

Non-Targeted Nuclear Magnetic Resonance Analysis for Food Authenticity: A Comparative Study on Tomato Samples

 ,
,  , , ,
, , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of Sample Preparation Protocol
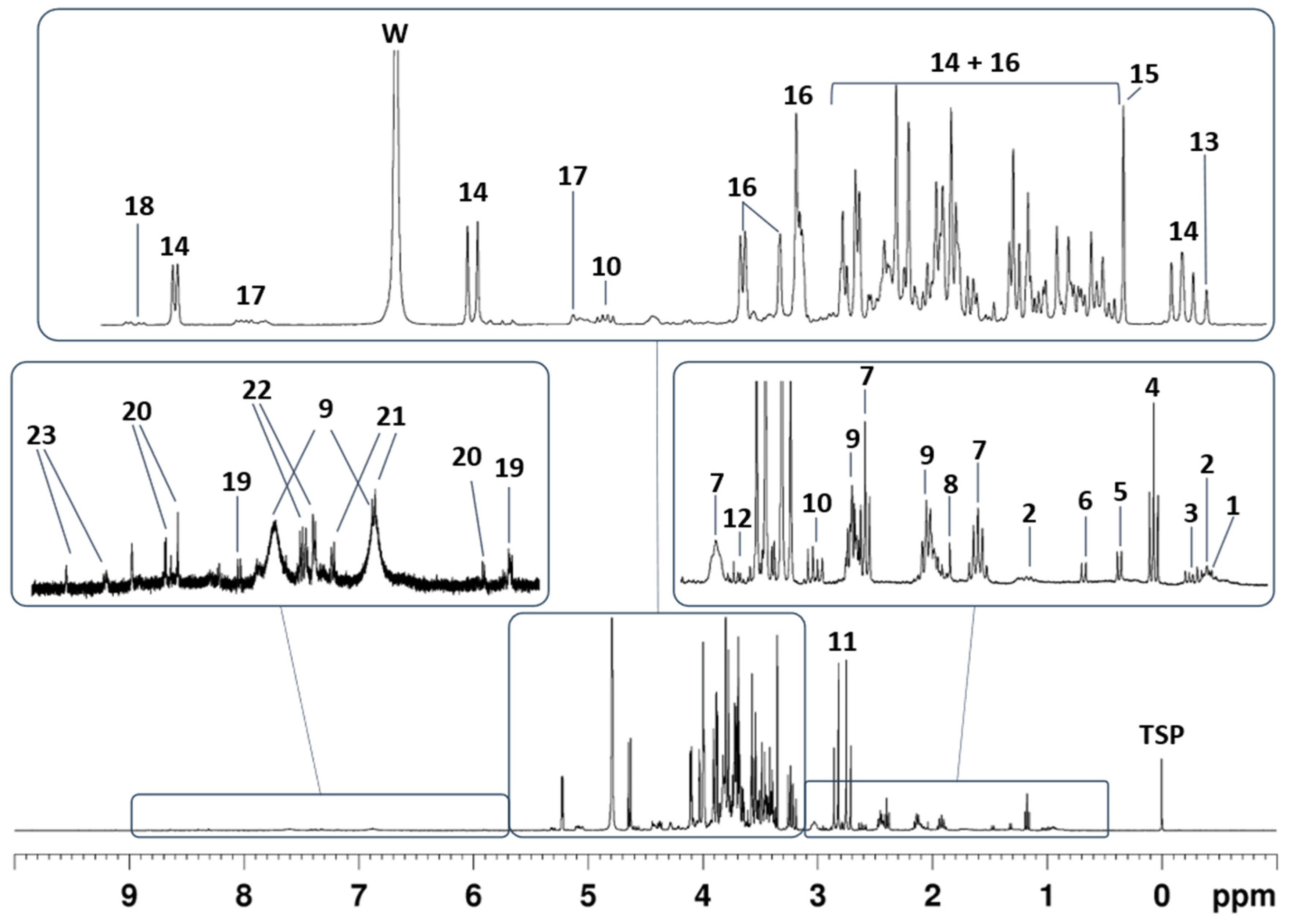
2.2. Metabolic Profile of Tomatoes Aqueous Extracts Following P3
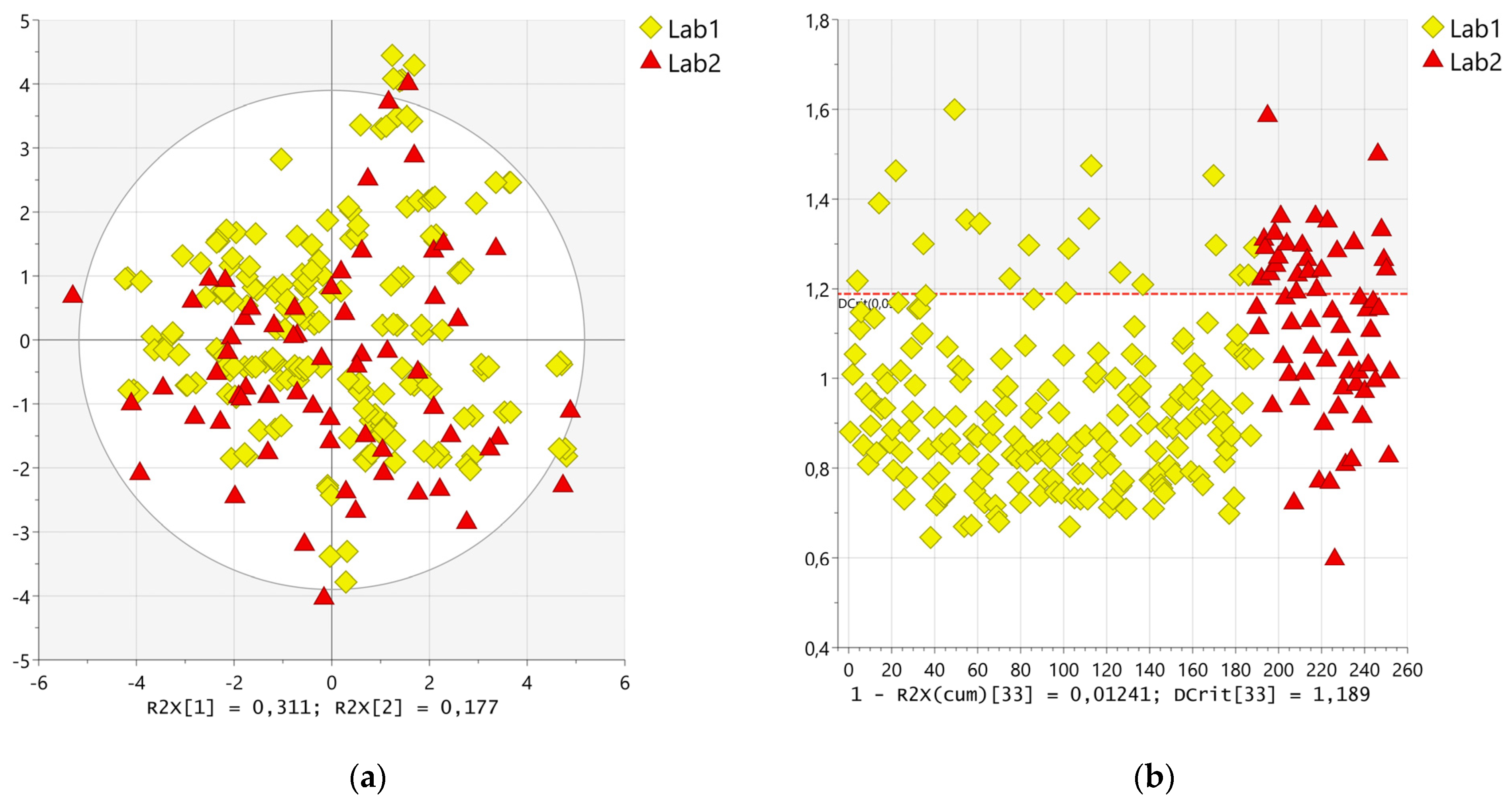
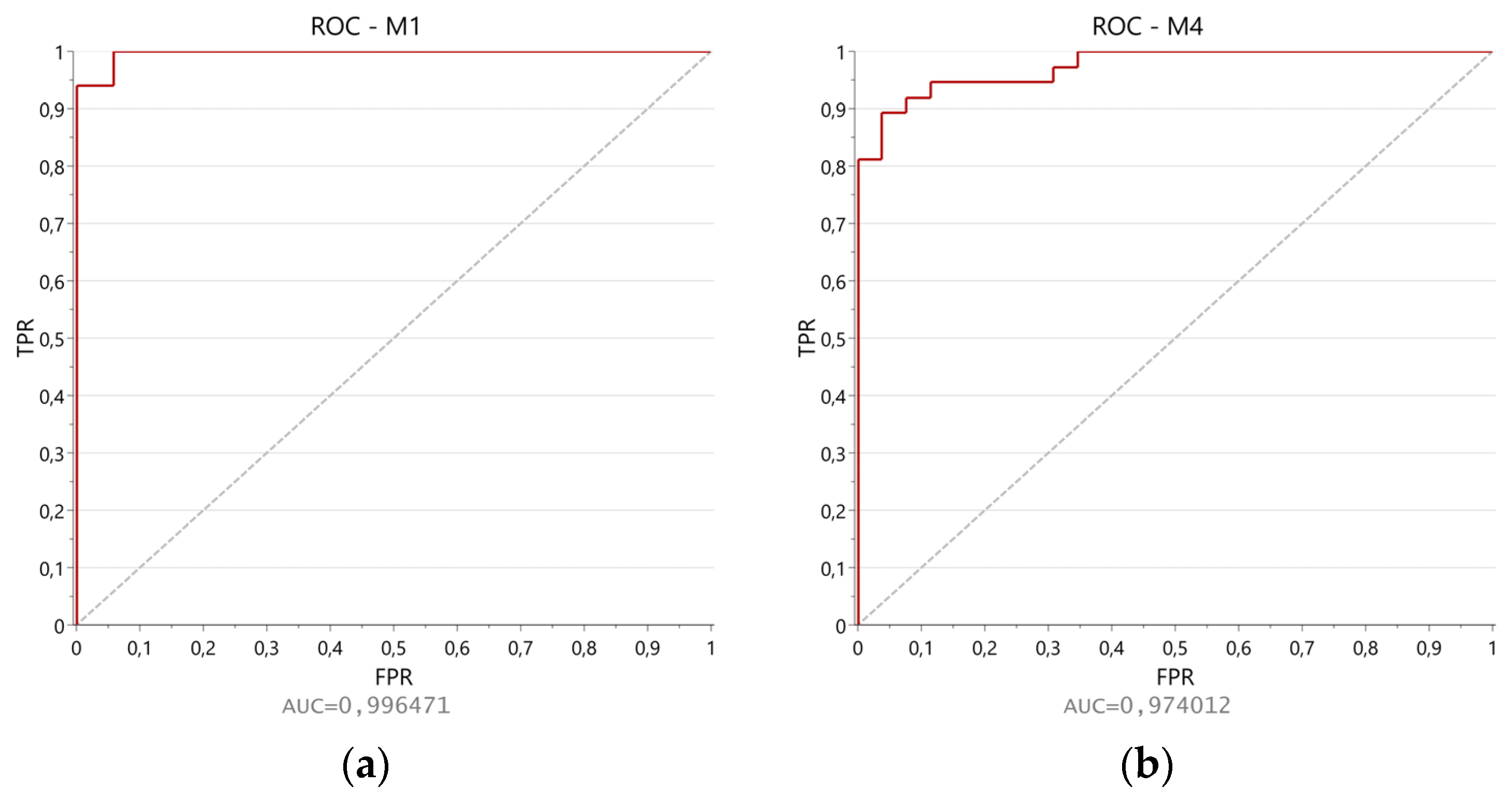
2.3. Statistical Analysis
3. Discussion
4. Materials and Methods
4.1. Materials
4.1.1. Method P1 [30]
4.1.2. Method P2 [50]
4.1.3. Method P3 [29]
4.2. NMR Measurements
4.3. Data Preprocessing and Chemometrics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hatzakis, E. Nuclear Magnetic Resonance (NMR) Spectroscopy in Food Science: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Abreu, A.C.; Fernández, I. NMR Metabolomics Applied on the Discrimination of Variables Influencing Tomato (Solanum lycopersicum). Molecules 2020, 25, 3738. [Google Scholar] [CrossRef] [PubMed]
- Tahir, H.E.; Arslan, M.; Komla Mahunu, G.; Adam Mariod, A.B.H.; Hashim, S.; Xiaobo, Z.; Jiyong, S.; El-Seedi, H.R.; Musa, T.H. The use of analytical techniques coupled with chemometrics for tracing the geographical origin of oils: A systematic review (2013–2020). Food Chem. 2022, 366, 130633. [Google Scholar] [CrossRef] [PubMed]
- Rifna, E.J.; Pandiselvam, R.; Kothakota, A.; Subba Rao, K.V.; Dwivedi, M.; Kumar, M.; Thirumdas, R.; Ramesh, S.V. Advanced process analytical tools for identification of adulterants in edible oils—A review. Food Chem. 2022, 369, 130898. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, M.; Saroja, S.G.; Khan, I.A. NMR technique and methodology in botanical health product analysis and quality control. J. Pharm. Biomed. Anal. 2022, 207, 114376. [Google Scholar] [CrossRef]
- Suh, J.H. Critical review: Metabolomics in dairy science—Evaluation of milk and milk product quality. Food Res. Int. 2022, 154, 110984. [Google Scholar] [CrossRef]
