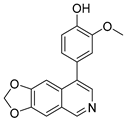
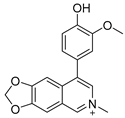
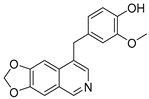
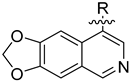
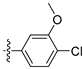
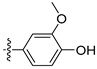
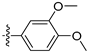
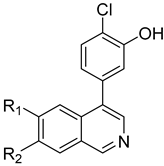
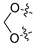
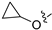
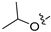
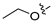
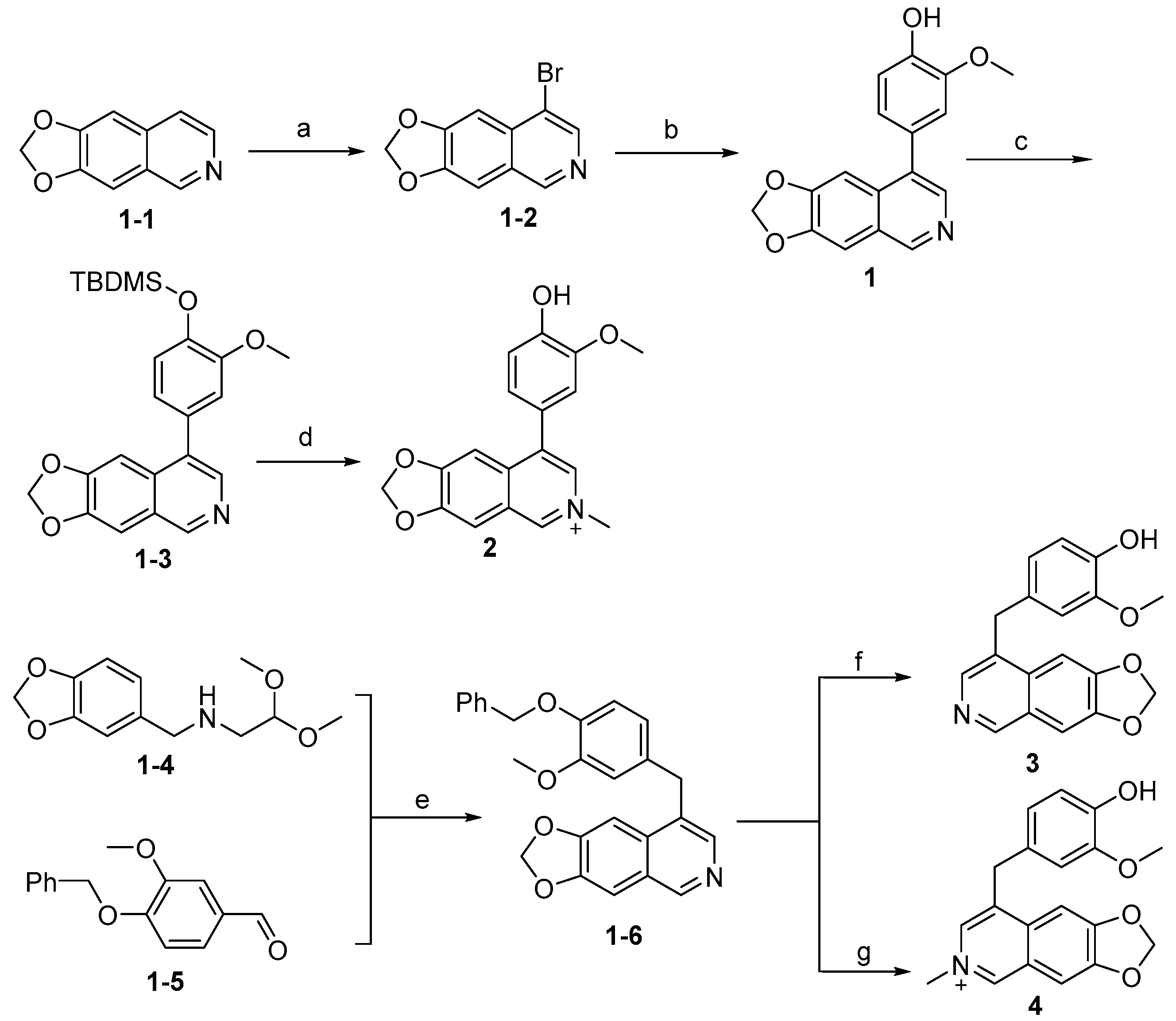
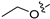3.2. Synthesis of Target Compounds
3.2.1. Synthesis of Intermediate (1-2)
Intermediate (
1-
1) was synthesized as described [
32]. To a solution of (
1-
1) (300 mg, 1.73 mmol) in CCl
4 (10 mL) was added NBS (617 mg, 3.47 mmol), and the reaction mixture was refluxed at 85 °C for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na
2SO
4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (10–20% EA in PE) to give (
1-
2) as a white solid (217 mg, 45%).
1H NMR (400 MHz, Chloroform-d) δ 8.89 (s, 1H), 8.56 (s, 1H), 7.46 (s, 1H), 7.19 (s, 1H), 6.16 (s, 1H). ESI-MS:
m/
z = 252.0 [M + H]
+.
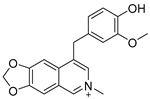
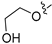
3.2.2. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-methoxyphenol (1)
Method A. To a solution of (1-2) (100 mg, 0.40 mmol) in 1,4-dioxane/H2O (10/1 mL) at room temperature under N2 atmosphere were added (4-hydroxy-3-methoxyphenyl) boronic acid (81 mg, 0.48 mmol), Pd(dppf)Cl2 (30 mg, 0.04 mmol), K2CO3 (110 mg, 0.8 mmol). After stirring at 85 °C for 12 h, the mixture was cooled to room temperature, and the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give 1 as a white solid (89 mg, 75%). M.p. 214–216 °C 1H NMR (500 MHz, Chloroform-d) δ 8.99 (s, 1H), 8.23 (s, 1H), 8.17 (s, 1H), 7.55 (s, 1H), 7.14 (s, 1H), 7.02 (d, J = 2.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.88 (dd, J = 8.0, 2.0 Hz, 1H), 6.20 (s, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ 151.1, 149.0, 148.0, 147.7, 146.5, 141.8, 132.4, 131.8, 127.7, 125.6, 122.2, 115.7, 113.7, 103.2, 102.0, 100.1, 55.7. HRMS (ESI+) calcd for C17H14NO4+ (M + H+): 296.0917, found: 296.0919. HPLC analysis: 9.706 min, 99.6% purity.
3.2.3. Synthesis of Intermediate (1-3)
To a solution of 1 (100 mg, 0.34 mmol) in dry DMF (5 mL) at 0 °C under N2 atmosphere were added imidazole (46 mg, 0.68 mmol) and TBDMSCl (62 mg, 0.41 mmol), and then the mixture was stirred at room temperature for 12 h. Upon completion of the reaction, the resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–15% EA in PE) to give (1-3) as a white solid (125 mg, 90%). 1H NMR (400 MHz, Chloroform-d) δ 8.95 (s, 1H), 8.32 (s, 1H), 7.24 (s, 1H), 7.19 (s, 1H), 6.97 (d, J = 7.9 Hz, 1H), 6.95–6.87 (m, 2H), 6.08 (s, 2H), 3.83 (s, 3H), 1.04 (s, 9H), 0.23 (s, 6H). ESI-MS: m/z = 410.2 [M + H]+.
3.2.4. 8-(4-Hydroxy-3-methoxyphenyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinolin-6-ium (2)
Method B. To a sealed tube containing a solution of (1-3) (125 mg, 0.31 mmol) in acetonitrile (5 mL) was added methyl iodide (66 mg, 0.47 mmol) and the mixture was stirred at 85 °C for 3 h. Subsequently, the reaction mixture was cooled to room temperature followed by evaporation of the volatiles under vacuum. The resulting mixture was dissolved in DCM/TFA (10/5 mL) at room temperature and stirred for 3 h, after which the volatiles were removed in vacuo again. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give 2 as a white solid (24 mg, 25%). 1H NMR (600 MHz, DMSO-d6) δ 9.48 (s, 1H), 8.53 (s, 1H), 8.42 (s, 1H), 7.83 (s, 1H), 7.41 (s, 1H), 7.10 (d, J = 2.0 Hz, 1H), 7.04 (d, J = 8.0 Hz, 1H), 6.97 (dd, J = 8.0, 2.0 Hz, 1H), 6.42 (s, 2H), 4.38 (s, 3H), 3.83 (s, 3H). ESI-MS: m/z = 311.1 [M + H]+.
3.2.5. Synthesis of Intermediate (1-6)
To a solution of (1-5) (1.5 g, 6.27 mmol) in the mixture of EtOH and Conc. HCl (20/20 mL) was added (1-4) (1.5 g, 6.27 mmol). The reaction mixture was stirred at 100 °C for 3 h. Upon completion of the reaction, NaHCO3 (aq) was added to adjust the pH to 8~9, and then the volatiles were evaporated under vacuum. The resulting mixture was diluted with EtOAc (60 mL) and water (30 mL). The organic layer was separated and washed with brine (30 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (5–20% EA in PE) to give (1-6) as a yellow solid (752 mg, 30%). 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 8.14 (s, 1H), 7.88 (s, 1H), 7.47 (s, 1H), 7.29–7.04 (m, 5H), 6.78–6.72 (m, 1H), 6.60 (m, J = 21.1 Hz, 2H), 6.18 (s, 2H), 5.14 (s, 2H), 4.13 (s, 2H), 3.52 (s, 3H). ESI-MS: m/z = 400.2 [M + H]+.
