
Recent Advances and Prospects of Nucleic Acid Therapeutics for Anti-Cancer Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Overview of Nucleic Acid Therapeutics (NATs)
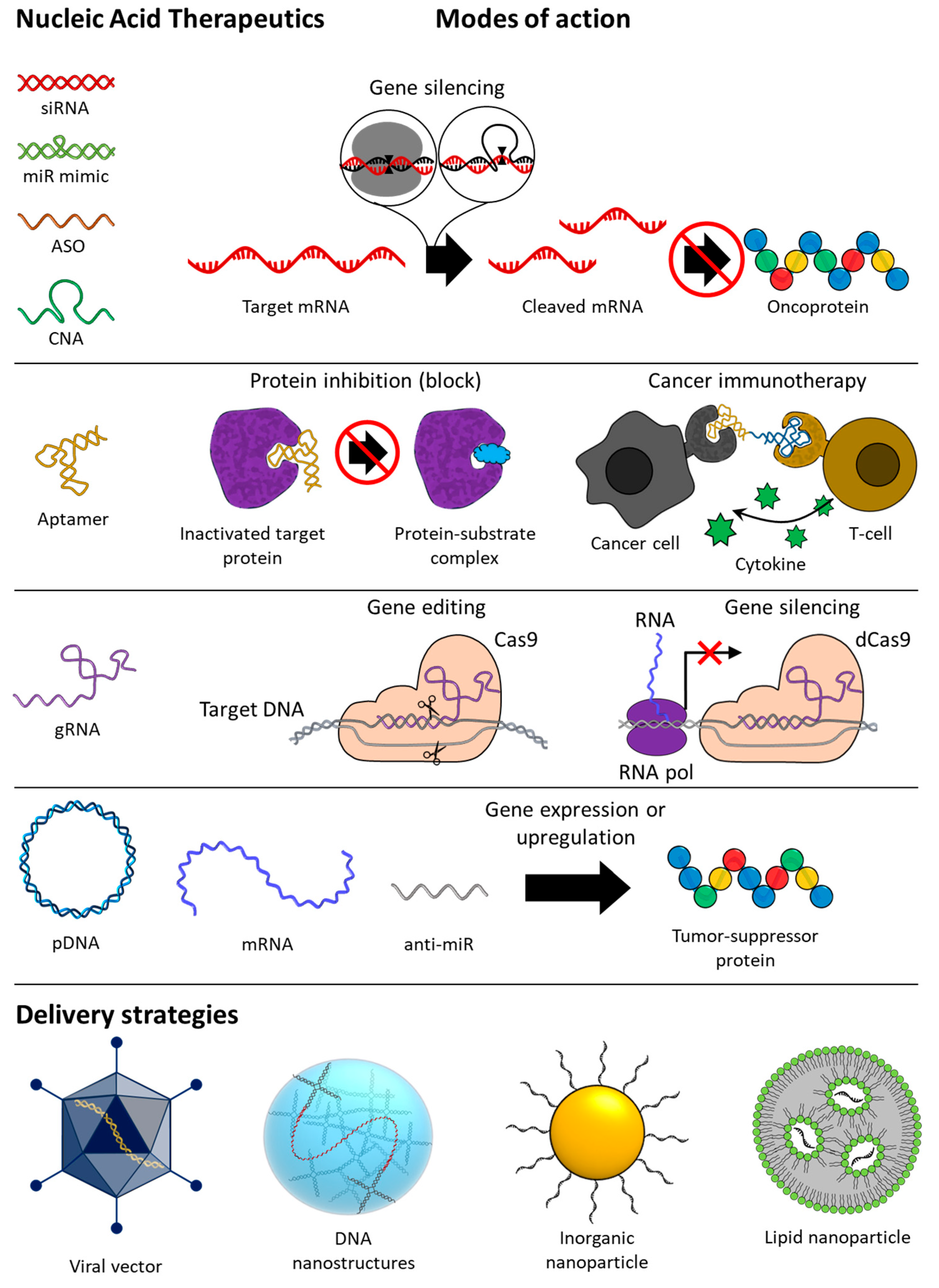
1.2. Nucleic Acid Therapeutics (NATs)
2. Gene Silencing
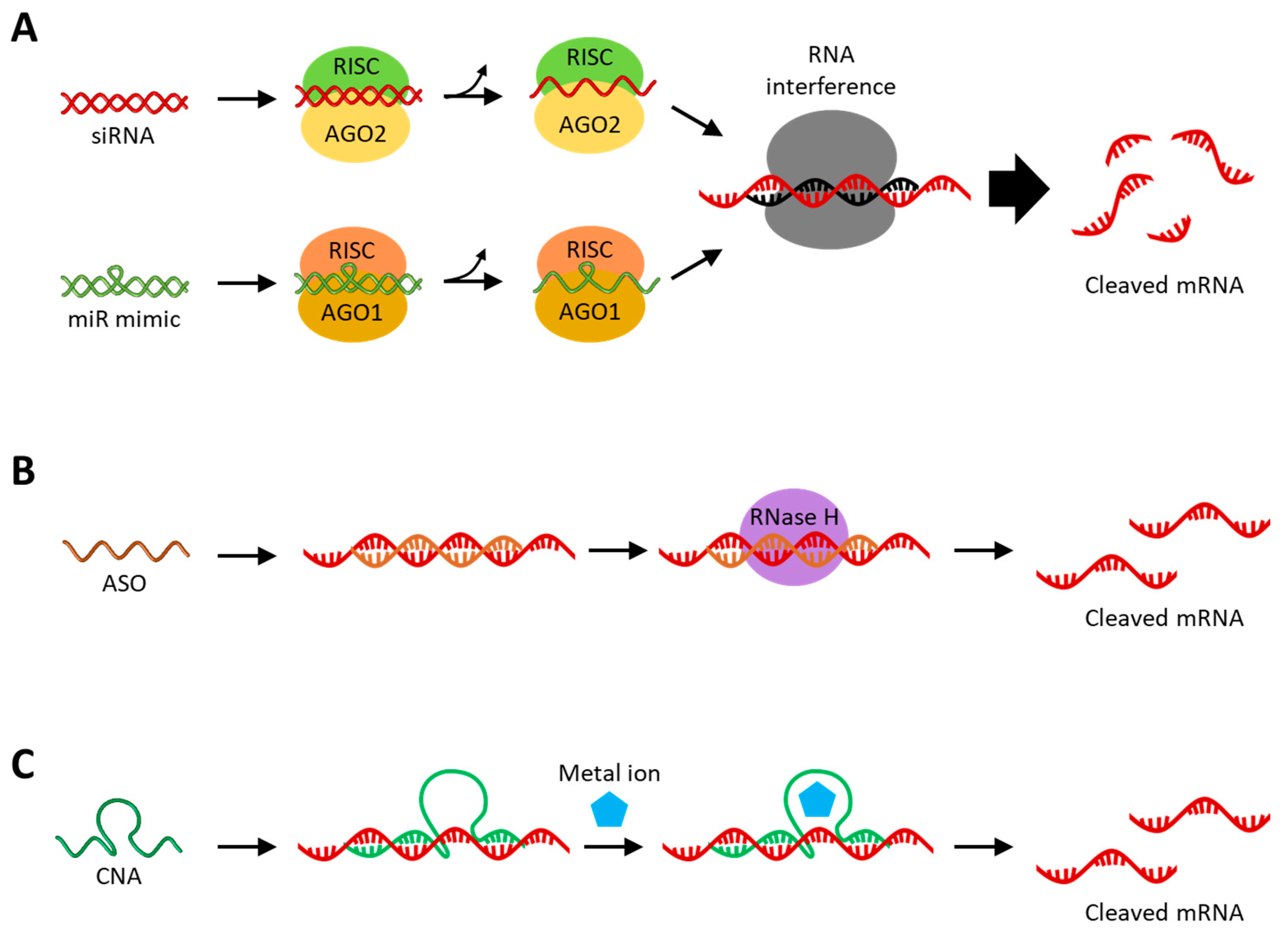
2.1. RNA Interference (RNAi)
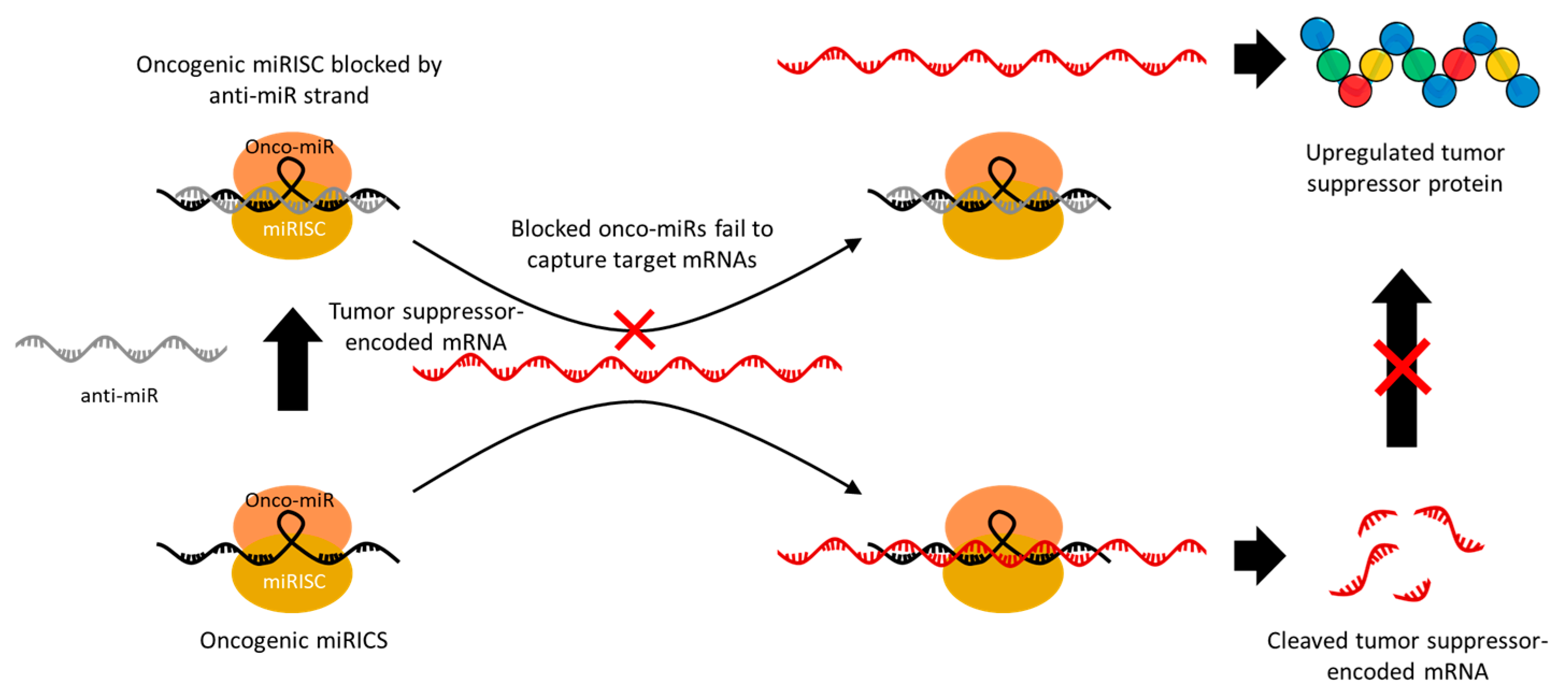
2.2. Antisense Oligonucleotide (ASO)
2.3. Catalytic Nucleic Acid (CNA)
3. Aptamer
3.1. Aptamer-Based Protein Inhibition

3.2. Aptamer-Based Immunotherapy

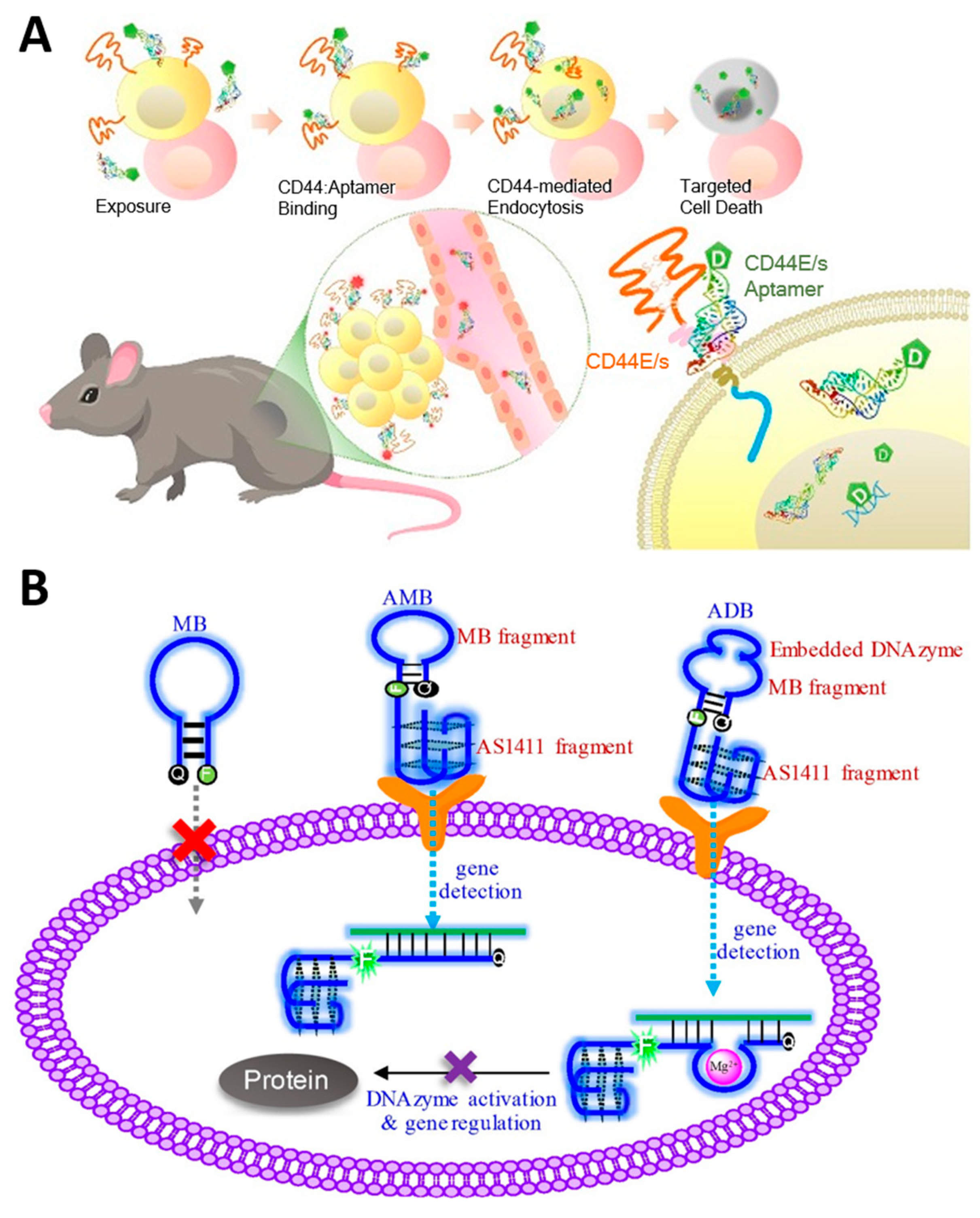
3.3. Aptamer-Based Targeted Therapy
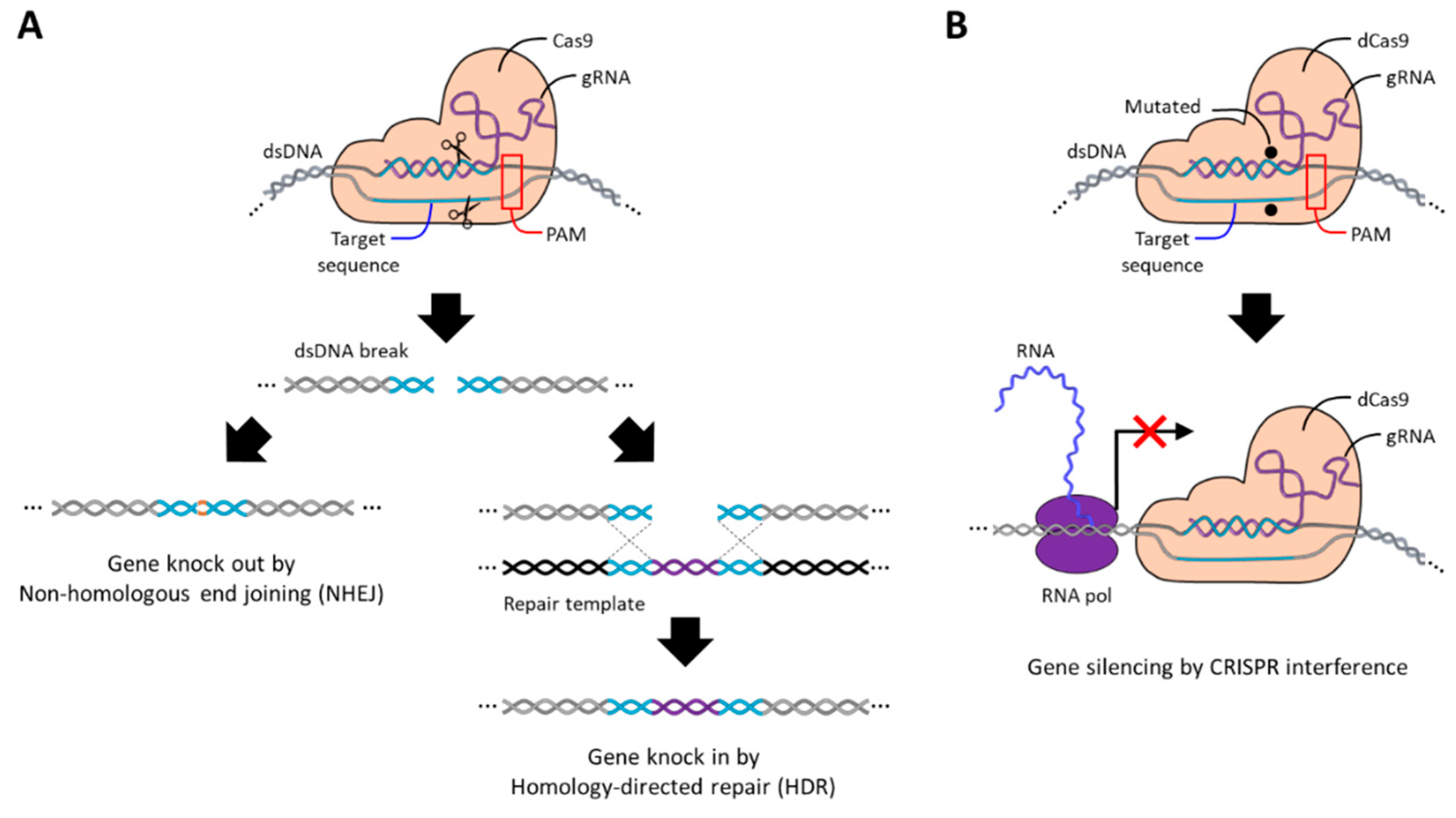
4. CRISPR Cas9 Guide RNA (gRNA)
5. Gene Expression
5.1. Plasmid DNA (pDNA)
5.2. Messenger RNA (mRNA)
6. Delivery Strategies
7. Conclusions and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gopal, S.; Sivaram, S.; Rajaraman, P.; Trimble, E.L. Think globally about cancer. Nat. Med. 2019, 25, 351. [Google Scholar]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer. 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lou, W.; Wang, J. Advances in nucleic acid therapeutics: Structures, delivery systems, and future perspectives in cancer treatment. Clin. Exp. Med. 2024, 24, 200. [Google Scholar] [CrossRef] [PubMed]
- Yahya, E.B.; Alqadhi, A.M. Recent trends in cancer therapy: A review on the current state of gene delivery. Life Sci. 2021, 269, 119087. [Google Scholar] [CrossRef]
- Das, S.K.; Menezes, M.E.; Bhatia, S.; Wang, X.; Emdad, L.; Sarkar, D.; Fisher, P.B. Gene Therapies for Cancer: Strategies, Challenges and Successes. J. Cell. Physiol. 2014, 230, 259–271. [Google Scholar] [CrossRef]
- Cesur-Ergün, B.; Demir-Dora, D. Gene therapy in cancer. J. Gene Med. 2023, 25, e3550. [Google Scholar] [CrossRef]
- Ahmadzada, T.; Reid, G.; McKenzie, D.R. Fundamentals of siRNA and miRNA therapeutics and a review of targeted nanoparticle delivery systems in breast cancer. Biophys. Rev. 2018, 10, 69–86. [Google Scholar] [CrossRef]
- Vetter, V.C.; Wagner, E. Targeting nucleic acid-based therapeutics to tumors: Challenges and strategies for polyplexes. J. Control. Release 2022, 346, 110–135. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; Van Der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Ingle, R.G.; Fang, W.-J. An Overview of the Stability and Delivery Challenges of Commercial Nucleic Acid Therapeutics. Pharmaceutics 2023, 15, 1158. [Google Scholar] [CrossRef]
- Wang, T.; Tang, Y.; Tao, Y.; Zhou, H.; Ding, D. Nucleic acid drug and delivery techniques for disease therapy: Present situation and future prospect. Interdiscip. Med. 2024, 2, e20230041. [Google Scholar] [CrossRef]
- Paterson, B.M.; Roberts, B.E.; Kuff, E.L. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc. Natl. Acad. Sci. USA 1977, 74, 4370–4374. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Chou, M.Y.; Inouye, M. A unique mechanism regulating gene expression: Translational inhibition by a complementary RNA transcript (micRNA). Proc. Natl. Acad. Sci. USA 1984, 81, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Kijima, H. Therapeutic applications of ribozymes. Pharmacol. Ther. 1995, 68, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Shreya; Pandey, A.K.; Bhandari, H.R. Gene Silencing: The Mechanism to Down Regulate the Target Gene. Int. J. Bio-Resour. Stress Manag. 2018, 9, 682–690. [Google Scholar]
- Ratcliff, F.; Harrison, B.D.; Baulcombe, D.C. A Similarity Between Viral Defense and Gene Silencing in Plants. Science 1997, 276, 1558–1560. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Lam, J.K.W.; Chow, M.Y.T.; Zhang, Y.; Leung, S.W.S. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef]
- Isenmann, M.; Stoddart, M.J.; Schmelzeisen, R.; Gross, C.; Della Bella, E.; Rothweiler, R.M. Basic Principles of RNA Interference: Nucleic Acid Types and In Vitro Intracellular Delivery Methods. Micromachines 2023, 14, 1321. [Google Scholar] [CrossRef]
- Mello, C.C.; Conte, D. Revealing the world of RNA interference. Nature 2004, 431, 338–342. [Google Scholar] [CrossRef]
- Takeda, A.; Iwasaki, S.; Watanabe, T.; Utsumi, M.; Watanabe, Y. The Mechanism Selecting the Guide Strand from Small RNA Duplexes is Different Among Argonaute Proteins. Plant Cell Physiol. 2008, 49, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Holen, T.; Amarzguioui, M.; Babaie, E.; Prydz, H. Similar behaviour of single-strand and double-strand siRNAs suggests they act through a common RNAi pathway. Nucleic Acids Res. 2003, 31, 2401–2407. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Cai, X.; Fan, Y.; Jin, M.; Xie, Y.; Jing, Z.; Zang, X.; Han, Y. Codelivery of Que and BCL-2 siRNA with Lipid–Copolymer Hybrid Nanocomplexes for Efficient Tumor Regression. ACS Biomater. Sci. Eng. 2023, 9, 4805–4820. [Google Scholar] [CrossRef]
- Vaidya, S.; Mohod, A.; Eedara, A.C.; Andugulapati, S.B.; Pabbaraja, S. Synthesis and Characterization of a New Cationic Lipid: Efficient siRNA Delivery and Anticancer Activity of Survivin-siRNA Lipoplexes for the Treatment of Lung and Breast Cancers. Chem. Med. Chem. 2023, 18, e202300097. [Google Scholar] [CrossRef]
- Heris, N.N.; Baghani, L.; Khonsari, F.; Varshochian, R.; Dinarvand, R.; Atyabi, F. Delivery of EGFR-siRNA to prostatic cancerous cells based on polydopamine coated gold nanoparticles. Int. J. Drug Deliv. Technol. 2023, 87, 104869. [Google Scholar] [CrossRef]
- Aslan, M.; Ozturk, S.; Shahbazi, R.; Bozdemir, Ö.; Zeybek, N.D.; Vargel, İ.; Eroğlu, İ.; Ulubayram, K. Therapeutic targeting of siRNA/anti-cancer drug delivery system for non-melanoma skin cancer. Part I: Development and gene silencing of JAK1siRNA/5-FU loaded liposome nanocomplexes. Eur. J. Pharm. Biopharm. 2024, 203, 114432. [Google Scholar] [CrossRef]
- Jain, D.; Yadav, A.K. Development of hyaluronic acid–anchored polycaprolactone nanoparticles for efficient delivery of PLK1 siRNA to breast cancer. Drug Deliv. Transl. Res. 2023, 13, 1730–1744. [Google Scholar] [CrossRef]
- Haghiralsadat, F.; Amoabediny, G.; Naderinezhad, S.; Zandieh-Doulabi, B.; Forouzanfar, T.; Helder, M.N. Codelivery of doxorubicin and JIP1 siRNA with novel EphA2-targeted PEGylated cationic nanoliposomes to overcome osteosarcoma multidrug resistance. Int. J. Nanomed. 2018, 13, 3853–3866. [Google Scholar] [CrossRef]