- Dimitrakopoulou, M.E.; Vantarakis, A. Does Traceability Lead to Food Authentication? A Systematic Review from A European Perspective. Food Rev. Int. 2021, 39, 537–559. [Google Scholar] [CrossRef]
- Colnago, L.A.; Wiesman, Z.; Pages, G.; Musse, M.; Monaretto, T.; Windt, C.W.; Rondeau-Mouro, C. Low field, time domain NMR in the agriculture and agrifood sectors: An overview of applications in plants, foods and biofuels. J. Magn. Reson. 2021, 323, 106899. [Google Scholar] [CrossRef] [PubMed]
- Laghi, L.; Picone, G.; Capozzi, F. Nuclear magnetic resonance for foodomics beyond food analysis. TrAC—Trends Anal. Chem. 2014, 59, 93–102. [Google Scholar] [CrossRef]
- Solovyev, P.A.; Fauhl-Hassek, C.; Riedl, J.; Esslinger, S.; Bontempo, L.; Camin, F. NMR spectroscopy in wine authentication: An official control perspective. Compr. Rev. Food Sci. Food Saf. 2021, 20, 2040–2062. [Google Scholar] [CrossRef]
- Palacios-Jordan, H.; Jané-Brunet, A.; Jané-Brunet, E.; Puiggròs, F.; Canela, N.; Rodríguez, M.A. Considerations on the Analysis of E-900 Food Additive: An NMR Perspective. Foods 2022, 11, 297. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.; Horn, B.; Raeke, J.; Fauhl-Hassek, C.; Hermann, A.; Brockmeyer, J.; Riedl, J. Towards harmonization of non-targeted 1H NMR spectroscopy-based wine authentication: Instrument comparison. Food Control 2022, 132, 108508. [Google Scholar] [CrossRef]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted pofiling: Quantitative analysis of1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Mounet, F.; Lemaire-Chamley, M.; Maucourt, M.; Cabasson, C.; Giraudel, J.L.; Deborde, C.; Lessire, R.; Gallusci, P.; Bertrand, A.; Gaudillère, M.; et al. Quantitative metabolic profiles of tomato flesh and seeds during fruit development: Complementary analysis with ANN and PCA. Metabolomics 2007, 3, 273–288. [Google Scholar] [CrossRef]