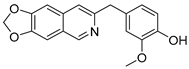
3.2.6. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-ylmethyl)-2-methoxyphenol (3)
To a solution of (1-6) (100 mg, 0.25 mmol) in MeOH (20 mL) was added Pd/C (10 mg). The reaction was stirred under H2 atmosphere at room temperature for 12 h. Upon completion of the reaction, the mixture was filtered to remove the solid residue, and the filtrate was evaporated under vacuum. And then, the resulting mixture was diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give 3 as a white solid (66 mg, 85%). 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 8.75 (s, 1H), 8.23 (s, 1H), 7.46 (s, 1H), 7.37 (s, 1H), 6.88 (d, J = 2.0 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.55 (dd, J = 8.0, 2.0 Hz, 1H), 6.17 (s, 2H), 4.16 (s, 2H), 3.70 (s, 3H). ESI-MS: m/z = 310.1 [M + H]+.
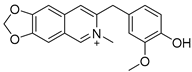
3.2.7. 8-(4-Hydroxy-3-methoxybenzyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinolin-6-ium (4)
Method B. Compound 4 was prepared from the intermediate (1-6) (100 mg, 0.25 mmol) and methyl iodide (53 mg, 0.38 mmol) in a sealed tube using the method for the synthesis of compound 2. This afforded a white solid (16 mg, 20%). 1H NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.98 (s, 1H), 8.45 (s, 1H), 7.74 (s, 1H), 7.72 (s, 1H), 6.97 (d, J = 1.9 Hz, 1H), 6.71–6.58 (m, 2H), 6.39 (s, 2H), 4.35 (s, 3H), 4.31 (s, 2H), 3.73 (s, 3H). ESI-MS: m/z = 324.1 [M + H]+.
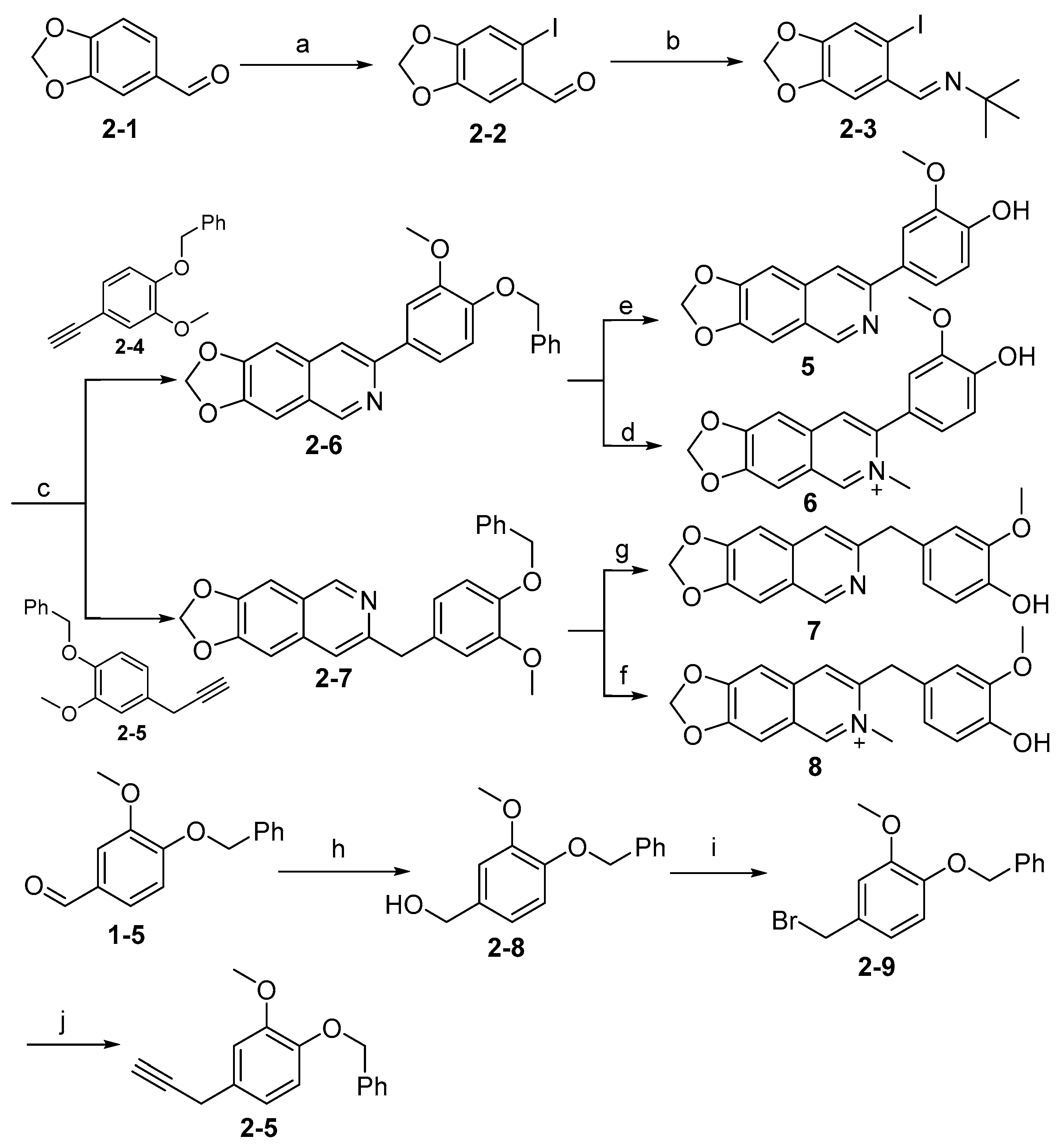
3.2.8. Synthesis of Intermediate (2-2)
To a solution of commercially available intermediate (2-1) (1 g, 6.7 mmol) in dry MeOH (30 mL) were added iodine (1.86 g, 7.3 mmol) and AgCO3 (1.14 g, 6.7 mmol). The reaction was stirred under N2 atmosphere at room temperature for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was diluted with EtOAc (60 mL) and water (30 mL). The organic layer was separated and washed with brine (30 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–20% EA in PE) to give (2-2) as a white solid (1.39 g, 75%). 1H NMR (400 MHz, DMSO-d6) δ 9.79 (s, 1H), 7.60 (s, 1H), 7.26 (s, 1H), 6.18 (s, 2H). ESI-MS: m/z = 276.9 [M + H]+.
3.2.9. Synthesis of Intermediate (2-3)
To a solution of intermediate (2-2) (1.39 g, 5.0 mmol) in water (30 mL) was added tert-butylamine (1.1 g, 15 mmol). The reaction was stirred under N2 atmosphere at room temperature for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was diluted with EtOAc (60 mL) and water (30 mL). The organic layer was separated and washed with brine (30 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford (2-3) as a white solid without further purification. (1.04 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ 8.27 (s, 1H), 7.45 (s, 1H), 7.35 (s, 1H), 6.10 (s, 2H), 1.23 (s, 9H). ESI-MS: m/z = 332.0 [M + H]+.
3.2.10. Synthesis of Intermediate (2-5)
To a solution of CuCl (16 mg, 0.15 mmol) in dry THF (5 mL) was added ethynyl magnesium bromide (126 mg, 0.98 mmol). The reaction mixture was stirred at room temperature for 10 min; intermediate (2-9) (150 mg, 0.49 mmol) was added and refluxed at 75 °C for 21 h. Upon completion of the reaction, the mixture was quenched by addition of 5 mL NH4Cl (aq), followed by dilution with EtOAc (20 mL) and water (5 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give (2-5) as a yellow solid (41 mg, 33%). 1H NMR (400 MHz, Chloroform-d) δ 7.48–7.28 (m, 5H), 6.91 (d, J = 1.6 Hz, 1H), 6.84–6.79 (m, 2H), 5.14 (s, 2H), 3.90 (s, 3H), 3.55 (d, J = 2.7 Hz, 2H), 2.18 (t, J = 2.7 Hz, 1H). ESI-MS: m/z = 253.1 [M + H]+.
3.2.11. Synthesis of Intermediate (2-6)
Intermediate (
2-
4) was synthesized as described [
34]. Dry Et
3N (2 mL), PdCl
2(PPh
3)
2 (21 mg, 0.03 mmol), (
2-
3) (100 mg, 0.3 mmol), (
2-
4) (86 mg, 0.36 mmol), and CuI (3 mg, 0.015 mmol) were placed in a two-neck reaction flask. The reaction was stirred at 55 °C under N
2 atmosphere for 6 h. After the reaction was completed, the reaction mixture was cooled to room temperature, the precipitates were filtered off and washed with ether, and the filtrate was evaporated under reduced pressure. The residue obtained was transferred to a two-dram vial and DMF (5 mL) and CuI (3 mg, 0.015 mmol) were added. The reaction was stirred at 100 °C under N
2 atmosphere for 12 h. Upon completion of the reaction, the mixture was diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na
2SO
4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to afford (
2-
6) as a white solid. (58 mg, 50%).
1H NMR (400 MHz, Chloroform-d) δ 9.03 (s, 1H), 7.82 (s, 1H), 7.73 (d,
J = 2.1 Hz, 1H), 7.53–7.45 (m, 6H), 7.20 (s, 1H), 7.10 (s, 1H), 6.98 (d,
J = 8.3 Hz, 1H), 6.10 (s, 2H), 5.23 (s, 2H), 4.03 (s, 3H). ESI-MS:
m/
z = 386.1 [M + H]
+.
3.2.12. Synthesis of Intermediate (2-7)
Dry Et3N (2 mL), PdCl2(PPh3)2 (21 mg, 0.03 mmol), (2-3) (100 mg, 0.3 mmol), (2-5) (91 mg, 0.36 mmol), and CuI (3 mg, 0.015 mmol) were placed in a two-neck reaction flask. The reaction was stirred at 55 °C under N2 atmosphere for 6 h. After the reaction was completed, the mixture was cooled to room temperature, the precipitates were filtered off and washed with ether, and the filtrate was evaporated under reduced pressure. The residue obtained was transferred to a two-dram vial and DMF (5 mL) and CuI (3 mg, 0.015 mmol) were added. The reaction was stirred at 100 °C under N2 atmosphere for 12 h. Upon completion of the reaction, the mixture was diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to afford (2-7) as a white solid. (64 mg, 53%). 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 7.39 (d, J = 4.1 Hz, 2H), 7.23–7.10 (m, 7H), 6.75 (s, 1H), 6.61 (s, 1H), 6.16 (s, 2H), 3.98 (s, 2H), 3.82 (s, 2H), 3.73 (s, 3H). ESI-MS: m/z = 400.1 [M + H]+.