- Mao, D.; Che, J.; Han, S.; Zhao, H.; Zhu, Y.; Zhu, H. RNAi-mediated knockdown of the CLN3 gene inhibits proliferation and promotes apoptosis in drug-resistant ovarian cancer cells. Mol. Med. Rep. 2015, 12, 6635–6641. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Liao, M.; Yu, S.; Yuan, B.; Jia, Z.; Zhou, L.; Tang, Y. Exosomes-delivered PD-L1 siRNA and CTLA-4 siRNA protect against growth and tumor immune escape in colorectal cancer. Genomics 2023, 115, 110646. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed]
- Denli, A.M.; Tops, B.B.J.; Plasterk, R.H.A.; Ketting, R.F.; Hannon, G.J. Processing of primary microRNAs by the Microprocessor complex. Nature 2004, 432, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; Görlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef]
- Yoshida, T.; Asano, Y.; Ui-Tei, K. Modulation of MicroRNA Processing by Dicer via Its Associated dsRNA Binding Proteins. Non-Coding RNA 2021, 7, 57. [Google Scholar] [CrossRef]
- Meijer, H.A.; Smith, E.M.; Bushell, M. Regulation of miRNA strand selection: Follow the leader? Biochem. Soc. Trans. 2014, 42, 1135–1140. [Google Scholar] [CrossRef]
- Hutvagner, G.; Simard, M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008, 9, 22–32. [Google Scholar] [CrossRef]
- Tristán-Ramos, P.; Rubio-Roldan, A.; Peris, G.; Sánchez, L.; Amador-Cubero, S.; Viollet, S.; Cristofari, G.; Heras, S.R. The tumor suppressor microRNA let-7 inhibits human LINE-1 retrotransposition. Nat. Commun. 2020, 11, 5712. [Google Scholar] [CrossRef]
- Lin, L.; Bao, Y.; Tian, M.; Ren, Q.; Zhang, W. miR-29 family inhibited the proliferation and migration of lung cancer cells by targeting SREBP-1. Mol. Cell. Toxicol. 2021, 18, 165–175. [Google Scholar] [CrossRef]
- Zhao, Y.; Ye, L.; Yu, Y. MicroRNA-126-5p suppresses cell proliferation, invasion and migration by targeting EGFR in liver cancer. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 865–873. [Google Scholar] [CrossRef]
- Kapadia, C.H.; Ioele, S.A.; Day, E.S. Layer-by-layer assembled PLGA nanoparticles carrying miR-34a cargo inhibit the proliferation and cell cycle progression of triple-negative breast cancer cells. J. Biomed. Mater. Res. Part A 2019, 108, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tan, J.; Ou, S.; Chen, J.; Chen, L. MicroRNA-101-3p suppresses proliferation and migration in hepatocellular carcinoma by targeting the HGF/c-Met pathway. Investig. New Drugs 2019, 38, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Yu, Y.J.; You, J.Y.; Rhee, W.J.; Kim, J.A. Exosome-mediated microRNA-497 delivery for anti-cancer therapy in a microfluidic 3D lung cancer model. Lab A Chip 2020, 20, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Prakash, T.P.; Corey, D.R. Argonaute 2-dependent Regulation of Gene Expression by Single-stranded miRNA Mimics. Mol. Ther. 2016, 24, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef]
- Van Rooij, E.; Purcell, A.L.; Levin, A.A. Developing MicroRNA Therapeutics. Circ. Res. 2012, 110, 496–507. [Google Scholar] [CrossRef]
- Hogan, D.J.; Vincent, T.M.; Fish, S.; Marcusson, E.G.; Bhat, B.; Chau, B.N.; Zisoulis, D.G. Anti-miRs Competitively Inhibit microRNAs in Argonaute Complexes. PLoS ONE 2014, 9, e100951. [Google Scholar] [CrossRef]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E.; Haque, F.; Xu, R.; Guo, P. Systemic Delivery of Anti-miRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef]
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261. [Google Scholar] [CrossRef]
- Sun, X.; Xu, H.; Huang, T.; Zhang, C.; Wu, J.; Luo, S. Simultaneous delivery of anti-miRNA and docetaxel with supramolecular self-assembled “chitosome” for improving chemosensitivity of triple negative breast cancer cells. Drug Deliv. Transl. Res. 2020, 11, 192–204. [Google Scholar] [CrossRef]
- Raniolo, S.; Unida, V.; Vindigni, G.; Stolfi, C.; Iacovelli, F.; Desideri, A.; Biocca, S. Combined and selective miR-21 silencing and doxorubicin delivery in cancer cells using tailored DNA nanostructures. Cell Death Dis. 2021, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther.—Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Rhim, J.; Baek, W.; Seo, Y.; Kim, J.H. From Molecular Mechanisms to Therapeutics: Understanding MicroRNA-21 in Cancer. Cells 2022, 11, 2791. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Duo, Y.; Bi, J.; Zeng, X.; Mei, L.; Bao, S.; He, L.; Shan, A.; Zhang, Y.; Yu, X. Targeted delivery of anti-miR-155 by functionalized mesoporous silica nanoparticles for colorectal cancer therapy. Int. J. Nanomed. 2018, 13, 1241–1256. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Rajendran, V.; Kulshreshtha, R.; Ghosh, P.C. Enhanced efficacy of anti-miR-191 delivery through stearylamine liposome formulation for the treatment of breast cancer cells. Int. J. Pharm. 2017, 530, 387–400. [Google Scholar] [CrossRef]
- Ru, P.; Steele, R.; Hsueh, E.C.; Ray, R.B. Anti-miR-203 Upregulates SOCS3 Expression in Breast Cancer Cells and Enhances Cisplatin Chemosensitivity. Genes Cancer 2011, 2, 720–727. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Bai, M.; Ning, T.; Ge, S.; Deng, T.; Liu, R.; Zhang, L.; Ying, G.; Ba, Y. Exosomes Serve as Nanoparticles to Deliver Anti-miR-214 to Reverse Chemoresistance to Cisplatin in Gastric Cancer. Mol. Ther. 2018, 26, 774–783. [Google Scholar] [CrossRef]
- Han, S.; Li, G.; Jia, M.; Zhao, Y.; He, C.; Huang, M.; Jiang, L.; Wu, M.; Yang, J.; Ji, X.; et al. Delivery of Anti-miRNA-221 for Colorectal Carcinoma Therapy Using Modified Cord Blood Mesenchymal Stem Cells-Derived Exosomes. Front. Mol. Biosci. 2021, 8, 743013. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense Oligonucleotides: Basic Concepts and Mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef]
- Yoo, B.H.; Bochkareva, E.; Bochkarev, A.; Mou, T.-C.; Gray, D.M. 2’-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Kawasaki, A.M.; Wancewicz, E.V.; Shen, L.; Monia, B.P.; Ross, B.S.; Bhat, B.; Manoharan, M. Comparing In Vitro and In Vivo Activity of 2′-O-[2-(Methylamino)-2-oxoethyl]- and 2′-O-Methoxyethyl-Modified Antisense Oligonucleotides. J. Med. Chem. 2008, 51, 2766–2776. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2017, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Ashizawa, A.T. Making Sense of Antisense Oligonucleotide Therapeutics Targeting Bcl-2. Pharmaceutics 2022, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ye, W.; Zhou, C.; Li, Y.; He, J. Effect of polyamide nanoparticles on apoptosis of colon cancer cells induced by survivin antisense oligonucleotide. Mater. Express 2020, 10, 1573–1580. [Google Scholar] [CrossRef]