- Corsaro, C.; Mallamace, D.; Vasi, S.; Ferrantelli, V.; Dugo, G.; Cicero, N. 1H HR-MAS NMR Spectroscopy and the Metabolite Determination of Typical Foods in Mediterranean Diet. J. Anal. Methods Chem. 2015, 2015, 175696. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Thomas, F.; Donarski, J.; Ingallina, C.; Circi, S.; Cesare Marincola, F.; Capitani, D.; Mannina, L. Use of NMR applications to tackle future food fraud issues. Trends Food Sci. Technol. 2019, 91, 347–353. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR spectroscopy. TrAC - Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Sobolev, A.P.; Mannina, L.; Proietti, N.; Carradori, S.; Daglia, M.; Giusti, A.M.; Antiochia, R.; Capitani, D. Untargeted NMR-based methodology in the study of fruit metabolites. Molecules 2015, 20, 4088–4108. [Google Scholar] [CrossRef]
- Fiorino, G.M.; Garino, C.; Arlorio, M.; Logrieco, A.F.; Losito, I.; Monaci, L. Overview on Untargeted Methods to Combat Food Frauds: A Focus on Fishery Products. J. Food Qual. 2018, 2018, 1581746. [Google Scholar] [CrossRef]
- Calò, F.; Girelli, C.R.; Wang, S.C.; Fanizzi, F.P. Geographical Origin Assessment of Extra Virgin Olive Oil via NMR and MS Combined with Chemometrics as Analytical Approaches. Foods 2022, 11, 113. [Google Scholar] [CrossRef]
- Oms-Oliu, G.; Hertog, M.L.A.T.M.; Van de Poel, B.; Ampofo-Asiama, J.; Geeraerd, A.H.; Nicolai, B.M. Metabolic characterization of tomato fruit during preharvest development, ripening, and postharvest shelf-life. Postharvest Biol. Technol. 2011, 62, 7–16. [Google Scholar] [CrossRef]
- Riswanto, F.D.O.; Windarsih, A.; Lukitaningsih, E.; Rafi, M.; Fadzilah, N.A.; Rohman, A. Metabolite Fingerprinting Based on1 H-NMR Spectroscopy and Liquid Chromatography for the Authentication of Herbal Products. Molecules 2022, 27, 1198. [Google Scholar] [CrossRef] [PubMed]
- Gallo, V.; Ragone, R.; Musio, B.; Todisco, S.; Rizzuti, A.; Mastrorilli, P.; Pontrelli, S.; Intini, N.; Scapicchio, P.; Triggiani, M.; et al. A Contribution to the Harmonization of Non-targeted NMR Methods for Data-Driven Food Authenticity Assessment. Food Anal. Methods 2020, 13, 530–541. [Google Scholar] [CrossRef]