3.2.13. Synthesis of Intermediate (2-8)
To a solution of (1-5) (1 g, 4.1 mmol) in MeOH (30 mL) at 0 °C was added NaBH4 (312 mg, 8.2 mmol). Subsequently, the reaction was recovered to room temperature and stirred for 6 h. Upon completion of the reaction, the solvent was removed under vacuum. The resulting mixture was then diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give (2-8) as a white solid (941 mg, 94%). 1H NMR (400 MHz, DMSO-d6) δ 7.59–7.32 (m, 5H), 7.01–6.45 (m, 4H), 5.07 (s, 2H), 4.41 (s, 2H), 3.77 (s, 3H). ESI-MS: m/z = 245.1 [M + H]+.
3.2.14. Synthesis of Intermediate (2-9)
To a solution of (2-8) (941 mg, 3.9 mmol) in dry THF (20 mL) at 0 °C under N2 atmosphere, PBr3 (2.1 g, 7.8 mmol) was slowly added dropwise. The reaction mixture was stirred at 0 °C for 0.5 h, then quenched by the addition of 5 mL water. The volatiles were removed under vacuum and the resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% EA in PE) to give (2-9) as a white solid (358 mg, 30%). 1H NMR (400 MHz, Chloroform-d) δ 7.47–7.29 (m, 5H), 6.94 (d, J = 2.0 Hz, 1H), 6.88 (dd, J = 8.2, 2.0 Hz, 1H), 6.82 (d, J = 8.2 Hz, 1H), 5.16 (s, 2H), 4.48 (s, 2H), 3.91 (s, 3H). ESI-MS: m/z = 307.3 [M + H]+.
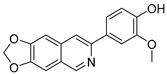
3.2.15. 4-([1,3]Dioxolo[4,5-g]isoquinolin-7-yl)-2-methoxyphenol (5)
To a solution of intermediate (2-6) (58 mg, 0.15 mmol) in MeOH (20 mL) was added Pd/C (10 mg). The reaction was stirred under H2 atmosphere at room temperature for 12 h. Upon completion of the reaction, the precipitates were filtered off and washed with MeOH (10 mL), and the filtrate was evaporated under vacuum. The resulting mixture was diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to afford 5 as a white solid. (37 mg, 83%). 1H NMR (600 MHz, Chloroform-d) δ 9.03 (s, 1H), 7.84 (s, 1H), 7.73 (d, J = 2.0 Hz, 1H), 7.53 (dd, J = 8.2, 2.0 Hz, 1H), 7.20 (s, 1H), 7.11 (s, 1H), 7.02 (d, J = 8.2 Hz, 1H), 6.11 (s, 2H), 4.03 (s, 3H). ESI-MS: m/z = 296.1 [M + H]+.
3.2.16. 7-(4-Hydroxy-3-methoxyphenyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinolin-6-ium (6)
Method B. Compound 6 was prepared from the intermediate (2-6) (100 mg, 0.26 mmol) and methyl iodide (54 mg, 0.39 mmol) in a sealed tube using the method for the synthesis of compound 2. This afforded a white solid (20 mg, 25%). 1H NMR (600 MHz, DMSO-d6) δ 9.61 (s, 1H), 8.48 (s, 1H), 8.19 (s, 1H), 7.74 (s, 1H), 7.64 (s, 1H), 7.23 (s, 1H), 7.07–6.91 (m, 2H), 6.44 (s, 2H), 4.14 (s, 3H), 3.82 (s, 3H). ESI-MS: m/z = 310.1 [M + H]+.
3.2.17. 4-([1,3]Dioxolo[4,5-g]isoquinolin-7-ylmethyl)-2-methoxyphenol (7)
To a solution of intermediate (2-7) (100 mg, 0.25 mmol) in MeOH (20 mL) was added Pd/C (10 mg). The reaction was stirred under H2 atmosphere at room temperature for 12 h. Upon completion of the reaction, the precipitates were filtered off and washed with MeOH (10 mL), and the filtrate was evaporated under vacuum. The resulting mixture was diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to afford 7 as a white solid (58 mg, 75%). 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.74 (s, 1H), 7.41 (s, 2H), 7.25 (s, 1H), 6.86 (d, J = 1.7 Hz, 1H), 6.72–6.62 (m, 2H), 6.17 (s, 2H), 4.02 (s, 2H), 3.71 (s, 3H). ESI-MS: m/z = 310.1 [M + H]+.
3.2.18. 7-(4-Hydroxy-3-methoxybenzyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinolin-6-ium (8)
Method B. Compound 8 was prepared from the intermediate (2-7) (100 mg, 0.25 mmol) and methyl iodide (55 mg, 0.38 mmol) in a sealed tube using the method for the synthesis of compound 2. This afforded a white solid (22 mg, 27%). 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.41 (s, 1H), 7.86 (s, 1H), 7.67 (s, 1H), 7.64 (s, 1H), 6.87 (s, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 6.39 (s, 2H), 4.36 (s, 2H), 4.25 (s, 3H), 3.73 (s, 3H). ESI-MS: m/z = 324.1 [M + H]+.
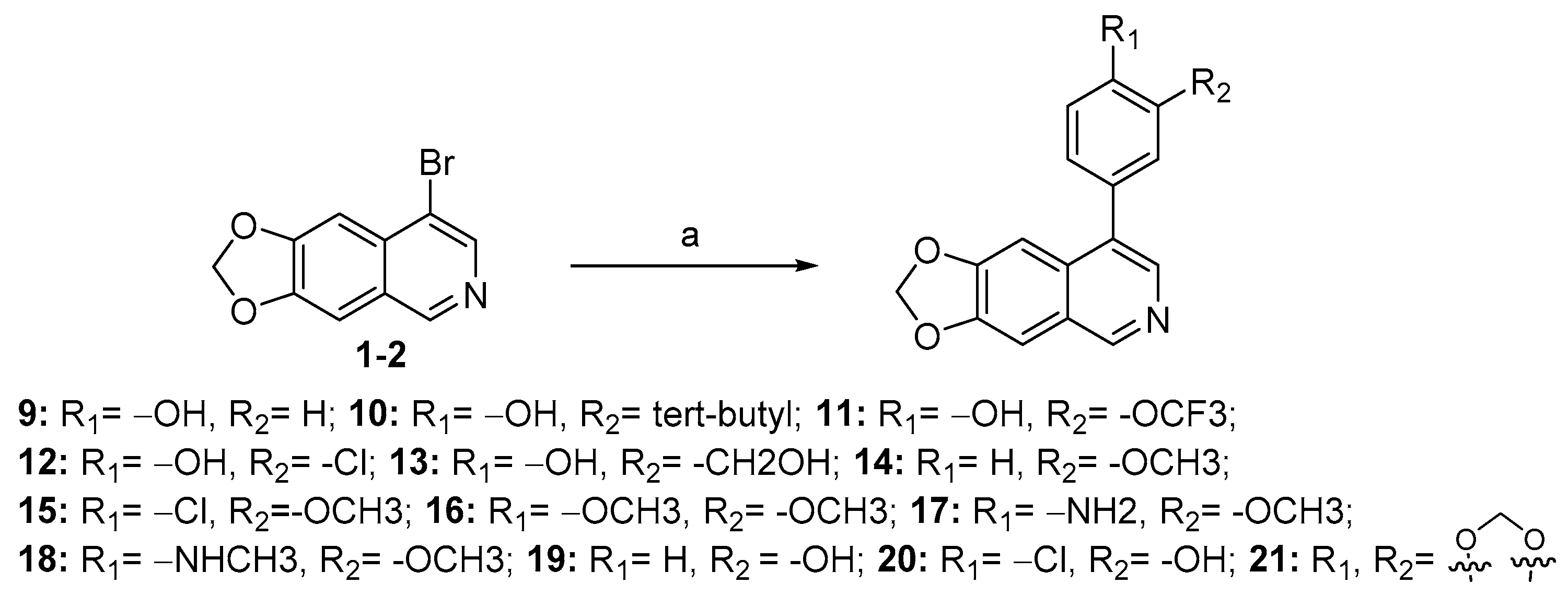
Compounds 9–21 were synthesized from the intermediate (1-2) with various substituted arylboronic acids or arylboronates through Suzuki coupling reaction.
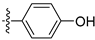
3.2.19. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)phenol (9)
Method A. White solid, yield 76%. 1H NMR (400 MHz, DMSO-d6) δ 9.69 (s, 1H), 8.99 (s, 1H), 8.18 (s, 1H), 7.54 (s, 1H), 7.30 (d, J = 8.5 Hz, 2H), 7.07 (s, 1H), 6.93 (d, J = 8.5 Hz, 2H), 6.20 (s, 2H). ESI-MS: m/z = 265.1 [M + H]+.
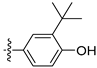
3.2.20. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-(tert-butyl)phenol (10)
Method A. White solid, yield 70%. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (s, 1H), 8.98 (s, 1H), 8.20 (s, 1H), 7.54 (s, 1H), 7.22 (d, J = 2.2 Hz, 1H), 7.15 (dd, J = 8.1, 2.2 Hz, 1H), 7.10 (s, 1H), 6.95 (d, J = 8.1 Hz, 1H), 6.20 (s, 2H), 1.39 (s, 9H). ESI-MS: m/z = 321.1 [M + H]+.