- Madanayake, T.W.; Welsh, E.A.; Darville, L.N.F.; Koomen, J.M.; Chalfant, C.E.; Haura, E.B.; Robinson, T.J. Inhibition of Epidermal Growth Factor Receptor Signaling by Antisense Oligonucleotides as a Novel Approach to Epidermal Growth Factor Receptor Inhibition. Nucleic Acid Ther. 2022, 32, 391–400. [Google Scholar] [CrossRef]
- Zhang, Q.-Y.; Ding, W.; Mo, J.-S.; Ou-Yang, S.-M.; Lin, Z.-Y.; Peng, K.-R.; Liu, G.-P.; Lu, J.-J.; Yue, P.-B.; Lei, J.-P.; et al. Novel STAT3 oligonucleotide compounds suppress tumor growth and overcome the acquired resistance to sorafenib in hepatocellular carcinoma. Acta Pharmacol. Sin. 2024, 45, 1701–1714. [Google Scholar] [CrossRef]
- Fernandez-Rodriguez, L.; Cianciaruso, C.; Bill, R.; Trefny, M.P.; Klar, R.; Kirchhammer, N.; Buchi, M.; Festag, J.; Michel, S.; Kohler, R.H.; et al. Dual TLR9 and PD-L1 targeting unleashes dendritic cells to induce durable antitumor immunity. J. ImmunoTherapy Cancer 2023, 11, e006714. [Google Scholar] [CrossRef]
- Liang, X.-H.; Vickers, T.A.; Guo, S.; Crooke, S.T. Efficient and specific knockdown of small non-coding RNAs in mammalian cells and in mice. Nucleic Acids Res. 2010, 39, e13. [Google Scholar] [CrossRef]
- Vickers, T.A.; Crooke, S.T. Antisense Oligonucleotides Capable of Promoting Specific Target mRNA Reduction via Competing RNase H1-Dependent and Independent Mechanisms. PLoS ONE 2014, 9, e108625. [Google Scholar] [CrossRef]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.R.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Zadrożna-Nowak, A.; Romanowicz, H. The Role of lncRNA in the Development of Tumors, including Breast Cancer. Int. J. Mol. Sci. 2021, 22, 8427. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lu, H.; Liu, J.; Wu, S.; Kim, P.; Zhou, X. lncRNAfunc: A knowledgebase of lncRNA function in human cancer. Nucleic Acids Res. 2021, 50, D1295–D1306. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-C.; Ni, J.-J.; Cui, W.-Y.; Wang, B.-Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar]
- Zhou, T.; Kim, Y.; MacLeod, A.R. Targeting Long Noncoding RNA with Antisense Oligonucleotide Technology as Cancer Therapeutics. Methods Mol. Biol. 2016, 1402, 199–213. [Google Scholar]
- Singh, N.N.; Luo, D.; Singh, R.N. Pre-mRNA Splicing Modulation by Antisense Oligonucleotides. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2018; pp. 415–437. [Google Scholar]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic Potential of Splice-Switching Oligonucleotides. Oligonucleotides 2009, 19, 1–13. [Google Scholar] [CrossRef]
- Liang, X.-H.; Shen, W.; Sun, H.; Migawa, M.T.; Vickers, T.A.; Crooke, S.T. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat. Biotechnol. 2016, 34, 875–880. [Google Scholar] [CrossRef]
- Lim, K.H.; Han, Z.; Jeon, H.Y.; Kach, J.; Jing, E.; Weyn-Vanhentenryck, S.; Downs, M.; Corrionero, A.; Oh, R.; Scharner, J.; et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020, 11, 3501. [Google Scholar] [CrossRef]
- Ward, W.L.; Plakos, K.; DeRose, V.J. Nucleic Acid Catalysis: Metals, Nucleobases, and Other Cofactors. Chem. Rev. 2014, 114, 4318–4342. [Google Scholar] [CrossRef]
- Ma, L.; Liu, J. Catalytic Nucleic Acids: Biochemistry, Chemical Biology, Biosensors, and Nanotechnology. iScience 2020, 23, 100815. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Saran, R.; Liu, J. Tandem DNAzymes for mRNA cleavage: Choice of enzyme, metal ions and the antisense effect. Bioorg. Med. Chem. Lett. 2015, 25, 1460–1463. [Google Scholar] [CrossRef] [PubMed]
- Kashani-Sabet, M. Ribozyme Therapeutics. J. Investig. Dermatol. Symp. Proc. 2002, 7, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-splicing RNA: Autoexcision and autocyclization of the ribosomal RNA intervening sequence of tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Wilson, D.S.; Szostak, J.W. In Vitro Selection of Functional Nucleic Acids. Annu. Rev. Biochem. 1999, 68, 611–647. [Google Scholar] [CrossRef]
- Silverman, S.K. Deoxyribozymes: Selection Design and Serendipity in the Development of DNA Catalysts. Acc. Chem. Res. 2009, 42, 1521–1531. [Google Scholar] [CrossRef]
- Scott, W.G.; Horan, L.H.; Martick, M. The Hammerhead Ribozyme: Structure, Catalysis, and Gene Regulation. Prog. Mol. Biol. Transl. Sci. 2013, 120, 1–23. [Google Scholar]
- Puerta-Fernández, E.; Romero-López, C.; Barroso-delJesus, A.; Berzal-Herranz, A. Ribozymes: Recent advances in the development of RNA tools. FEMS Microbiol. Rev. 2003, 27, 75–97. [Google Scholar] [CrossRef]
- Tokunaga, T.; Tsuchida, T.; Kijima, H.; Okamoto, K.; Oshika, Y.; Sawa, N.; Ohnishi, Y.; Yamazaki, H.; Miura, S.; Ueyama, Y.; et al. Ribozyme-mediated inactivation of mutant K-ras oncogene in a colon cancer cell line. Br. J. Cancer 2000, 83, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Aigner, A.; Renneberg, H.; Bojunga, J.; Apel, J.; Nelson, P.S.; Czubayko, F. Ribozyme-targeting of a secreted FGF-binding protein (FGF-BP) inhibits proliferation of prostate cancer cells in vitro and in vivo. Oncogene 2002, 21, 5733–5742. [Google Scholar] [CrossRef] [PubMed]
- Nagata, J.; Kijima, H.; Hatanaka, H.; Asai, S.; Miyachi, H.; Takagi, A.; Miwa, T.; Mine, T.; Yamazaki, H.; Nakamura, M.; et al. Reversal of Cisplatin and Multidrug Resistance by Ribozyme-Mediated Glutathione Suppression. Biochem. Biophys. Res. Commun. 2001, 286, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhou, G.-Y.; Guo, L.-L.; Zhang, Q.-H.; Zhen, J.-H.; Fang, A.-J.; Lin, X.-Y. Reversal of drug resistance in breast carcinoma cells by anti-mdr1 ribozyme regulated by the tumor-specific MUC-1 promoter. Cancer Lett. 2007, 256, 81–89. [Google Scholar] [CrossRef]
- Han, S.R.; Lee, C.H.; Im, J.Y.; Kim, J.H.; Kim, J.H.; Kim, S.J.; Cho, Y.W.; Kim, E.; Kim, Y.; Ryu, J.-H.; et al. Targeted suicide gene therapy for liver cancer based on ribozyme-mediated RNA replacement through post-transcriptional regulation. Mol. Ther.—Nucleic Acids 2021, 23, 154–168. [Google Scholar] [CrossRef]
- Huang, X.; Wang, M.; Liu, Y.; Gui, Y. Synthesis of RNA-based gene regulatory devices for redirecting cellular signaling events mediated by p53. Theranostics 2021, 11, 4688–4698. [Google Scholar] [CrossRef]
- Huo, W.; Li, X.; Wang, B.; Zhang, H.; Zhang, J.; Yang, X.; Jin, Y. Recent advances of DNAzyme-based nanotherapeutic platform in cancer gene therapy. Biophys. Rep. 2020, 6, 256–265. [Google Scholar] [CrossRef]