- Zailer, E.; Holzgrabe, U.; Diehl, B.W.K. Interlaboratory Comparison Test as an Evaluation of Applicability of an Alternative Edible Oil Analysis by 1H NMR Spectroscopy. J. AOAC Int. 2017, 100, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.A.A.; Magalhães, A.; Ferreira, M.M.C. Optimized bucketing for NMR spectra: Three case studies. Chemom. Intell. Lab. Syst. 2013, 122, 93–102. [Google Scholar] [CrossRef]
- Karaman, I. Preprocessing and pretreatment of metabolomics data for statistical analysis. In Advances in Experimental Medicine and Biology; Springer New York LLC: New York, NY, USA, 2017; Volume 965, pp. 145–161. [Google Scholar]
- Mulder, F.A.A.; Tenori, L.; Licari, C.; Luchinat, C. Practical considerations for rapid and quantitative NMR-based metabolomics. J. Magn. Reson. 2023, 352, 107462. [Google Scholar] [CrossRef]
- Ragone, R.; Todisco, S.; Triggiani, M.; Pontrelli, S.; Latronico, M.; Mastrorilli, P.; Intini, N.; Ferroni, C.; Musio, B.; Gallo, V. Development of a food class-discrimination system by non-targeted NMR analyses using different magnetic field strengths. Food Chem. 2020, 332, 127339. [Google Scholar] [CrossRef]
- Deborde, C.; Fontaine, J.X.; Jacob, D.; Botana, A.; Nicaise, V.; Richard-Forget, F.; Lecomte, S.; Decourtil, C.; Hamade, K.; Mesnard, F.; et al. Optimizing 1D 1H-NMR profiling of plant samples for high throughput analysis: Extract preparation, standardization, automation and spectra processing. Metabolomics 2019, 15, 28. [Google Scholar] [CrossRef]
- Musio, B.; Ragone, R.; Todisco, S.; Rizzuti, A.; Latronico, M.; Mastrorilli, P.; Pontrelli, S.; Intini, N.; Scapicchio, P.; Triggiani, M.; et al. A community-built calibration system: The case study of quantification of metabolites in grape juice by qNMR spectroscopy. Talanta 2020, 214, 120855. [Google Scholar] [CrossRef]
- Le Gall, G.; Colquhoun, I.J.; Davis, A.L.; Collins, G.J.; Verhoeyen, M.E. Metabolite profiling of tomato (Lycopersicon esculentum) using 1H NMR spectroscopy as a tool to detect potential unintended effects following a genetic modification. J. Agric. Food Chem. 2003, 51, 2447–2456. [Google Scholar] [CrossRef]