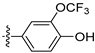
3.2.21. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-(trifluoromethoxy)phenol (11)
Method A. White solid, yield 72%. 1H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 8.22 (s, 2H), 7.57 (s, 1H), 7.38–7.33 (m, 2H), 7.19 (d, J = 8.3 Hz, 1H), 7.05 (s, 1H), 6.21 (s, 2H). ESI-MS: m/z = 349.1 [M + H]+.
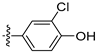
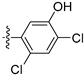
3.2.22. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-chlorophenol (12)
Method A. White solid, yield 60%. 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.01 (s, 1H), 8.20 (s, 1H), 7.55 (s, 1H), 7.44 (d, J = 2.2 Hz, 1H), 7.27 (dd, J = 8.3, 2.2 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 7.05 (s, 1H), 6.21 (s, 2H). ESI-MS: m/z = 299.0 [M + H]+.
3.2.23. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-(hydroxymethyl)phenol (13)
Method A. White solid, yield 65%. 1H NMR (400 MHz, DMSO-d6) δ 9.65 (s, 1H), 8.99 (s, 1H), 8.18 (s, 1H), 7.54 (s, 1H), 7.40 (d, J = 2.3 Hz, 1H), 7.18 (dd, J = 8.1, 2.3 Hz, 1H), 7.12 (s, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.20 (s, 2H), 5.04 (t, J = 5.7 Hz, 1H), 4.56 (d, J = 5.4 Hz, 2H). ESI-MS: m/z = 295.1 [M + H]+.
3.2.24. 8-(3-Methoxyphenyl)-[1,3]dioxolo[4,5-g]isoquinoline (14)
Method A. White solid, yield 80%. 1H NMR (400 MHz, Chloroform-d) δ 8.99 (s, 1H), 8.33 (s, 1H), 7.46–7.36 (m, 1H), 7.19 (s, 1H), 7.02–6.97 (m, 2H), 6.09 (s, 2H), 3.87 (s, 3H). ESI-MS: m/z = 279.1 [M + H]+.
3.2.25. 8-(4-Chloro-3-methoxyphenyl)-[1,3]dioxolo[4,5-g]isoquinoline (15)
Method A. White solid, yield 81%. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.28 (s, 1H), 7.62–7.54 (m, 2H), 7.24 (d, J = 1.9 Hz, 1H), 7.11 (s, 1H), 7.06 (dd, J = 7.9, 1.9 Hz, 1H), 6.22 (s, 2H), 3.90 (s, 3H). ESI-MS: m/z = 313.1 [M + H]+.
3.2.26. 8-(3,4-Dimethoxyphenyl)-[1,3]dioxolo[4,5-g]isoquinoline (16)
Method A. White solid, yield 82%. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.24 (s, 1H), 7.56 (s, 1H), 7.14–7.10 (m, 2H), 7.05 (d, J = 2.0 Hz, 1H), 7.01 (dd, J = 8.1, 2.0 Hz, 1H), 6.21 (s, 2H), 3.83 (s, 3H), 3.80 (s, 3H). ESI-MS: m/z = 309.1 [M + H]+.
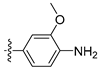
3.2.27. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-methoxyaniline (17)
Method A. White solid, yield 60%. 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.21 (s, 1H), 7.53 (s, 1H), 7.19 (s, 1H), 6.90 (d, J = 1.8 Hz, 1H), 6.83–6.77 (m, 2H), 6.21 (s, 2H), 4.95 (s, 2H), 3.81 (s, 3H). ESI-MS: m/z = 294.1 [M + H]+.
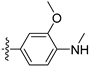
3.2.28. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-methoxy-N-methylaniline (18)
Method A. White solid, yield 63%. 1H NMR (400 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.23 (s, 1H), 7.53 (s, 1H), 7.21 (s, 1H), 7.00–6.89 (m, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.20 (s, 2H), 5.28–5.20 (m, 1H), 3.83 (s, 3H), 2.79 (d, J = 4.3 Hz, 3H). ESI-MS: m/z = 308.1 [M + H]+.
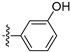
3.2.29. 3-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)phenol (19)
Method A. White solid, yield 70%. M.p. 262–264 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H), 9.03 (s, 1H), 8.21 (s, 1H), 7.56 (s, 1H), 7.34 (t, J = 7.8 Hz, 1H), 7.07 (s, 1H), 6.91–6.81 (m, 3H), 6.21 (s, 2H). 13C NMR (126 MHz, DMSO) δ 157.6, 151.2, 149.5, 148.1, 141.5, 138.1, 132.2, 131.5, 129.9, 125.6, 120.3, 116.5, 114.9, 103.3, 102.1, 99.8. HRMS (ESI+) calcd for C16H12NO3+ (M + H+): 266.0812, found: 266.0812.
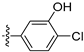
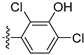
3.2.30. 4-([1,3]Dioxolo[4,5-g]isoquinolin-8-yl)-2-chlorophenol (20)
Method A. White solid, yield 65%. M.p. 229–231 °C. 1H NMR (500 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.21 (s, 1H), 7.57 (s, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.09 (s, 1H), 7.06 (d, J = 2.0 Hz, 1H), 6.92 (dd, J = 8.1, 2.0 Hz, 1H), 6.22 (s, 2H). 13C NMR (126 MHz, DMSO) δ 153.2, 151.4, 149.8, 148.1, 141.5, 136.7, 131.4, 131.2, 130.2, 125.6, 121.3, 119.5, 117.7, 103.4, 102.2, 99.8. HRMS (ESI+) calcd for C16H11ClNO3+ (M + H+): 300.0422, found: 300.0424. HPLC analysis: 12.868 min, 96.5% purity.
3.2.31. 8-(Benzo[d][1,3]dioxol-5-yl)-[1,3]dioxolo[4,5-g]isoquinoline (21)
Method A. White solid, yield 78%. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (s, 1H), 8.21 (s, 1H), 7.56 (s, 1H), 7.12–7.04 (m, 3H), 6.94 (dd, J = 7.9, 1.8 Hz, 1H), 6.21 (s, 2H), 6.12 (s, 2H). ESI-MS: m/z = 293.1 [M + H]+.
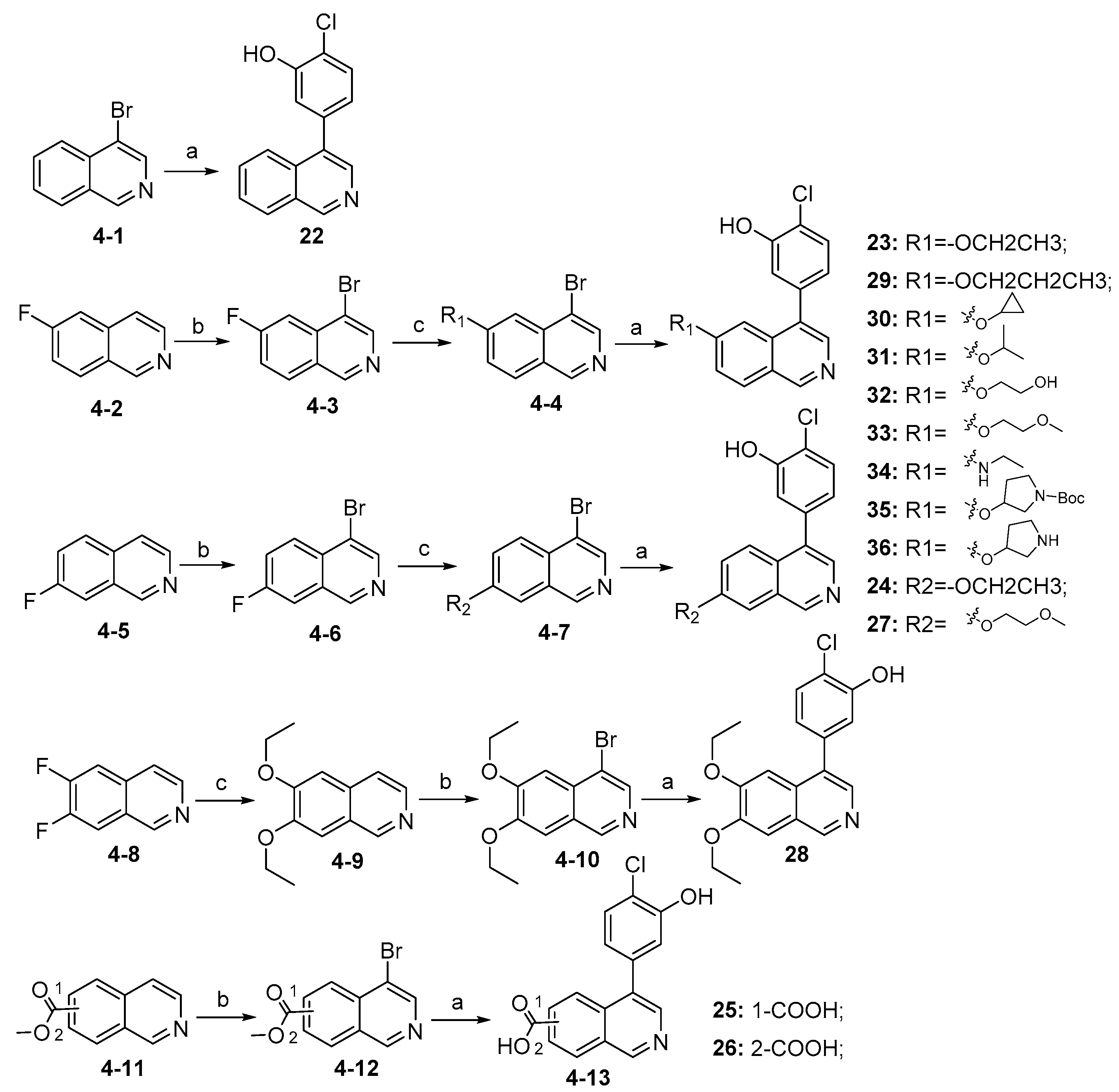
3.2.32. 2-Chloro-5-(isoquinolin-4-yl)phenol (22)
Method A. Compound 22 was synthesized from commercially available reagent (4-1); white solid, yield 70%. 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 9.35 (s, 1H), 8.42 (s, 1H), 8.23 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.85–7.80 (m, 1H), 7.78–7.71 (m, 1H), 7.52 (dd, J = 8.0, 1.1 Hz, 1H), 7.11 (s, 1H), 7.00–6.92 (m, 1H). ESI-MS: m/z = 255.1 [M + H]+.
