Osma, J.A.; Lara-Sánchez, A.; Rojas, R.S.; Otero, A. Ring-Opening Polymerization and Copolymerization of Cyclic Esters Catalyzed by Amidinate Aluminum Complexes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2397–2407. [Google Scholar] [CrossRef]
- Nimitsiriwat, N.; Gibson, V.C.; Marshall, E.L.; White, A.J.P.; Dale, S.H.; Elsegood, M.R.J. Tert-Butylamidinate Tin(II) Complexes: High Activity, Single-Site Initiators for the Controlled Production of Polylactide. Dalton Trans. 2007, 4464. [Google Scholar] [CrossRef] [PubMed]
- Phomphrai, K.; Pongchan-o, C.; Thumrongpatanaraks, W.; Sangtrirutnugul, P.; Kongsaeree, P.; Pohmakotr, M. Synthesis of High-Molecular-Weight Poly(ε-Caprolactone) Catalyzed by Highly Active Bis(Amidinate) Tin(II) Complexes. Dalton Trans. 2011, 40, 2157–2159. [Google Scholar] [CrossRef] [PubMed]
- Lentz, N.; Sodreau, A.; Acuña, A.; Mallet-Ladeira, S.; Maerten, E.; Sotiropoulos, J.-M.; Rojas Guerrero, R.S.; Madec, D. Synthesis and structures of homoleptic germylenes and stannylenes with sulfonimidamide ligands. Dalton Trans. 2023, 52, 6841–6846. [Google Scholar] [CrossRef]
- Foley, S.R.; Zhou, Y.; Yap, G.P.A.; Richeson, D.S. Synthesis of MII[N(SiMe3)2][Me3SiNC(tBu)NSiMe3] (M = Sn, Ge) from Amidinate Precursors: Active Catalysts for Phenyl Isocyanate Cyclization. Inorg. Chem. 2000, 39, 924–929. [Google Scholar] [CrossRef]
- Chlupatý, T.; Růžičková, Z.; Horáček, M.; Alonso, M.; De Proft, F.; Kampová, H.; Brus, J.; Růžička, A. Oxidative Additions of Homoleptic Tin(II) Amidinate. Organometallics 2015, 34, 606–615. [Google Scholar] [CrossRef]
- Nakata, N.; Hosoda, N.; Takahashi, S.; Ishii, A. Chlorogermylenes and -Stannylenes Stabilized by Diimidosulfinate Ligands: Synthesis, Structures, and Reactivity. Dalton Trans. 2018, 47, 481–490. [Google Scholar] [CrossRef]
- Okazaki, R.; Tokitoh, N. Heavy Ketones, the heavier Element Congeners of a Ketone. Acc. Chem. Res. 2000, 33, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Foley, S.R.; Yap, G.P.A.; Richeson, D.S. Oxidative Addition to M(II) (M = Ge, Sn) Amidinate Complexes: Routes to Group 14 Chalcogenolates with Hypervalent Coordination Environments. J. Chem. Soc. Dalton Trans. 2000, 10, 1663–1668. [Google Scholar] [CrossRef]
- Ahmet, I.Y.; Hill, M.S.; Raithby, P.R.; Johnson, A.L. Tin Guanidinato Complexes: Oxidative Control of Sn, SnS, SnSe and SnTe Thin Film Deposition. Dalton Trans. 2018, 47, 5031–5048. [Google Scholar] [CrossRef] [PubMed]
- Ahmet, I.Y.; Thompson, J.R.; Johnson, A.L. Oxidative Addition to Sn(II) Guanidinate Complexes: Precursors to Tin(II) Chalcogenide Nanocrystals. Eur. J. Inorg. Chem. 2018, 2018, 1670–1678. [Google Scholar] [CrossRef]
- Foley, S.R.; Yap, G.P.A.; Richeson, D.S. Formation of Novel Tetrasulfido Tin Complexes and Their Ability to Catalyze the Cyclotrimerization of Aryl Isocyanates. Organometallics 1999, 18, 4700–4705. [Google Scholar] [CrossRef]
- Zhou, Y.; Richeson, D.S. Multiple Bonds between Sn and S: Synthesis and Structural Characterization of (CyNC(tBu)NCy)2SnS and [(CyNC(Me)NCy)2Sn(μ-S)]2. J. Am. Chem. Soc. 1996, 118, 10850–10852. [Google Scholar] [CrossRef]
- Sen, N.; Pal, S.; Khade, V.V.; Khan, S. Cyclic Four-Membered Stanna Thio and Seleno Compounds from 2-Aminopyridinato Stannylenes. Eur. J. Inorg. Chem. 2019, 2019, 4450–4454. [Google Scholar] [CrossRef]
- Shan, Y.-L.; Leong, B.-X.; Xi, H.-W.; Ganguly, R.; Li, Y.; Lim, K.H.; So, C.-W. Reactivity of an Amidinato Silylene and Germylene toward Germanium(ii), Tin(ii) and Lead(ii) Halides. Dalton Trans. 2017, 46, 3642–3648. [Google Scholar] [CrossRef]
- Watson, I.C.; Ferguson, M.J.; Rivard, E. Zinc-Mediated Transmetalation as a Route to Anionic N-Heterocyclic Olefin Complexes in the p-Block. Inorg. Chem. 2021, 60, 18347–18359. [Google Scholar] [CrossRef]
- Brazeau, A.L.; DiLabio, G.A.; Kreisel, K.A.; Monillas, W.; Yap, G.P.A.; Barry, S.T. Theoretical and experimental investigations of ligand exchange in guanidinate ligand systems for group 13 metals. Dalton Trans. 2007, 3297–3304. [Google Scholar] [CrossRef]
- Boeré, R.T.; Klassen, V.; Wolmershäuser, G. Synthesis of Some Very Bulky N,N′-Disubstituted Amidines and Initial Studies of Their Coordination Chemistry. J. Chem. Soc. Dalton Trans. 1998, 24, 4147–4154. [Google Scholar] [CrossRef]
- Davidson, P.; Harris, D.; Lappert, M. Subvalent Group 4B metal alkyls and amides. Part I. The synthesis and physical properties of kinetically stable bis[bis(trimethysilyl)methyl]- germanium(II), -tin(II), and -lead(II). J. Chem. Soc. Chem. Dalton Trans. 1976, 2268–2274. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Van der Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Cryst. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comp. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Andrae, D.; Häussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Theory and Applications of Computational Chemistry: The First 40 Years; Dykstra, D.C.E., Frenking, G., Kim, K.S., Scuseria, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).