- Zhang, G.; Abdulla, W. On honey authentication and adulterant detection techniques. Food Control 2022, 138, 108992. [Google Scholar] [CrossRef]
- ElNaker, N.A.; Daou, M.; Ochsenkühn, M.A.; Amin, S.A.; Yousef, A.F.; Yousef, L.F. A metabolomics approach to evaluate the effect of lyophilization versus oven drying on the chemical composition of plant extracts. Sci. Rep. 2021, 11, 22679. [Google Scholar] [CrossRef]
- Beteinakis, S.; Papachristodoulou, A.; Mikros, E.; Halabalaki, M. From sample preparation to NMR-based metabolic profiling in food commodities: The case of table olives. Phytochem. Anal. 2022, 33, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, Y.H.; Verpoorte, R. NMR-based metabolomic analysis of plants. Nat. Protoc. 2010, 5, 536–549. [Google Scholar] [CrossRef]
- Chamley, M.L.; Mounet, F.; Deborde, C.; Maucourt, M.; Jacob, D.; Moing, A. NMR-based tissular and developmental metabolomics of tomato fruit. Metabolites 2019, 9, 93. [Google Scholar] [CrossRef]
- Hohmann, M.; Christoph, N.; Wachter, H.; Holzgrabe, U. 1H NMR profiling as an approach to differentiate conventionally and organically grown tomatoes. J. Agric. Food Chem. 2014, 62, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Human Metabolome Database: 1H NMR Spectrum (1D, 500 MHz, H2O, Experimental) (HMDB0000641). Available online: https://hmdb.ca/spectra/nmr_one_d/1452 (accessed on 13 September 2024).
- Tjandra, N.; Bax, A. Solution NMR Measurement of Amide Proton Chemical Shift Anisotropy in 15N-Enriched Proteins. Correlation with Hydrogen Bond Length§. J. Am. Chem. Soc. 1997, 119, 8076–8082. [Google Scholar] [CrossRef]
- Bauer, M.; Bertario, A.; Boccardi, G.; Fontaine, X.; Rao, R.; Verrier, D. Reproducibility of 1H-NMR integrals: A collaborative study. J. Pharm. Biomed. Anal. 1998, 17, 419–425. [Google Scholar] [CrossRef]
- Chen, Z.; Lian, X.; Zhou, M.; Zhang, X.; Wang, C. Quantitation of L-cystine in Food Supplements and Additives Using 1H qNMR: Method Development and Application. Foods 2023, 12, 2421. [Google Scholar] [CrossRef]