3.2.33. Synthesis of Intermediate (4-3)
To a solution of commercially available reagent (4-2) (500 mg, 3.4 mmol) in acetic acid (30 mL) was added NBS (787 mg, 4.4 mmol), and the reaction was stirred at 90 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature. The solvent was removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8–9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford (4-3) as a yellow solid (306 mg, 40%). 1H NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.78 (s, 1H), 8.45–8.30 (m, 1H), 7.80–7.63 (m, 2H). ESI-MS: m/z = 226.0 [M + H]+.
3.2.34. Synthesis of Intermediate (4-6)
To a solution of commercially available reagent (4-5) (500 mg, 3.4 mmol) in acetic acid (30 mL) was added NBS (787 mg, 4.4 mmol), and the reaction was stirred at 90 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature. The solvent was removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8–9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford (4-6) as a yellow solid (344 mg, 45%). 1H NMR (400 MHz, DMSO-d6) δ 9.31 (s, 1H), 8.75 (s, 1H), 8.18 (dd, J = 9.3, 5.2 Hz, 1H), 8.07 (dd, J = 9.2, 2.7 Hz, 1H), 7.90 (td, J = 9.1, 2.6 Hz, 1H). ESI-MS: m/z = 226.0 [M + H]+.
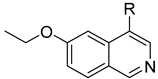
3.2.35. 2-Chloro-5-(6-ethoxyisoquinolin-4-yl)phenol (23)
To a solution of (4-3) (100 mg, 0.44 mmol) in dry EtOH (10 mL) was added NaOEt (60 mg, 0.88 mmol). The reaction was stirred at room temperature for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (10–50% EA in PE) to give 4-bromo-6-ethoxyisoquinoline (4-4-23) as a yellow solid (67 mg, 60%). 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.66 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.42 (dd, J = 8.9, 2.4 Hz, 1H), 7.31 (d, J = 2.4 Hz, 1H), 4.27 (q, J = 7.0 Hz, 2H), 1.43 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 252.0 [M + H]+. Method A. The intermediate (4-4-23) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 23 as a white solid (65%). M.p. 196–198 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.30 (s, 1H), 8.22 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 9.0 Hz, 1H), 7.16–7.10 (m, 2H), 6.99 (d, J = 8.1 Hz, 1H), 4.07 (q, J = 7.0 Hz, 2H), 1.35 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.3, 153.3, 151.3, 142.8, 136.6, 134.9, 130.3, 123.8, 121.3, 120.0, 119.5, 117.7, 102.8, 63.5, 14.4. HRMS (ESI+) calcd for C17H15ClNO2+ (M + H+): 300.0786, found: 300.0788. HPLC analysis: 14.827 min, 99.1% purity.
3.2.36. 2-Chloro-5-(7-ethoxyisoquinolin-4-yl)phenol (24)
To a solution of (4-6) (100 mg, 0.44 mmol) in dry EtOH (10 mL) was added NaOEt (60 mg, 0.88 mmol). The reaction was stirred at room temperature for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (10–50% EA in PE) to give 4-bromo-6-ethoxyisoquinoline (4-7-24) as a white solid (61 mg, 55%). 1H NMR (400 MHz, Chloroform-d) δ 9.23 (s, 1H), 9.14 (s, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.35 (dd, J = 9.0, 2.5 Hz, 1H), 7.21 (d, J = 2.5 Hz, 1H), 4.18 (q, J = 7.0 Hz, 2H), 1.50 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 252.0 [M + H]+. Method A. The intermediate (4-7-24) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 24 as a white solid (60%). 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 9.30 (s, 1H), 7.87 (d, J = 9.2 Hz, 1H), 7.69 (s, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.55–7.48 (m, 1H), 7.17 (s, 1H), 7.07–6.99 (m, 1H), 4.30 (q, J = 7.0 Hz, 2H), 1.51 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 300.07 [M + H]+.
3.2.37. 4-(4-Chloro-3-hydroxyphenyl)isoquinoline-6-carboxylic Acid (25)
To a solution of commercially available reagent methyl isoquinoline-6-carboxylate (200 mg, 1.1 mmol) in acetic acid (30 mL) was added NBS (393 mg, 2.2 mmol); the reaction was stirred at 90 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature. The solvent was removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8–9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford methyl 4-bromoisoquinoline-6-carboxylate (4-12-25) as a yellow solid (88 mg, 30%). 1H NMR (400 MHz, DMSO-d6) δ 9.46 (s, 1H), 8.89 (s, 1H), 8.69 (s, 1H), 8.38 (d, J = 8.6 Hz, 1H), 8.26 (d, J = 8.6 Hz, 1H), 3.98 (s, 3H). ESI-MS: m/z = 266.0 [M + H]+. Method A. The intermediate (4-12-25) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 25 as a white solid (30%). 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H), 9.45 (s, 1H), 8.51 (s, 1H), 8.47 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.18 (d, J = 8.5 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.12 (d, J = 2.1 Hz, 1H), 6.99 (dd, J = 8.1, 2.1 Hz, 1H). ESI-MS: m/z = 300.0 [M + H]+.
3.2.38. 4-(4-Chloro-3-hydroxyphenyl)isoquinoline-7-carboxylic Acid (26)
To a solution of commercially available reagent methyl 4-bromoisoquinoline-7-carboxylate (200 mg, 1.1 mmol) in acetic acid (30 mL) was added NBS (393 mg, 2.2 mmol); the reaction was stirred at 90 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature. The solvent was removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8–9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford methyl 4-bromoisoquinoline-6-carboxylate (4-12-26) as a yellow solid (103 mg, 35%). 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H), 8.92 (s, 1H), 8.90 (s, 1H), 8.40 (d, J = 8.9 Hz, 1H), 8.22 (d, J = 8.9 Hz, 1H), 3.96 (s, 3H). ESI-MS: m/z = 266.0 [M + H]+. Method A. The intermediate (4-12-26) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 26 as a white solid (33%). 1H NMR (400 MHz, DMSO-d6) δ 10.52 (s, 1H), 9.53 (s, 1H), 8.87 (d, J = 1.7 Hz, 1H), 8.52 (s, 1H), 8.24 (dd, J = 8.9, 1.8 Hz, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.11 (d, J = 2.0 Hz, 1H), 6.99 (dd, J = 8.1, 2.0 Hz, 1H). ESI-MS: m/z = 300.0 [M + H]+.
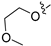
3.2.39. 2-Chloro-5-(7-(2-methoxyethoxy)isoquinolin-4-yl)phenol (27)
Method C. To a solution of NaH (41 mg, 1.7 mmol) in dry DMF at 0 °C was added 2-methoxyethan-1-ol (100 mg, 1.3 mmol). After stirring for 0.5h at the same temperature, the intermediate (4-6) (326 mg, 1.4 mmol) was added followed by recovery to room temperature and stirring for 12 h. Upon completion of the reaction, cold water (5 mL) was added to quench the mixture, and EtOAc (30 mL) and water (15 mL) were added. The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford 4-bromo-7-(2-methoxyethoxy)isoquinoline (4-7-27) as a yellow solid (257 mg, 65%). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.59 (s, 1H), 8.07–7.99 (m, 2H), 7.67–7.59 (m, 1H), 4.29 (t, J = 4.6 Hz, 2H), 3.75 (t, J = 4.6 Hz, 2H), 3.35 (s, 3H). ESI-MS: m/z = 282.0 [M + H]+. Method A. The intermediate (4-7-27) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 27 as a white solid (70%). 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.36 (s, 1H), 8.26 (s, 1H), 7.78 (d, J = 9.2 Hz, 1H), 7.62 (d, J = 2.7 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.46 (dd, J = 9.2, 2.7 Hz, 1H), 7.08 (d, J = 2.1 Hz, 1H), 6.94 (dd, J = 8.1, 2.1 Hz, 1H), 4.28 (t, J = 4.0 Hz, 2H), 3.75 (t, J = 4.0 Hz, 2H), 3.34 (s, 3H). ESI-MS: m/z = 330.1 [M + H]+
3.2.40. Synthesis of Intermediate (4-9)
To a solution of commercially available (4-8) (100 mg, 0.61 mmol) in dry EtOH (10 mL) was added NaOEt (92 mg, 1.34 mmol). The reaction was stirred at room temperature for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–20% EA in PE) to give (4-9) as a white solid (100 mg, 75%). 1H NMR (400 MHz, Chloroform-d) δ 9.01 (s, 1H), 8.35 (d, J = 5.7 Hz, 1H), 7.48 (d, J = 5.7 Hz, 1H), 7.19 (s, 1H), 7.05 (s, 1H), 4.28–4.16 (m, 4H), 1.56 (td, J = 7.0, 1.4 Hz, 6H). ESI-MS: m/z = 218.1 [M + H]+.
3.2.41. Synthesis of Intermediate (4-10)
To a solution of intermediate (4-9) (100 mg, 0.45 mmol) in acetic acid (30 mL) was added NBS (122 mg, 0.68 mmol); the reaction was stirred at 90 °C for 12 h. Upon completion of the reaction, the mixture was cooled to room temperature. The solvent was removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8–9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure to afford (4-10) as a yellow solid (60 mg, 45%). 1H NMR (400 MHz, Chloroform-d) δ 9.05 (s, 1H), 8.48 (d, J = 5.9 Hz, 1H), 7.90 (d, J = 5.9 Hz, 1H), 7.23 (s, 1H), 4.29–4.21 (m, 4H), 1.53–1.45 (m, 6H). ESI-MS: m/z = 296.0 [M + H]+.