- Li, J.; Zheng, W.; Kwon, A.H.; Lu, Y. In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000, 28, 481–488. [Google Scholar] [CrossRef]
- Santoro, S.W.; Joyce, G.F. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262. [Google Scholar] [CrossRef]
- Khachigian, L.M. DNAzymes: Cutting a path to a new class of therapeutics. Curr. Opin. Mol. Ther. 2002, 4, 119–121. [Google Scholar]
- Cairns, M.J.; King, A.; Sun, L.Q. Optimisation of the 10-23 DNAzyme-substrate pairing interactions enhanced RNA cleavage activity at purine-cytosine target sites. Nucleic Acids Res. 2003, 31, 2883–2889. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, Z.; Zhang, L.; He, J.; Sun, L.-Q. Selection and antitumor activity of anti-Bcl-2 DNAzymes. Biochem. Biophys. Res. Commun. 2016, 479, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, Y.-F.; Luo, S. Ani-survivin DNAzymes Inhibit Cell Proliferation and Migration in Breast Cancer Cell Line MCF-7. Asian Pac. J. Cancer Prev. 2012, 13, 6233–6237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Niu, J.; Zhao, C.; Wang, X.; Ren, J.; Qu, X. A Bimetallic Metal–Organic Framework Encapsulated with DNAzyme for Intracellular Drug Synthesis and Self-Sufficient Gene Therapy. Angew. Chem. 2021, 133, 12539–12545. [Google Scholar] [CrossRef]
- De Bock, C.E.; Lin, Z.; Itoh, T.; Morris, D.; Murrell, G.; Wang, Y. Inhibition of urokinase receptor gene expression and cell invasion by anti-uPAR DNAzymes in osteosarcoma cells. FEBS J. 2005, 272, 3572–3582. [Google Scholar] [CrossRef]
- Du, S.; Chen, C.; Qu, S.; Song, H.; Yang, J.; Li, Y.; Liu, K.; Lu, Q.; Luo, W.; Wang, R.; et al. DNAzyme-Assisted Nano-Herb Delivery System for Multiple Tumor Immune Activation. Small 2022, 18, 2203942. [Google Scholar] [CrossRef]
- Cai, H.; Santiago, F.S.; Prado-Lourenco, L.; Wang, B.; Patrikakis, M.; Davenport, M.P.; Maghzal, G.J.; Stocker, R.; Parish, C.R.; Chong, B.H.; et al. DNAzyme Targeting c- jun Suppresses Skin Cancer Growth. Sci. Transl. Med. 2012, 4, 139ra82. [Google Scholar] [CrossRef]
- Zhang, M.; Drummen, G.P.C.; Luo, S. Anti-insulin-like growth factor-IIP3 DNAzymes inhibit cell proliferation and induce caspase-dependent apoptosis in human hepatocarcinoma cell lines. Drug Des. Dev. Ther. 2013, 7, 1089–1102. [Google Scholar]
- Schubert, S. RNA Cleaving “10–23” DNAzymes with Enhanced Stability and Activity. Nucleic Acids Res. 2003, 31, 5982–5992. [Google Scholar] [CrossRef]
- Vester, B.; Lundberg, L.B.; Sørensen, M.D.; Babu, B.R.; Douthwaite, S.; Wengel, J. LNAzymes: Incorporation of LNA-Type Monomers into DNAzymes Markedly Increases RNA Cleavage. J. Am. Chem. Soc. 2002, 124, 13682–13683. [Google Scholar] [CrossRef]
- Thai, H.B.D.; Levi-Acobas, F.; Yum, S.-Y.; Jang, G.; Hollenstein, M.; Ahn, D.-R. Tetrahedral DNAzymes for enhanced intracellular gene-silencing activity. Chem. Commun. 2018, 54, 9410–9413. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Kim, M.; Lee, M.; Kim, S.; Park, N. Nanosized DNA Hydrogel Functionalized with a DNAzyme Tetrahedron for Highly Efficient Gene Silencing. Biomacromolecules 2024, 25, 4913–4924. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Chen, X.; Feng, C.; Ding, L.; Liu, X.; Chen, T.; Zhang, F.; Li, Y.; Ma, Z.; Tian, B.; et al. Visualized and cascade-enhanced gene silencing by smart DNAzyme-graphene nanocomplex. Nano Res. 2020, 13, 2165–2174. [Google Scholar] [CrossRef]
- He, M.; He, M.; Nie, C.; Yi, J.; Zhang, J.; Chen, T.; Chu, X. mRNA-Activated Multifunctional DNAzyme Nanotweezer for Intracellular mRNA Sensing and Gene Therapy. ACS Appl. Mater. Interfaces 2021, 13, 8015–8025. [Google Scholar] [CrossRef] [PubMed]
- Hermann, T.; Patel, D.J. Adaptive Recognition by Nucleic Acid Aptamers. Science 2000, 287, 820–825. [Google Scholar] [CrossRef]
- Hayashi, T.; Oshima, H.; Mashima, T.; Nagata, T.; Katahira, M.; Kinoshita, M. Binding of an RNA aptamer and a partial peptide of a prion protein: Crucial importance of water entropy in molecular recognition. Nucleic Acids Res. 2014, 42, 6861–6875. [Google Scholar] [CrossRef]
- Partha, R.; Kristi, D.V.; Erin, E.S.; Rebecca, S. Application of aptamers for targeted theraputics. Arch. Immunol. Ther. Exp. 2013, 61, 255–271. [Google Scholar]
- Dunn, M.R.; Jimenez, R.M.; Chaput, J.C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 2017, 1, 0076. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Mol. Ther.—Nucleic Acids 2014, 3, e169. [Google Scholar] [CrossRef]
- Ellington, A.; Szostak, J. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Bock, L.C.; Griffin, L.C.; Latham, J.A.; Vermaas, E.H.; Toole, J.J. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature 1992, 355, 564–566. [Google Scholar] [CrossRef] [PubMed]
- Ni, S.; Zhuo, Z.; Pan, Y.; Yu, Y.; Li, F.; Liu, J.; Wang, L.; Wu, X.; Li, D.; Wan, Y.; et al. Recent Progress in Aptamer Discoveries and Modifications for Therapeutic Applications. ACS Appl. Mater. Interfaces 2021, 13, 9500–9519. [Google Scholar] [CrossRef] [PubMed]
- Dua, P.; Kim, S.; Lee, D.-K. Nucleic acid aptamers targeting cell-surface proteins. Methods 2011, 54, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Seo, J.-M.; Shin, K.-J.; Yang, S.-G. Design and clinical developments of aptamer-drug conjugates for targeted cancer therapy. Biomater. Res. 2021, 25, 42. [Google Scholar] [CrossRef] [PubMed]
- Poolsup, S.; Kim, C.-Y. Therapeutic applications of synthetic nucleic acid aptamers. Curr. Opin. Biotechnol. 2017, 48, 180–186. [Google Scholar] [CrossRef]
- Vu, C.Q.; Rotkrua, P.; Soontornworajit, B.; Tantirungrotechai, Y. Effect of PDGF-B aptamer on PDGFRβ/PDGF-B interaction: Molecular dynamics study. J. Mol. Graph. Model. 2018, 82, 145–156. [Google Scholar] [CrossRef]
- Sae-Lim, S.; Soontornworajit, B.; Rotkrua, P. Inhibition of Colorectal Cancer Cell Proliferation by Regulating Platelet-Derived Growth Factor B Signaling with a DNA Aptamer. Asian Pac. J. Cancer Prev. 2019, 20, 487–494. [Google Scholar] [CrossRef]
- Liu, S.-C.; Alomran, R.; Chernikova, S.B.; Lartey, F.; Stafford, J.; Jang, T.; Merchant, M.; Zboralski, D.; Zöllner, S.; Kruschinski, A.; et al. Blockade of SDF-1 after irradiation inhibits tumor recurrences of autochthonous brain tumors in rats. Neuro-Oncol. 2013, 16, 21–28. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef]