- Okaru, A.O.; Scharinger, A.; Rajcic de Rezende, T.; Teipel, J.; Kuballa, T.; Walch, S.G.; Lachenmeier, D.W. Validation of a Quantitative Proton Nuclear Magnetic Resonance Spectroscopic Screening Method for Coffee Quality and Authenticity (NMR Coffee Screener). Foods 2020, 9, 47. [Google Scholar] [CrossRef]
- Bourafai-Aziez, A.; Jacob, D.; Charpentier, G.; Cassin, E.; Rousselot, G.; Moing, A.; Deborde, C. Development, Validation, and Use of 1H-NMR Spectroscopy for Evaluating the Quality of Acerola-Based Food Supplements and Quantifying Ascorbic Acid. Molecules 2022, 27, 5614. [Google Scholar] [CrossRef] [PubMed]
- Piccinonna, S.; Ragone, R.; Stocchero, M.; Del Coco, L.; De Pascali, S.A.; Schena, F.P.; Fanizzi, F.P. Robustness of NMR-based metabolomics to generate comparable data sets for olive oil cultivar classification. An inter-laboratory study on Apulian olive oils. Food Chem. 2016, 199, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.L.; Baker, J.M.; Miller, S.J.; Deborde, C.; Maucourt, M.; Biais, B.; Rolin, D.; Moing, A.; Moco, S.; Vervoort, J.; et al. An inter-laboratory comparison demonstrates that [1H]-NMR metabolite fingerprinting is a robust technique for collaborative plant metabolomic data collection. Metabolomics 2010, 6, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Commission Regulation (EU)-889/2008-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/IT/TXT/?uri=CELEX%3A32008R0889 (accessed on 16 September 2024).
- Commission Regulation (EU)-1235/2008-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32008R1235 (accessed on 16 September 2024).
- Commission Regulation (EU)-178/2010-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX:32010R0178 (accessed on 16 September 2024).
- Commission Regulation (EU)-401/2006-EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32006R0401 (accessed on 16 September 2024).