3.2.42. 2-Chloro-5-(7-(2-methoxyethoxy)isoquinolin-4-yl)phenol (28)
Method A. The intermediate (4-10) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 28 as a white solid (72%). M.p. 195–197 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.32 (s, 1H), 9.16 (s, 1H), 8.25 (d, J = 5.6 Hz, 1H), 7.64 (s, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.16 (d, J = 5.6 Hz, 1H), 6.90 (s, 1H), 6.76 (d, J = 8.4 Hz, 1H), 4.26 (q, J = 7.0 Hz, 2H), 3.95 (q, J = 7.0 Hz, 2H), 1.46 (t, J = 7.0 Hz, 3H), 1.06 (t, J = 7.0 Hz, 3H). HRMS (ESI+) calcd for C18H16NO4+ (M + H+): 310.1074, found: 310.1072.
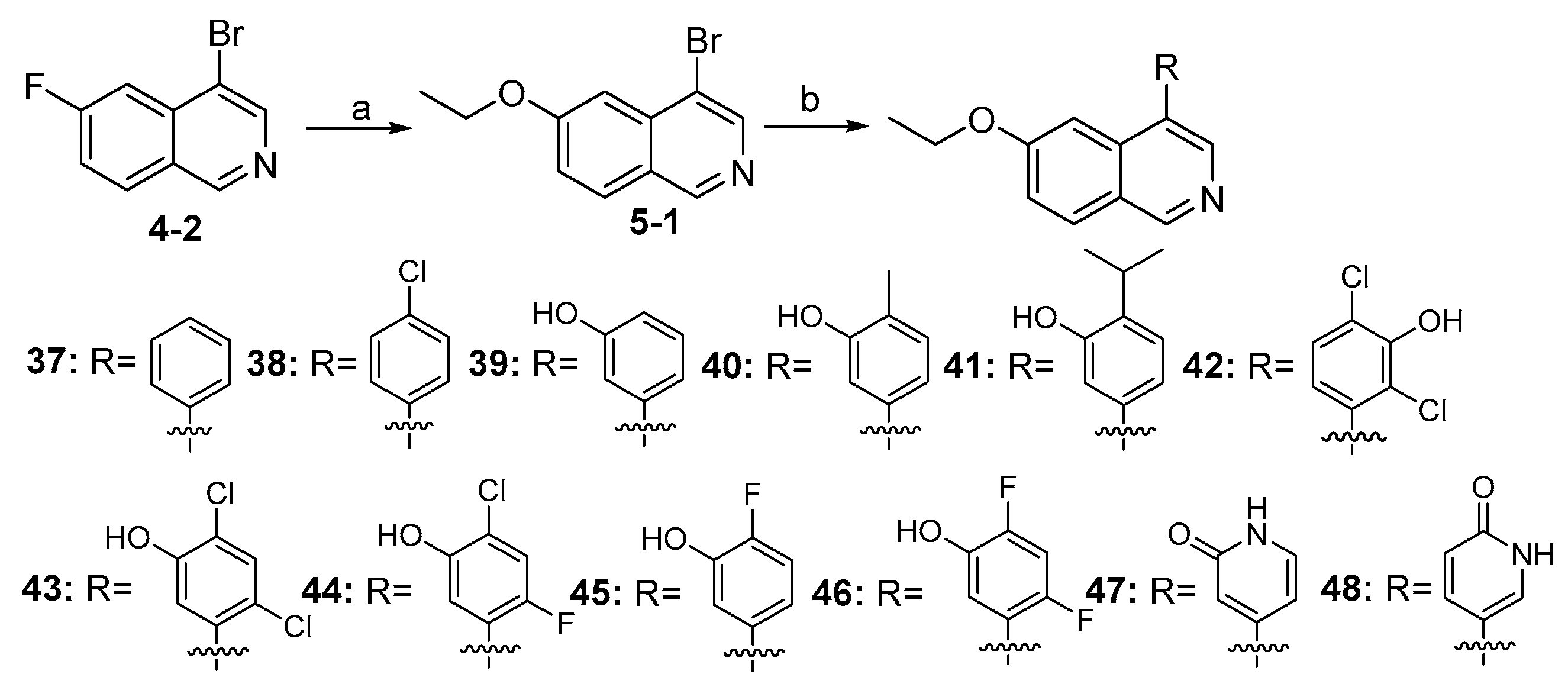
3.2.43. 2-Chloro-4-(6-propoxyisoquinolin-4-yl)phenol (29)
Method C. Intermediate 4-bromo-6-propoxyisoquinoline (4-4-29) was obtained as a white solid (yield 60%). 1H NMR (400 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.66 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.43 (dd, J = 9.0, 2.4 Hz, 1H), 7.31 (d, J = 2.4 Hz, 1H), 4.17 (t, J = 6.5 Hz, 2H), 1.90–1.76 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H). ESI-MS: m/z = 266.0 [M + H]+. Method A. The intermediate (4-4-29) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 29 as a white solid (70%). 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.33 (s, 1H), 8.30 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.37 (dd, J = 9.0, 2.4 Hz, 1H), 7.16–7.11 (m, 2H), 6.98 (dd, J = 8.1, 2.1 Hz, 1H), 3.98 (t, J = 6.4 Hz, 2H), 1.79–1.72 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). ESI-MS: m/z = 314.1 [M + H]+.
3.2.44. 2-Chloro-4-(6-cyclopropoxyisoquinolin-4-yl)phenol (30)
Method C. Intermediate 4-bromo-6-cyclopropoxyisoquinoline (4-4-30) was obtained as a white solid (yield 50%). 1H NMR (400 MHz, Chloroform-d) δ 9.01 (s, 1H), 8.64 (s, 1H), 7.85 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.30–7.22 (m, 1H), 3.97–3.90 (m, 1H), 0.97–0.91 (m, 2H), 0.89–0.80 (m, 2H). ESI-MS: m/z = 264.0 [M + H]+. Method A. The intermediate (4-4-30) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 30 as a white solid (65%). 1H NMR (400 MHz, DMSO-d6) δ 10.48 (s, 1H), 9.19 (s, 1H), 8.33 (s, 1H), 8.15 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 2.4 Hz, 1H), 7.39 (dd, J = 8.9, 2.4 Hz, 1H), 7.14 (d, J = 2.1 Hz, 1H), 7.01 (dd, J = 8.1, 2.1 Hz, 1H), 3.95–3.86 (m, 1H), 0.81–0.76 (m, 2H), 0.73–0.68 (m, 2H). ESI-MS: m/z = 312.1 [M + H]+.
3.2.45. 2-Chloro-4-(6-isopropoxyisoquinolin-4-yl)phenol (31)
Method C. Intermediate 4-bromo-6-isopropoxyisoquinoline (4-4-31) was obtained as a white solid (yield 55%). 1H NMR (400 MHz, Chloroform-d) δ 9.01 (s, 1H), 8.64 (s, 1H), 7.85 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.30–7.22 (m, 1H), 4.78–4.68 (m, 1H), 1.42 (d, J = 6.0 Hz, 2H). ESI-MS: m/z = 266.0 [M + H]+. Method A. The intermediate (4-4-31) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 31 as a white solid (70%). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 9.16 (s, 1H), 8.29 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.56–7.48 (m, 1H), 7.38–7.30 (m, 1H), 7.10 (s, 2H), 6.97 (d, J = 7.2 Hz, 1H), 4.69–4.60 (m, 1H), 1.29 (d, J = 5.9 Hz, 6H). ESI-MS: m/z = 314.1 [M + H]+.
3.2.46. 2-Chloro-4-(6-(2-hydroxyethoxy)isoquinolin-4-yl)phenol (32)
Method C. Intermediate 2-((4-bromoisoquinolin-6-yl)oxy)ethan-1-ol (4-4-32) was obtained as a white solid (yield 35%). 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.68 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.44 (dd, J = 9.0, 2.4 Hz, 1H), 7.33 (d, J = 2.4 Hz, 1H), 5.02 (s, 1H), 4.23 (t, J = 4.8 Hz, 2H), 3.82 (t, J = 4.8 Hz, 2H). ESI-MS: m/z = 268.0 [M + H]+. Method A. The intermediate (4-4-32) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 32 as a white solid (50%). 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.31 (s, 1H), 8.19 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.39 (dd, J = 9.0, 2.4 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.13 (d, J = 2.1 Hz, 1H), 6.99 (dd, J = 8.1, 2.1 Hz, 1H), 4.92 (s, 1H), 4.04 (t, J = 4.9 Hz, 2H), 3.74 (t, J = 4.9 Hz, 2H). ESI-MS: m/z = 316.1 [M + H]+.
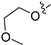
3.2.47. 2-Chloro-4-(6-(2-methoxyethoxy)isoquinolin-4-yl)phenol (33)
Method C. Intermediate 4-bromo-6-(2-methoxyethoxy)isoquinoline (4-4-33) was obtained as a white solid (yield 50%). 1H NMR (400 MHz, Chloroform-d) δ 9.02 (s, 1H), 8.65 (s, 1H), 7.87 (d, J = 8.9 Hz, 1H), 7.39 (d, J = 2.6 Hz, 2H), 7.34 (dd, J = 8.9, 2.6 Hz, 1H), 4.41–4.27 (m, 2H), 3.96–3.76 (m, 2H), 3.50 (s, 3H). ESI-MS: m/z = 282.0 [M + H]+. Method A. The intermediate (4-4-33) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 33 as a white solid (55%). 1H NMR (400 MHz, DMSO-d6) δ 10.42 (s, 1H), 9.18 (s, 1H), 8.31 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.1 Hz, 1H), 7.39 (dd, J = 9.0, 2.4 Hz, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.13 (d, J = 2.1 Hz, 1H), 6.99 (dd, J = 8.1, 2.1 Hz, 1H), 4.22–4.07 (m, 2H), 3.72–3.57 (m, 2H), 3.30 (s, 3H). ESI-MS: m/z = 330.1 [M + H]+.