- Hoellenriegel, J.; Zboralski, D.; Maasch, C.; Rosin, N.Y.; Wierda, W.G.; Keating, M.J.; Kruschinski, A.; Burger, J.A. The Spiegelmer NOX-A12, a novel CXCL12 inhibitor, interferes with chronic lymphocytic leukemia cell motility and causes chemosensitization. Blood 2014, 123, 1032–1039. [Google Scholar] [CrossRef]
- Gijs, M.; Penner, G.; Blackler, G.; Impens, N.; Baatout, S.; Luxen, A.; Aerts, A. Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals 2016, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, G.; Maron, R.; Mancini, M.; Schechter, B.; Sela, M.; Yarden, Y. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl. Acad. Sci. USA 2013, 110, 8170–8175. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, Y.; Wang, Y.; Xie, M.; Dai, J.; Qu, Z.; Zhou, M.; Cao, S.; Shi, J.; Wang, L.; et al. Directing Multivalent Aptamer-Receptor Binding on the Cell Surface with Programmable Atom-Like Nanoparticles. Angew. Chem. Int. Ed. 2022, 61, e202117168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jiao, P.; Yang, L.; Li, X.; Yan, B. Enhancing Cell Recognition by Scrutinizing Cell Surfaces with a Nanoparticle Array. J. Am. Chem. Soc. 2010, 133, 680–682. [Google Scholar] [CrossRef]
- Chen, S.; Xu, Z.; Yang, W.; Lin, X.; Li, J.; Li, J.; Yang, H. Logic-Gate-Actuated DNA-Controlled Receptor Assembly for the Programmable Modulation of Cellular Signal Transduction. Angew. Chem. 2019, 131, 18354–18358. [Google Scholar] [CrossRef]
- Wang, L.; Liang, H.; Sun, J.; Liu, Y.; Li, J.; Li, J.; Li, J.; Yang, H. Bispecific Aptamer Induced Artificial Protein-Pairing: A Strategy for Selective Inhibition of Receptor Function. J. Am. Chem. Soc. 2019, 141, 12673–12681. [Google Scholar] [CrossRef]
- Miao, Y.; Gao, Q.; Mao, M.; Zhang, C.; Yang, L.; Yang, Y.; Han, D. Bispecific Aptamer Chimeras Enable Targeted Protein Degradation on Cell Membranes. Angew. Chem. 2021, 133, 11367–11371. [Google Scholar] [CrossRef]
- Hoshiyama, J.; Okada, Y.; Cho, S.; Ueki, R.; Sando, S. Apt-clean: Aptamer-mediated cleavage of extracellular antigens for the inhibition of membrane protein functions. Biomater. Sci. 2023, 11, 445–449. [Google Scholar] [CrossRef]
- Varadé, J.; Magadán, S.; González-Fernández, Á. Human immunology and immunotherapy: Main achievements and challenges. Cell. Mol. Immunol. 2020, 18, 805–828. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Kejamurthy, P.; Devi, K.T.R. Immune checkpoint inhibitors and cancer immunotherapy by aptamers: An overview. Med. Oncol. 2023, 41, 40. [Google Scholar] [CrossRef] [PubMed]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.B.; Cydzik, M.; Gariépy, J. Targeting the PD-1/PD-L1 immune evasion axis with DNA aptamers as a novel therapeutic strategy for the treatment of disseminated cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zheng, Y.; Lin, Z.; Liu, X.; Li, J.; Yang, H.; Tan, W. Equipping Natural Killer Cells with Specific Targeting and Checkpoint Blocking Aptamers for Enhanced Adoptive Immunotherapy in Solid Tumors. Angew. Chem. 2020, 132, 12120–12126. [Google Scholar] [CrossRef]
- Huang, B.T.; Lai, W.Y.; Chang, Y.C.; Wang, J.W.; Yeh, S.D.; Lin, E.P.Y.; Yang, P.C. A CTLA-4 Antagonizing DNA Aptamer with Antitumor Effect. Mol. Ther.—Nucleic Acids 2017, 8, 520–528. [Google Scholar] [CrossRef]
- Boltz, A.; Piater, B.; Toleikis, L.; Guenther, R.; Kolmar, H.; Hock, B. Bi-specific Aptamers Mediating Tumor Cell Lysis. J. Biol. Chem. 2011, 286, 21896–21905. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, X.; Xu, J.; Cui, C.; Yazd, H.S.; Pan, X.; Zhu, Y.; Chen, X.; Li, X.; Li, J.; et al. Circular Bispecific Aptamer-Mediated Artificial Intercellular Recognition for Targeted T Cell Immunotherapy. ACS Nano 2020, 14, 9562–9571. [Google Scholar] [CrossRef]
- Zheng, A.; Du, Y.; Wang, Y.; Zheng, Y.; Ning, Z.; Wu, M.; Zhang, C.; Zhang, D.; Liu, J.; Liu, X. CD16/PD-L1 bi-specific aptamer for cancer immunotherapy through recruiting NK cells and acting as immunocheckpoint blockade. Mol. Ther.—Nucleic Acids 2022, 27, 998–1009. [Google Scholar] [CrossRef]
- Thomas, B.J.; Porciani, D.; Burke, D.H. Cancer immunomodulation using bispecific aptamers. Mol. Ther.—Nucleic Acids 2022, 27, 894–915. [Google Scholar] [CrossRef]
- Mahmoudian, F.; Ahmari, A.; Shabani, S.; Sadeghi, B.; Fahimirad, S.; Fattahi, F. Aptamers as an approach to targeted cancer therapy. Cancer Cell Int. 2024, 24, 108. [Google Scholar] [CrossRef]
- Kroschinsky, F.; Stölzel, F.; Von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. New drugs, new toxicities: Severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.W.-S.; Chan, C.K.W.; Yu, J.; He, M.; Choi, C.H.J.; Lau, J.Y.W.; Wong, N. Development of CD44E/s dual-targeting DNA aptamer as nanoprobe to deliver treatment in hepatocellular carcinoma. Nanotheranostics 2022, 6, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.Y.; Kim, H.; Lee, M.; Hong, J.; Lee, J.H.; Kim, J.; Choi, M.J.; Park, Y.S.; Kim, S.-C. Development of HER2-Specific Aptamer-Drug Conjugate for Breast Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 9764. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Kołat, D.; Kałuzińska-Kołat, Ż.; Celik, I.; Kontek, R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells 2023, 12, 659. [Google Scholar] [CrossRef]
- Yao, F.; An, Y.; Li, X.; Li, Z.; Duan, J.; Yang, X.-D. Targeted Therapy of Colon Cancer by Aptamer-Guided Holliday Junctions Loaded with Doxorubicin. Int. J. Nanomed. 2020, 15, 2119–2129. [Google Scholar] [CrossRef]
- Tan, Y.; Li, Y.; Qu, Y.-X.; Su, Y.; Peng, Y.; Zhao, Z.; Fu, T.; Wang, X.-Q.; Tan, W. Aptamer-Peptide Conjugates as Targeted Chemosensitizers for Breast Cancer Treatment. ACS Appl. Mater. Interfaces 2020, 13, 9436–9444. [Google Scholar] [CrossRef]
- Omer, M.; Andersen, V.L.; Nielsen, J.S.; Wengel, J.; Kjems, J. Improved Cancer Targeting by Multimerizing Aptamers on Nanoscaffolds. Mol. Ther.—Nucleic Acids 2020, 22, 994–1003. [Google Scholar] [CrossRef]
- Alves, L.N.; Missailidis, S.; Lage, C.A.; De Almeida, C.E.B. Anti-MUC1 Aptamer as Carrier Tool of the Potential Radiosensitizer 1,10 Phenanthroline in MCF-7 Breast Cancer Cells. Anticancer Res. 2019, 39, 1859–1867. [Google Scholar] [CrossRef]
- Ghahremani, F.; Kefayat, A.; Shahbazi-Gahrouei, D.; Motaghi, H.; Mehrgardi, M.A.; Haghjooy-Javanmard, S. AS1411 aptamer-targeted gold nanoclusters effect on the enhancement of radiation therapy efficacy in breast tumor-bearing mice. Nanomedicine 2018, 13, 2563–2578. [Google Scholar] [CrossRef]