- Jlilat, A.; Ragone, R.; Gualano, S.; Santoro, F.; Gallo, V.; Varvaro, L.; Mastrorilli, P.; Saponari, M.; Nigro, F.; D’Onghia, A.M. A non-targeted metabolomics study on Xylella fastidiosa infected olive plants grown under controlled conditions. Sci. Rep. 2021, 11, 1070. [Google Scholar] [CrossRef]
- Chemistry, C. Related compounds. J. Med. Pharm. Chem. 2001, 2, 1941–1944. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| Statistic Parameter | P1 | P2 | P3 |
|---|---|---|---|
| Median | 0.202 | 0.142 | 0.303 |
| Mean (μ) | 0.200 | 0.142 | 0.304 |
| Standard deviation (σ) | 0.005 | 0.003 | 0.004 |
| %RSD a | 2.68 | 1.83 | 1.32 |
| Model | Training Set | Validation Set |
|---|---|---|
| M1 | Lab1 = 126 | Lab1 = 63 |
| Lab2 = 42 | Lab2 = 21 | |
| Tot. = 168 | Tot. = 84 | |
| M2 | Lab1 avg = 42 | Lab1 avg = 21 |
| Lab2 = 42 | Lab2 = 21 | |
| Tot. = 84 | Tot. = 42 | |
| M3 | Lab1 med = 42 | Lab1 med = 21 |
| Lab2 = 42 | Lab2 = 21 | |
| Tot. = 84 | Tot. = 42 | |
| M4 | Lab1 = 189 | Lab2 = 63 |
| M5 | Lab1 avg = 63 | Lab2 = 63 |
| M6 | Lab1 med = 63 | Lab2 = 63 |
| M7 | Lab2 = 63 | Lab1 = 189 |
| M8 | Lab2 = 63 | Lab1 avg = 63 |
| M9 | Lab2 = 63 | Lab1 med = 63 |
| Model | No a | R2X (cum) b | R2Y (cum) c | Q2 (cum) d | Q2 Intercept | R2 Intercept | F-Value e | p-Value f | Correct Prediction |
|---|---|---|---|---|---|---|---|---|---|
| M1 | 1P + 8O | 0.850 | 0.767 | 0.619 | −0.444 | 0.254 | 13.4641 | 4.9424 × 10−23 | 97.62% |
| M2 | 1P + 6O | 0.811 | 0.741 | 0.505 | −0.630 | 0.365 | 5.0329 | 2.3550 × 10−6 | 90.48% |
| M3 | 1P + 6O | 0.810 | 0.748 | 0.521 | −0.645 | 0.371 | 5.3575 | 9.2400 × 10−7 | 92.86% |
| M4 | 1P + 8O | 0.861 | 0.802 | 0.718 | −0.351 | 0.193 | 24.0347 | 1.4347 × 10−37 | 87.30% |
| M5 | 1P + 2O | 0.608 | 0.567 | 0.419 | −0.357 | 0.215 | 6.7444 | 2.0513 × 10−5 | 80.95% |
| M6 | 1P + 2O | 0.606 | 0.555 | 0.399 | −0.361 | 0.223 | 8.9320 | 1.4372 × 10−13 | 80.95% |
| M7 | 1P + 2O | 0.565 | 0.505 | 0.308 | −0.363 | 0.242 | 4.1578 | 1.5945 × 10−3 | 85.19% |
| M8 | 85.71% | ||||||||
| M9 | 85.71% |
| Model | Class | Prediction Set | Predicted as SI | Predicted as LA | Correct Prediction % | Fisher’s Prob. |