3.2.48. 2-Chloro-5-(6-(ethylamino)isoquinolin-4-yl)phenol (34)
Method C. Intermediate 4-bromo-6-(2-methoxyethoxy)isoquinoline (4-4-34) was obtained as a white solid (yield 52%). M.p. 215-217 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.42 (s, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.13 (dd, J = 8.9, 2.2 Hz, 1H), 6.92 (t, J = 5.0 Hz, 1H), 6.66 (d, J = 2.2 Hz, 1H), 3.23–3.09 (m, 2H), 1.25 (t, J = 7.2 Hz, 3H). ESI-MS: m/z = 251.0 [M + H]+. Method A. The intermediate (4-4-34) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 34 as a white solid (54%). 1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.86 (s, 1H), 8.06 (s, 1H), 7.83 (d, J = 8.9 Hz, 1H), 7.46 (d, J = 8.1 Hz, 1H), 7.13–6.98 (m, 2H), 6.93 (dd, J = 8.1, 2.0 Hz, 1H), 6.63–6.47 (m, 2H), 3.07–2.92 (m, 2H), 1.16 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 153.6, 151.1, 151.0, 143.0, 138.0, 136.2, 130.5, 129.7, 129.5, 122.2, 121.7, 119.4, 119.0, 118.1, 97.9, 37.4, 14.3. HRMS (ESI+) calcd for C17H16ClN2O+ (M + H+): 299.0947, found: 299.0946.
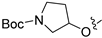
3.2.49. Tert-Butyl 3-((4-(4-chloro-3-hydroxyphenyl)isoquinolin-6-yl)oxy)pyrrolidine-1-carboxylate (35)
Method C. Intermediate tert-butyl 3-((4-bromoisoquinolin-6-yl)oxy)pyrrolidine-1-carboxylate (4-4-35) was obtained as a white solid (yield 50%). 1H NMR (400 MHz, Chloroform-d) δ 9.02 (s, 1H), 8.65 (s, 1H), 7.88 (d, J = 8.9 Hz, 1H), 7.33 (d, J = 2.3 Hz, 1H), 7.27–7.18 (m, 1H), 5.14 (s, 1H), 3.67–3.54 (m, 2H), 2.39–2.19 (m, 2H), 1.80–1.62 (m, 2H), 1.47 (s, 9H). ESI-MS: m/z = 393.1 [M + H]+. Method A. The intermediate (4-4-35) was reacted with (4-chloro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 35 as a white solid (50%). 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H), 9.20 (s, 1H), 8.32 (s, 1H), 8.17 (d, J = 9.0 Hz, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.41 (dd, J = 9.1, 2.1 Hz, 1H), 7.13–7.07 (m, 2H), 6.97 (dd, J = 8.2, 2.1 Hz, 1H), 5.16–4.92 (m, 1H), 3.59–3.51 (m, 1H), 3.45–3.37 (m, 3H), 2.19–1.98 (m, 2H), 1.39 (s, 9H). ESI-MS: m/z = 441.2 [M + H]+.
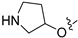
3.2.50. 2-Chloro-5-(6-(pyrrolidin-3-yloxy)isoquinolin-4-yl)phenol (36)
To a solution of 35 (100 mg, 0.23 mmol) in EtOAc (10 mL) at 0 °C was added 5 mL of HCl (aq, 4M in EtOAc). The reaction was stirred at room temperature for 2 h. Upon completion of the reaction, the volatiles were removed in vacuo, and NaHCO3 (aq) was added to adjust the pH to 8~9. The resulting mixture was diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (15 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–5% MeOH in DCM) to give 36 as a white solid (71 mg, 90%). 1H NMR (400 MHz, DMSO-d6) δ 9.68 (s, 1H), 9.60–9.48 (m, 2H), 8.53 (s, 1H), 7.69 (dd, J = 9.2, 2.3 Hz, 1H), 7.57 (d, J = 8.1 Hz, 1H), 7.30–7.25 (m, 2H), 7.04 (dd, J = 8.1, 2.0 Hz, 1H), 5.45–5.38 (m, 1H), 3.83–3.74 (m, 2H), 3.18–3.12 (m, 2H), 2.33–2.12 (m, 2H). ESI-MS: m/z = 341.2 [M + H]+.
3.2.51. 6-Ethoxy-4-phenylisoquinoline (37)
Intermediate (5-1) was prepared as previously described for the synthesis of compound 23. Method A. The intermediate (5-1) was reacted with phenylboronic acid through Suzuki coupling reaction to afford 37 as a white solid (80%). M.p. 56–58 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.33 (s, 1H), 8.14 (d, J = 8.9 Hz, 1H), 7.62–7.51 (m, 5H), 7.36 (dd, J = 9.0, 2.3 Hz, 1H), 7.10 (d, J = 2.3 Hz, 1H), 4.03 (q, J = 6.9 Hz, 2H), 1.33 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.2, 151.1, 143.0, 136.7, 135.1, 131.6, 130.1, 129.7, 128.8, 128.0, 123.8, 119.9, 102.9, 63.4, 14.4. HRMS (ESI+) calcd for C17H16NO+ (M + H+): 250.1226, found: 250.1229.
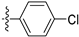
3.2.52. 4-(4-Chlorophenyl)-6-ethoxyisoquinoline (38)
Method A. The intermediate (5-1) was reacted with (4-chlorophenyl)boronic acid through Suzuki coupling reaction to afford 38 as a white solid (82%). M.p. 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.33 (d, J = 2.0 Hz, 1H), 8.15 (dd, J = 9.0, 2.0 Hz, 1H), 7.68–7.56 (m, 4H), 7.37 (d, J = 9.0 Hz, 1H), 7.07 (s, 1H), 4.06 (q, J = 6.9 Hz, 2H), 1.35 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.3, 151.4, 143.1, 135.6, 134.9, 132.8, 131.5, 130.4, 130.2, 128.9, 123.8, 120.0, 102.6, 63.5, 14.4. HRMS (ESI+) calcd for C17H15ClNO+ (M + H+): 284.0837, found: 284.0838.
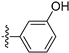
3.2.53. 3-(6-Ethoxyisoquinolin-4-yl)phenol (39)
Method A. The intermediate (5-1) was reacted with (3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 39 as a white solid (65%). M.p. 210–212 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.66 (s, 1H), 9.16 (s, 1H), 8.30 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.41–7.28 (m, 2H), 7.17 (s, 1H), 6.98–6.85 (m, 3H), 4.06 (q, J = 7.2 Hz, 2H), 1.39–1.27 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.1, 157.6, 151.0, 142.7, 138.0, 135.1, 131.6, 130.1, 129.9, 123.8, 120.3, 119.8, 116.4, 114.9, 103.1, 63.4, 14.4. HRMS (ESI+) calcd for C17H16NO2+ (M + H+): 266.1176, found: 266.1177.
3.2.54. 5-(6-Ethoxyisoquinolin-4-yl)-2-methylphenol (40)
Method A. The intermediate (5-1) was reacted with (3-hydroxy-4-methylphenyl)boronic acid through Suzuki coupling reaction to afford 40 as a white solid (63%). M.p. 199–201 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 9.14 (s, 1H), 8.27 (s, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.34 (dd, J = 9.0, 2.3 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 7.20 (s, 1H), 6.95 (s, 1H), 6.88 (d, J = 7.7 Hz, 1H), 4.05 (q, J = 6.9 Hz, 2H), 1.35 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.5, 156.0, 151.2, 143.2, 135.6, 135.5, 132.1, 131.5, 130.5, 124.3, 124.2, 120.6, 120.3, 116.2, 103.5, 63.9, 16.3, 14.9. HRMS (ESI+) calcd for C18H18NO2+ (M + H+): 280.1332, found: 280.1333.
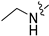
3.2.55. Synthesis of 5-Bromo-2-isopropylphenol (41-a)
To a solution of 4-bromo-1-isopropyl-2-methoxybenzene (100 mg, 0.44 mmol) in dry DCM (10 mL) at 0 °C was added BBr3 (133 mg, 0.53 mmol), and then the reaction was stirred at room temperature for 5 h. Upon completion of the reaction, the mixture was poured into ice-cold water (10 mL), and the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–30% EA in PE) to give (41-a) as a white solid (43 mg, 45%). 1H NMR (400 MHz, DMSO-d6) δ 9.76 (d, J = 1.4 Hz, 1H), 7.04 (dd, J = 8.1, 1.4 Hz, 1H), 6.98–6.86 (m, 2H), 3.21–3.00 (m, 1H), 1.13 (d, J = 1.4 Hz, 6H). ESI-MS: m/z = 214.0 [M + H]+.
3.2.56. Synthesis of 2-Isopropyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (41-b)
To a solution of (41-a) (43 mg, 0.2 mmol) in 1,4-dioxane (10 mL) at room temperature under N2 atmosphere were added 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) (61 mg, 0.24 mmol), Pd(dppf)Cl2 (15 mg, 0.02 mmol), potassium acetate (39 mg, 0.4 mmol). After stirring at 85 °C for 12 h, the reaction mixture was cooled to room temperature, and the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–30% EA in PE) to give 41-b as a white solid (34 mg, 65%). 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 7.14–7.05 (m, 3H), 3.26–3.12 (m, 1H), 1.26 (s, 12H), 1.14 (d, J = 6.8 Hz, 6H). ESI-MS: m/z = 285.2 [M + Na]+.
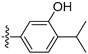
3.2.57. 5-(6-Ethoxyisoquinolin-4-yl)-2-isopropylphenol (41)
Method A. The intermediate (5-1) was reacted with (41-b) through Suzuki coupling reaction to afford 41 as a white solid (60%). 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 9.14 (s, 1H), 8.29 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.36 (dd, J = 9.0, 2.3 Hz, 1H), 7.29 (d, J = 7.7 Hz, 1H), 7.24 (d, J = 2.3 Hz, 1H), 7.01–6.87 (m, 2H), 4.07 (q, J = 6.9 Hz, 2H), 3.28–3.22 (m, 1H), 1.36 (t, J = 6.9 Hz, 3H), 1.24 (d, J = 6.9 Hz, 6H). ESI-MS: m/z = 308.2 [M + H]+.