- Dassie, J.P.; Liu, X.-Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ding, J.; Zhou, W. An aptamer-tethered, DNAzyme-embedded molecular beacon for simultaneous detection and regulation of tumor-related genes in living cells. Analyst 2019, 144, 5098–5107. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, X.; Liu, H.; Wu, D.; She, J.-X. Co-targeting EGFR and survivin with a bivalent aptamer-dual siRNA chimera effectively suppresses prostate cancer. Sci. Rep. 2016, 6, 30346. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Sun, N.; Liu, M.; Wang, J.; Pei, R. Building a chimera of aptamer–antisense oligonucleotide for silencing galectin-1 gene. RSC Adv. 2016, 6, 112445–112450. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef]
- Martinez-Lage, M.; Puig-Serra, P.; Menendez, P.; Torres-Ruiz, R.; Rodriguez-Perales, S. CRISPR/Cas9 for Cancer Therapy: Hopes and Challenges. Biomedicines 2018, 6, 105. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Chen, H.; Choi, J.; Bailey, S. Cut Site Selection by the Two Nuclease Domains of the Cas9 RNA-guided Endonuclease. J. Biol. Chem. 2014, 289, 13284–13294. [Google Scholar] [CrossRef]
- Stefanoudakis, D.; Kathuria-Prakash, N.; Sun, A.W.; Abel, M.; Drolen, C.E.; Ashbaugh, C.; Zhang, S.; Hui, G.; Tabatabaei, Y.A.; Zektser, Y.; et al. The Potential Revolution of Cancer Treatment with CRISPR Technology. Cancers 2023, 15, 1813. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2015, 17, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, Y.; Yu, B.; Hu, Y.; Zhang, N.; Zheng, Y.; Yang, M.; Xu, F. A Lactose-Derived CRISPR/Cas9 Delivery System for Efficient Genome Editing In Vivo to Treat Orthotopic Hepatocellular Carcinoma. Adv. Sci. 2020, 7, 2001424. [Google Scholar] [CrossRef]
- He, Z.-Y.; Zhang, Y.-G.; Yang, Y.-H.; Ma, C.-C.; Wang, P.; Du, W.; Li, L.; Xiang, R.; Song, X.-R.; Zhao, X.; et al. In Vivo Ovarian Cancer Gene Therapy Using CRISPR-Cas9. Hum. Gene Ther. 2018, 29, 223–233. [Google Scholar] [CrossRef]
- Koo, T.; Yoon, A.-R.; Cho, H.-Y.; Bae, S.; Yun, C.-O.; Kim, J.-S. Selective disruption of an oncogenic mutant allele by CRISPR/Cas9 induces efficient tumor regression. Nucleic Acids Res. 2017, 45, 7897–7908. [Google Scholar] [CrossRef]
- Hussen, B.M.; Rasul, M.F.; Abdullah, S.R.; Hidayat, H.J.; Faraj, G.S.H.; Ali, F.A.; Salihi, A.; Baniahmad, A.; Ghafouri-Fard, S.; Rahman, M.; et al. Targeting miRNA by CRISPR/Cas in cancer: Advantages and challenges. Mil. Med. Res. 2023, 10, 32. [Google Scholar] [CrossRef]
- Yoshino, H.; Yonemori, M.; Miyamoto, K.; Tatarano, S.; Kofuji, S.; Nohata, N.; Nakagawa, M.; Enokida, H. microRNA-210-3p depletion by CRISPR/Cas9 promoted tumorigenesis through revival of TWIST1 in renal cell carcinoma. Oncotarget 2017, 8, 20881–20894. [Google Scholar] [CrossRef]
- De Santa-Inez, D.C.; Fuziwara, C.S.; Saito, K.C.; Kimura, E.T. Targeting the Highly Expressed microRNA miR-146b with CRISPR/Cas9n Gene Editing System in Thyroid Cancer. Int. J. Mol. Sci. 2021, 22, 7992. [Google Scholar] [CrossRef]
- Jiang, F.; Liang, Y.; Wei, W.; Zou, C.; Chen, G.; Wan, Y.; Liu, Z.; Yang, Y.; Han, Z.; Zhu, J.; et al. Functional classification of prostate cancer-associated miRNAs through CRISPR/Cas9-mediated gene knockout. Mol. Med. Rep. 2020, 5, 3777–3784. [Google Scholar] [CrossRef]
- Vaghari-Tabari, M.; Hassanpour, P.; Sadeghsoltani, F.; Malakoti, F.; Alemi, F.; Qujeq, D.; Asemi, Z.; Yousefi, B. CRISPR/Cas9 gene editing: A new approach for overcoming drug resistance in cancer. Cell. Mol. Biol. Lett. 2022, 27, 49. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Yang, Y.; Qi, H.; Cui, W.; Zhang, L.; Fu, X.; He, X.; Liu, M.; Li, P.-F.; Yu, T. CRISPR/Cas9 therapeutics: Progress and prospects. Signal Transduct. Target. Ther. 2023, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Ansori, A.N.; Antonius, Y.; Susilo, R.J.; Hayaza, S.; Kharisma, V.D.; Parikesit, A.A.; Zainul, R.J.; Jakhmola, V.; Saklani, T.; Rebezov, M.; et al. Application of CRISPR-Cas9 genome editing technology in various fields: A review. Narra J. 2023, 3, e184. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Lester, G.M.S.; Petishnok, L.C.; Dean, D.A. Cytoplasmic transport and nuclear import of plasmid DNA. Biosci. Rep. 2017, 37, BSR20160616. [Google Scholar] [CrossRef]
- Martínez-Puente, D.H.; Pérez-Trujillo, J.J.; Zavala-Flores, L.M.; García-García, A.; Villanueva-Olivo, A.; Rodríguez-Rocha, H.; Valdés, J.; Saucedo-Cárdenas, O.; De Oca-Luna, R.M.; De Jesús Loera-Arias, M. Plasmid DNA for Therapeutic Applications in Cancer. Pharmaceutics 2022, 14, 1861. [Google Scholar] [CrossRef]
- Liu, N. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Gómez-Navarro, J.; Arafat, W.; Xiang, J. Gene therapy for carcinoma of the breast: Pro-apoptotic gene therapy. Breast Cancer Res. 1999, 2, 32. [Google Scholar] [CrossRef]
- Neves, A.R.; Sousa, A.; Faria, R.; Albuquerque, T.; Queiroz, J.A.; Costa, D. Cancer gene therapy mediated by RALA/plasmid DNA vectors: Nitrogen to phosphate groups ratio (N/P) as a tool for tunable transfection efficiency and apoptosis. Colloids Surf. B Biointerfaces 2020, 185, 110610. [Google Scholar] [CrossRef]
- Singh, S.; Asal, R.; Bhagat, S. Multifunctional antioxidant nanoliposome-mediated delivery of PTEN plasmids restore the expression of tumor suppressor protein and induce apoptosis in prostate cancer cells. J. Biomed. Mater. Res. Part A 2018, 106, 3152–3164. [Google Scholar] [CrossRef]
- Hong, S.-H.; Lee, J.-H.; Jiang, H.-L.; Kim, J.-E.; Lee, A.Y.; Kim, S.; Cho, C.-S.; Cho, M.-H. Dual Expression of shAkt1 and Pdcd4 Suppresses Lung Tumorigenesis in K-rasLA1 Mice. Anticancer Res. 2015, 35, 2015–2019. [Google Scholar] [PubMed]
- Zarovni, N.; Vago, R.; Soldà, T.; Monaco, L.; Fabbrini, M.S. Saporin as a novel suicide gene in anticancer gene therapy. Cancer Gene Ther. 2006, 14, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, B.L.; Yu, Z.; Chung, W.G.; Weiss, R.; Cui, Z. Replicase-based plasmid DNA shows anti-tumor activity. BMC Cancer 2011, 11, 110. [Google Scholar] [CrossRef]
- Mizrahi, A.; Czerniak, A.; Levy, T.; Amiur, S.; Gallula, J.; Matouk, I.; Abu-Lail, R.; Sorin, V.; Birman, T.; De Groot, N.; et al. Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J. Transl. Med. 2009, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Danishmalik, S.N.; Hwang, H.; Sin, J.; Oh, J.; Cho, Y.; Lee, H.; Jeong, M.; Kim, S.; Hong, H.J. Gene therapy using plasmid DNA-encoded anti-HER2 antibody for cancers that overexpress HER2. Cancer Gene Ther. 2016, 23, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Marnin, L.; Kudchodkar, S.B.; Perales-Puchalt, A.; Choi, H.; Agarwal, S.; Scott, V.L.; Reuschel, E.L.; Zaidi, F.I.; Duperret, E.K.; et al. Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immunol. Immunother. 2017, 66, 1577–1588. [Google Scholar] [CrossRef]