|---|---|---|---|---|---|---|
| M1 | SI | 34 | 32 | 2 | 97.62 | 3.6 × 10−21 |
| LA | 50 | 0 | 50 | |||
| M2 | SI | 19 | 16 | 3 | 90.48 | 8.8 × 10−8 |
| LA | 23 | 1 | 22 | |||
| M3 | SI | 19 | 16 | 3 | 92.86 | 5.8 × 10−9 |
| LA | 23 | 0 | 23 | |||
| M4 | SI | 26 | 20 | 6 | 87.3 | 3 × 10−9 |
| LA | 37 | 2 | 35 | |||
| M5 | SI | 26 | 15 | 11 | 80.95 | 7.9 × 10−7 |
| LA | 37 | 1 | 36 | |||
| M6 | SI | 26 | 15 | 11 | 80.95 | 7.9 × 10−7 |
| LA | 37 | 1 | 36 | |||
| M7 | SI | 78 | 60 | 18 | 85.19 | 1.5 × 10−22 |
| LA | 111 | 10 | 101 | |||
| M8 | SI | 26 | 20 | 6 | 85.71 | 2.0 × 10−8 |
| LA | 37 | 3 | 34 | |||
| M9 | SI | 26 | 20 | 6 | 85.71 | 2.0 × 10−8 |
| LA | 37 | 3 | 34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musio, B.; Ragone, R.; Todisco, S.; Rizzuti, A.; Iorio, E.; Chirico, M.; Pisanu, M.E.; Meloni, N.; Mastrorilli, P.; Gallo, V. Non-Targeted Nuclear Magnetic Resonance Analysis for Food Authenticity: A Comparative Study on Tomato Samples. Molecules 2024, 29, 4441. https://doi.org/10.3390/molecules29184441
Musio B, Ragone R, Todisco S, Rizzuti A, Iorio E, Chirico M, Pisanu ME, Meloni N, Mastrorilli P, Gallo V. Non-Targeted Nuclear Magnetic Resonance Analysis for Food Authenticity: A Comparative Study on Tomato Samples. Molecules. 2024; 29(18):4441. https://doi.org/10.3390/molecules29184441
Chicago/Turabian StyleMusio, Biagia, Rosa Ragone, Stefano Todisco, Antonino Rizzuti, Egidio Iorio, Mattea Chirico, Maria Elena Pisanu, Nadia Meloni, Piero Mastrorilli, and Vito Gallo. 2024. "Non-Targeted Nuclear Magnetic Resonance Analysis for Food Authenticity: A Comparative Study on Tomato Samples" Molecules 29, no. 18: 4441. https://doi.org/10.3390/molecules29184441
APA StyleMusio, B., Ragone, R., Todisco, S., Rizzuti, A., Iorio, E., Chirico, M., Pisanu, M. E., Meloni, N., Mastrorilli, P., & Gallo, V. (2024). Non-Targeted Nuclear Magnetic Resonance Analysis for Food Authenticity: A Comparative Study on Tomato Samples. Molecules, 29(18), 4441. https://doi.org/10.3390/molecules29184441