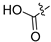
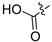
3.2.58. Synthesis of (2,4-Dichloro-3-hydroxyphenyl)boronic Acid (42-a)
To a solution of (4-chloro-3-hydroxyphenyl)boronic acid (200 mg, 1.16 mmol) in acetonitrile (20 mL) was added N-chlorosuccinimide (186 mg, 1.39 mmol); the reaction was stirred at 80 °C for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–30% EA in PE) to give (42-a) as a yellow solid (65 mg, 27%). 1H NMR (400 MHz, DMSO-d6) δ 9.82 (s, 1H), 8.31 (s, 2H), 7.27 (d, J = 7.9 Hz, 1H), 6.84 (d, J = 7.9 Hz, 1H). ESI-MS: m/z = 207.0 [M + H]+.
3.2.59. 2,6-Dichloro-3-(6-ethoxyisoquinolin-4-yl)phenol (42)
Method A. The intermediate (5-1) was reacted with (42-a) through Suzuki coupling reaction to afford 42 as a white solid (60%). 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.25 (s, 1H), 8.13 (d, J = 9.0 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.34 (dd, J = 9.0, 2.5 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.66 (d, J = 2.5 Hz, 1H), 3.98 (q, J = 6.9 Hz, 2H), 1.32 (t, J = 6.9 Hz, 3H). ESI-MS: m/z = 334.0 [M + H]+.
3.2.60. Synthesis of (2,4-Dichloro-5-hydroxyphenyl)boronic Acid (43-a)
To a solution of (4-chloro-3-hydroxyphenyl)boronic acid (200 mg, 1.16 mmol) in acetonitrile (20 mL) was added N-chlorosuccinimide (186 mg, 1.39 mmol); the reaction was stirred at 80 °C for 12 h. Upon completion of the reaction, the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (30 mL) and water (15 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–30% EA in PE) to give (43-a) as a yellow solid (55 mg, 23%). 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.31 (s, 2H), 7.34 (s, 1H), 6.97 (s, 1H). ESI-MS: m/z = 207.0 [M + H]+.
3.2.61. 2,4-Dichloro-5-(6-ethoxyisoquinolin-4-yl)phenol (43)
Method A. The intermediate (5-1) was reacted with (43-a) through Suzuki coupling reaction to afford 43 as a white solid (55%). M.p. 198–200 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.85 (s, 1H), 9.22 (s, 1H), 8.27 (s, 1H), 8.14 (d, J = 8.9 Hz, 1H), 7.69 (s, 1H), 7.35 (dd, J = 8.9, 2.4 Hz, 1H), 7.06 (s, 1H), 6.67 (d, J = 2.4 Hz, 1H), 4.02 (q, J = 7.0, 2H), 1.32 (t, J = 7.0 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.7, 152.9, 152.3, 143.6, 135.7, 135.3, 130.6, 129.1, 124.0, 123.2, 121.1, 120.6, 119.9, 103.4, 64.08, 14.8. HRMS (ESI+) calcd for C17H14Cl2NO2+ (M + H+): 334.0396, found: 334.0397.
3.2.62. Synthesis of (4-Chloro-2-fluoro-5-hydroxyphenyl)boronic Acid (44-a)
To a solution of (4-chloro-2-fluoro-5-methoxyphenyl)boronic acid (100 mg, 0.49 mmol) in dry DCM (10 mL) at 0 °C was added BBr3 (160 mg, 0.64 mmol); the reaction was stirred at room temperature for 5 h. Upon completion of the reaction, the mixture was poured into ice-cold water (10 mL), and the volatiles were removed in vacuo. The resulting mixture was then diluted with EtOAc (20 mL) and water (10 mL). The organic layer was separated and washed with brine (10 mL × 2), dried over Na2SO4, filtrated, and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (0–30% EA in PE) to give (44-a) as a yellow solid (71 mg, 76%). 1H NMR (400 MHz, DMSO-d6) δ 9.94 (s, 1H), 8.19 (s, 2H), 7.15 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 5.5 Hz, 1H). ESI-MS: m/z = 191.0 [M + H]+.
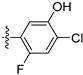
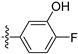
3.2.63. 2-Chloro-5-(6-ethoxyisoquinolin-4-yl)-4-fluorophenol (44)
Method A. The intermediate (5-1) was reacted with (44-a) through Suzuki coupling reaction to afford 44 as a yellow solid (60%). 1H NMR (400 MHz, Chloroform-d) δ 9.10 (s, 1H), 8.44 (s, 1H), 7.93 (d, J = 9.1 Hz, 1H), 7.37–7.20 (m, 3H), 7.09 (d, J = 6.5 Hz, 1H), 6.91 (s, 1H), 4.06 (q, J = 7.0 Hz, 2H), 1.44 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 318.1 [M + H]+.
3.2.64. 5-(6-Ethoxyisoquinolin-4-yl)-2-fluorophenol (45)
Method A. The intermediate (5-1) was reacted with (4-fluoro-3-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 45 as a white solid (65%). M.p. 209–211 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.16 (s, 1H), 8.29 (s, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.35 (dd, J = 9.0, 2.6 Hz, 1H), 7.31 (dd, 3JF-H = 11.4 Hz, 3JH-H = 8.4 Hz, 1H), 7.12 (d, J = 2.6 Hz, 1H), 7.09 (dd, 4JF-H = 9.5 Hz, 4JH-H = 2.2 Hz, 1H), 6.95 (ddd, 3JH-H = 8.3 Hz, 4JF-H = 4.3 Hz, 4JH-H = 2.2 Hz, 1H), 4.06 (q, J = 6.9 Hz, 2H), 1.35 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 160.2, 151.1, 150.8 (d, 1J C-F = 240.3 Hz), 145.0 (d, 2J C-F = 12.6 Hz), 142.8, 135.1, 133.2, 130.8, 130.1, 123.8, 120.7, 119.9, 119.0, 116.5 (d, 2J C-F = 17.6 Hz), 102.9, 63.5, 14.4. HRMS (ESI+) calcd for C17H15FNO2+ (M + H+): 284.1081, found: 284.1081.
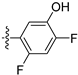
3.2.65. 5-(6-Ethoxyisoquinolin-4-yl)-2,4-difluorophenol (46)
Method A. The intermediate (5-1) was reacted with (2,4-difluoro-5-hydroxyphenyl)boronic acid through Suzuki coupling reaction to afford 46 as a white solid (45%). M.p. 230–232 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.32 (s, 1H), 8.28 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.41 (dd, 3JF-H = 11.2, 9.6 Hz, 1H), 7.36 (dd, J = 9.0, 2.4 Hz, 1H), 7.03 (dd, 4JF-H = 9.7, 7.3 Hz, 1H), 6.83 (d, J = 2.5 Hz, 1H), 4.05 (q, J = 6.9 Hz, 2H), 1.34 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 160.3, 151.6 (dd, 1JC-F = 132.3 Hz, 2JC-F = 10.1 Hz), 151.8, 150.5 (dd, 1JC-F = 205.4 Hz, 2JC-F = 10.1 Hz), 143.7, 141.9 (dd, 2J C-F = 12.2 Hz, 3JC-F = 2.8 Hz), 135.5, 130.2, 125.4, 123.6, 120.1, 119.7 (dd, 2J = 17.7 Hz, 3J = 3.4 Hz), 119.5, 105.1 (dd, 2JC-F = 22.7 Hz, 3JC-F = 18.9 Hz), 102.9, 63.6, 14.4. HRMS (ESI+) calcd for C17H14F2NO2+ (M + H+): 302.0987, found: 302.0986. HPLC analysis: 14.260 min, 99.5% purity.
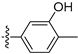
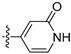
3.2.66. 4-(6-Ethoxyisoquinolin-4-yl)pyridin-2(1H)-one (47)
Method A. The intermediate (5-1) was reacted with (2-oxo-1,2-dihydropyridin-4-yl)boronic acid through Suzuki coupling reaction to afford 47 as a white solid (50%). 1H NMR (400 MHz, DMSO-d6) δ 11.79 (s, 1H), 9.21 (s, 1H), 8.35 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.51 (d, J = 6.6 Hz, 1H), 7.38 (dd, J = 9.0, 2.4 Hz, 1H), 7.14 (d, J = 2.4 Hz, 1H), 6.45 (d, J = 1.7 Hz, 1H), 6.37 (dd, J = 6.6, 1.7 Hz, 1H), 4.12 (q, J = 6.9 Hz, 2H), 1.37 (t, J = 6.9 Hz, 3H). ESI-MS: m/z = 267.1 [M + H]+.
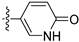
3.2.67. 5-(6-Ethoxyisoquinolin-4-yl)pyridin-2(1H)-one (48)
Method A. The intermediate (5-1) was reacted with (6-oxo-1,6-dihydropyridin-3-yl)boronic acid through Suzuki coupling reaction to afford 48 as a white solid (55%). 1H NMR (400 MHz, DMSO-d6) δ 11.90 (s, 1H), 9.14 (s, 1H), 8.31 (s, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.66 (dd, J = 9.4, 2.7 Hz, 1H), 7.58 (d, J = 2.4 Hz, 1H), 7.35 (dd, J = 9.0, 2.4 Hz, 1H), 7.06 (d, J = 2.4 Hz, 1H), 6.49 (dd, J = 9.4, 0.7 Hz, 1H), 4.12 (q, J = 7.0 Hz, 2H), 1.37 (t, J = 7.0 Hz, 3H). ESI-MS: m/z = 267.1 [M + H]+.



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}