- Pandya, A.; Shah, Y.; Kothari, N.; Postwala, H.; Shah, A.; Parekh, P.; Chorawala, M.R. The future of cancer immunotherapy: DNA vaccines leading the way. Med. Oncol. 2023, 40, 200. [Google Scholar] [CrossRef]
- Kawano, H.; Nishikawa, M.; Mitsui, M.; Takahashi, Y.; Kako, K.; Yamaoka, K.; Watanabe, Y.; Takakura, Y. Improved anti-cancer effect of interferon gene transfer by sustained expression using CpG-reduced plasmid DNA. Int. J. Cancer 2007, 121, 401–406. [Google Scholar] [CrossRef]
- Pang, J.; Zhuang, B.; Zhang, L.-M. A co-carrier for plasmid DNA and curcumin delivery to treat pancreatic cancer via dendritic poly(l-lysine) modified amylose. Int. J. Biol. Macromol. 2023, 253, 127467. [Google Scholar] [CrossRef]
- Chen, J.; Liu, H.; Jehng, T.; Li, Y.; Chen, Z.; Lee, K.-D.; Shen, H.-T.; Jones, L.; Huang, X.F.; Chen, S.-Y. A Novel Anti-PD-L1 Vaccine for Cancer Immunotherapy and Immunoprevention. Cancers 2019, 11, 1909. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Y.; Zheng, X.; Liu, M.; Wang, G. Enhancedanti-tumor efficacy through a combination of intramuscularly expressed DNA vaccine and plasmid-encoded PD-1 antibody. Front. Immunol. 2023, 14, 1169850. [Google Scholar] [CrossRef] [PubMed]
- Duperret, E.K.; Wise, M.C.; Trautz, A.; Villarreal, D.O.; Ferraro, B.; Walters, J.; Yan, J.; Khan, A.; Masteller, E.; Humeau, L.; et al. Synergy of Immune Checkpoint Blockade with a Novel Synthetic Consensus DNA Vaccine Targeting TERT. Mol. Ther. 2018, 26, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Kos, S.; Lopes, A.; Preat, V.; Cemazar, M.; Tratar, U.L.; Ucakar, B.; Vanvarenberg, K.; Sersa, G.; Vandermeulen, G. Intradermal DNA vaccination combined with dual CTLA-4 and PD-1 blockade provides robust tumor immunity in murine melanoma. PLoS ONE 2019, 14, e0217762. [Google Scholar] [CrossRef] [PubMed]
- Taxman, D.J.; Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2010; pp. 139–156. [Google Scholar]
- Künnapuu, K.; Veiman, K.-L.; Porosk, L.; Rammul, E.; Kiisholts, K.; Langel, Ü.; Kurrikoff, K. Tumor gene therapy by systemic delivery of plasmid DNA with cell-penetrating peptides. FASEB BioAdvances 2018, 1, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Seraj, S.; Cho, Y.J.; Lee, J.-W.; Ahn, H.J. Cytoplasmic expression of EGFR shRNA using a modified T7 autogene-based hybrid mRNA/DNA system induces long-term EGFR silencing and prolongs antitumor effects. Biochem. Pharmacol. 2020, 171, 113735. [Google Scholar] [CrossRef]
- Zhao, L.; Luo, Y.; Huang, Q.; Cao, Z.; Yang, X. Photo-Enhanced CRISPR/Cas9 System Enables Robust PD-L1 Gene Disruption in Cancer Cells and Cancer Stem-Like Cells for Efficient Cancer Immunotherapy. Small 2020, 16, 2004879. [Google Scholar] [CrossRef]
- Yip, B. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a Messenger RNA Polynucleotide Vaccine Vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef]
- Youn, H.; Chung, J.-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348. [Google Scholar] [CrossRef]
- Whitley, J.; Zwolinski, C.; Denis, C.; Maughan, M.; Hayles, L.; Clarke, D.; Snare, M.; Liao, H.; Chiou, S.; Marmura, T.; et al. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl. Res. 2022, 242, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.; Xiao, Y.; Shi, S.; Ji, X.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-X.; Wang, Y.; Ding, J.; Jiang, A.; Wang, J.; Yu, M.; Blake, S.; Liu, S.; Bieberich, C.J.; Farokhzad, O.C.; et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci. Transl. Med. 2021, 13, eaba9772. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Tang, Y.; Chen, J.; Muriph, R.; Ye, Z.; Huang, C.; Evans, J.; Henske, E.P.; Xu, Q. Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2116271119. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Feng, C.; Yuan, Q.; Liu, C.; Ge, B.; Sun, F.; Zhu, X. Lantern-shaped flexible RNA origami for Smad4 mRNA delivery and growth suppression of colorectal cancer. Nat. Commun. 2023, 14, 1307. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Van Lint, S.; Roose, K.; Van Parys, A.; Vandenabeele, P.; Grooten, J.; Tavernier, J.; De Koker, S.; Saelens, X. Treatment with mRNA coding for the necroptosis mediator MLKL induces antitumor immunity directed against neo-epitopes. Nat. Commun. 2018, 9, 3417. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Sayour, E.J.; Boczkowski, D.; Mitchell, D.A.; Nair, S.K. Cancer mRNA vaccines: Clinical advances and future opportunities. Nat. Rev. Clin. Oncol. 2024, 21, 489–500. [Google Scholar] [CrossRef]
- Liu, J.-Q.; Zhang, C.; Zhang, X.; Yan, J.; Zeng, C.; Talebian, F.; Lynch, K.; Zhao, W.; Hou, X.; Du, S.; et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. J. Control. Release 2022, 345, 306–313. [Google Scholar] [CrossRef]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef]
- Hochmann, S.; Mittermeir, M.; Santic, R.; Koszik, F.; Griessner, L.; Sonderegger, A.S.; Hoffmann, T.; Russe, E.; Scheiblhofer, S.; Weiss, R.; et al. Evaluation of modified Interferon alpha mRNA constructs for the treatment of non-melanoma skin cancer. Sci. Rep. 2018, 8, 12954. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-N.; Li, M.; Luo, Y.-L.; Chen, Q.; Wang, L.; Zhang, H.-B.; Shen, S.; Gu, Z.; Wang, J. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater. Sci. 2018, 6, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Guo, J.; Zhao, Y.; Ma, S.; Hou, Y.; Yang, J. Intranasal delivery of cationic liposome-protamine complex mRNA vaccine elicits effective anti-tumor immunity. Cell. Immunol. 2020, 354, 104143. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, Y.; Miao, L.; Liu, Q.; Musetti, S.; Li, J.; Huang, L. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol. Ther. 2018, 26, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, W.; Tian, J.; Qi, C.; Cai, Z.; Yan, W.; Xuan, S.; Shang, A. Engineered mRNA-expressed bispecific antibody prevent intestinal cancer via lipid nanoparticle delivery. Bioengineered 2021, 12, 12383–12393. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, Y.; Zi, G.; Chen, J.; Peng, B. Co-delivery of Cas9 mRNA and guide RNAs for editing of LGMN gene represses breast cancer cell metastasis. Sci. Rep. 2024, 14, 8095. [Google Scholar] [CrossRef]
- Reyes-Darias, J.A.; Berzal-Herranz, A. Detection of immune response activation by exogenous nucleic acids by a multiplex RT-PCR method. Mol. Cell. Probes 2014, 28, 181–185. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Ghosh, S.; Brown, A.M.; Jenkins, C.; Campbell, K. Viral Vector Systems for Gene Therapy: A Comprehensive Literature Review of Progress and Biosafety Challenges. Appl. Biosaf. 2020, 25, 7–18. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Zhu, Y.; Li, F. DNA Functional Nanomaterials for Controlled Delivery of Nucleic Acid-Based Drugs. Front. Bioeng. Biotechnol. 2021, 9, 720291. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Luo, S.; Wang, J.; Shen, Z.; Wu, Z.-S. Nuclease-resistant signaling nanostructures made entirely of DNA oligonucleotides. Nanoscale 2021, 13, 7034–7051. [Google Scholar] [CrossRef] [PubMed]
- Morya, V.; Walia, S.; Mandal, B.B.; Ghoroi, C.; Bhatia, D. Functional DNA Based Hydrogels: Development, Properties and Biological Applications. ACS Biomater. Sci. Eng. 2020, 6, 6021–6035. [Google Scholar] [CrossRef]
- Fu, X.; Chen, T.; Song, Y.; Feng, C.; Chen, H.; Zhang, Q.; Chen, G.; Zhu, X. mRNA Delivery by a pH-Responsive DNA Nano-Hydrogel. Small 2021, 17, 2101224. [Google Scholar] [CrossRef]
- Song, J.; Lee, M.; Kim, T.; Na, J.; Jung, Y.; Jung, G.Y.; Kim, S.; Park, N. A RNA producing DNA hydrogel as a platform for a high performance RNA interference system. Nat. Commun. 2018, 9, 4331. [Google Scholar] [CrossRef]
- Choi, C.H.J.; Hao, L.; Narayan, S.P.; Auyeung, E.; Mirkin, C.A. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc. Natl. Acad. Sci. USA 2013, 110, 7625–7630. [Google Scholar] [CrossRef]
- Brodin, J.D.; Sprangers, A.J.; McMillan, J.R.; Mirkin, C.A. DNA-Mediated Cellular Delivery of Functional Enzymes. J. Am. Chem. Soc. 2015, 137, 14838–14841. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Y.; Wu, Q.; Lou, B.; Liu, Z. DNA nanostructures for stimuli-responsive drug delivery. Smart Mater. Med. 2021, 3, 66–84. [Google Scholar] [CrossRef]
- Han, X.; Xu, X.; Wu, Z.; Wu, Z.; Qi, X. Synchronous conjugation of i-motif DNA and therapeutic siRNA on the vertexes of tetrahedral DNA nanocages for efficient gene silence. Acta Pharm. Sin. B 2021, 11, 3286–3296. [Google Scholar] [CrossRef]
- Jiang, Y.; Huo, S.; Hardie, J.; Liang, X.-J.; Rotello, V.M. Progress and perspective of inorganic nanoparticle-based siRNA delivery systems. Expert Opin. Drug Deliv. 2016, 13, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Thayumanavan, S. Noncationic Material Design for Nucleic Acid Delivery. Adv. Ther. 2020, 3, 1900206. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, X.; Luo, G.; Jiao, J. DNA-functionalized gold nanoparticles: Modification, characterization, and biomedical applications. Front. Chem. 2022, 10, 1095488. [Google Scholar] [CrossRef]
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef]
- Kus-Liśkiewicz, M.; Fickers, P.; Tahar, I.B. Biocompatibility and Cytotoxicity of Gold Nanoparticles: Recent Advances in Methodologies and Regulations. Int. J. Mol. Sci. 2021, 22, 10952. [Google Scholar] [CrossRef]
- Luther, D.C.; Huang, R.; Jeon, T.; Zhang, X.; Lee, Y.-W.; Nagaraj, H.; Rotello, V.M. Delivery of drugs, proteins, and nucleic acids using inorganic nanoparticles. Adv. Drug Deliv. Rev. 2020, 156, 188–213. [Google Scholar] [CrossRef]
- Torres-Vanegas, J.D.; Cruz, J.C.; Reyes, L.H. Delivery Systems for Nucleic Acids and Proteins: Barriers, Cell Capture Pathways and Nanocarriers. Pharmaceutics 2021, 13, 428. [Google Scholar] [CrossRef]
- Dong, S.; Feng, Z.; Ma, R.; Zhang, T.; Jiang, J.; Li, Y.; Zhang, Y.; Li, S.; Liu, X.; Liu, X.; et al. Engineered Design of a Mesoporous Silica Nanoparticle-Based Nanocarrier for Efficient mRNA Delivery in Vivo. Nano Lett. 2023, 23, 2137–2147. [Google Scholar] [CrossRef]
- Zhang, J.; Lei, W.; Meng, Y.; Zhou, C.; Zhang, B.; Yuan, J.; Wang, M.; Xu, D.; Meng, X.; Chen, W. Expression of PEI-coated gold nanoparticles carrying exogenous gene in periwinkle mesophyll cells and its practice in huanglongbing research. iScience 2022, 25, 104479. [Google Scholar] [CrossRef]
- Aschmann, D.; Knol, R.A.; Kros, A. Lipid-Based Nanoparticle Functionalization with Coiled-Coil Peptides for In Vitro and In Vivo Drug Delivery. Acc. Chem. Res. 2024, 57, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Kiaie, S.H.; Zolbanin, N.M.; Ahmadi, A.; Bagherifar, R.; Valizadeh, H.; Kashanchi, F.; Jafari, R. Recent advances in mRNA-LNP therapeutics: Immunological and pharmacological aspects. J. Nanobiotechnology 2022, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, S.; Koide, H.; Asai, T. Recent advances in siRNA delivery mediated by lipid-based nanoparticles. Adv. Drug Deliv. Rev. 2020, 154–155, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Heyes, J.; Lutwyche, P. Lipid nanoparticles for nucleic acid delivery: Current perspectives. Adv. Drug Deliv. Rev. 2020, 154–155, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, C.H.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef]
- Nsairat, H.; Mahmoud, I.S.; Odeh, F.; Abuarqoub, D.; Al-Azzawi, H.; Zaza, R.; Qadri, M.I.; Ismail, S.; Bawab, A.A.; Awidi, A.; et al. Grafting of anti-nucleolin aptamer into preformed and remotely loaded liposomes through aptamer-cholesterol post-insertion. RSC Adv. 2020, 10, 36219–36229. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Lee, M.; Song, Y.; Kim, S.; Park, N. Recent Advances and Prospects of Nucleic Acid Therapeutics for Anti-Cancer Therapy. Molecules 2024, 29, 4737. https://doi.org/10.3390/molecules29194737
Lee M, Lee M, Song Y, Kim S, Park N. Recent Advances and Prospects of Nucleic Acid Therapeutics for Anti-Cancer Therapy. Molecules. 2024; 29(19):4737. https://doi.org/10.3390/molecules29194737
Chicago/Turabian StyleLee, Minhyuk, Minjae Lee, Youngseo Song, Sungjee Kim, and Nokyoung Park. 2024. "Recent Advances and Prospects of Nucleic Acid Therapeutics for Anti-Cancer Therapy" Molecules 29, no. 19: 4737. https://doi.org/10.3390/molecules29194737
APA StyleLee, M., Lee, M., Song, Y., Kim, S., & Park, N. (2024). Recent Advances and Prospects of Nucleic Acid Therapeutics for Anti-Cancer Therapy. Molecules, 29(19), 4737. https://doi.org/10.3390/molecules29194737