
Conformational Analysis and Organocatalytic Activity of Helical Stapled Peptides Containing α-Carbocyclic α,α-Disubstituted α-Amino Acids
 ,
,
Abstract
1. Introduction
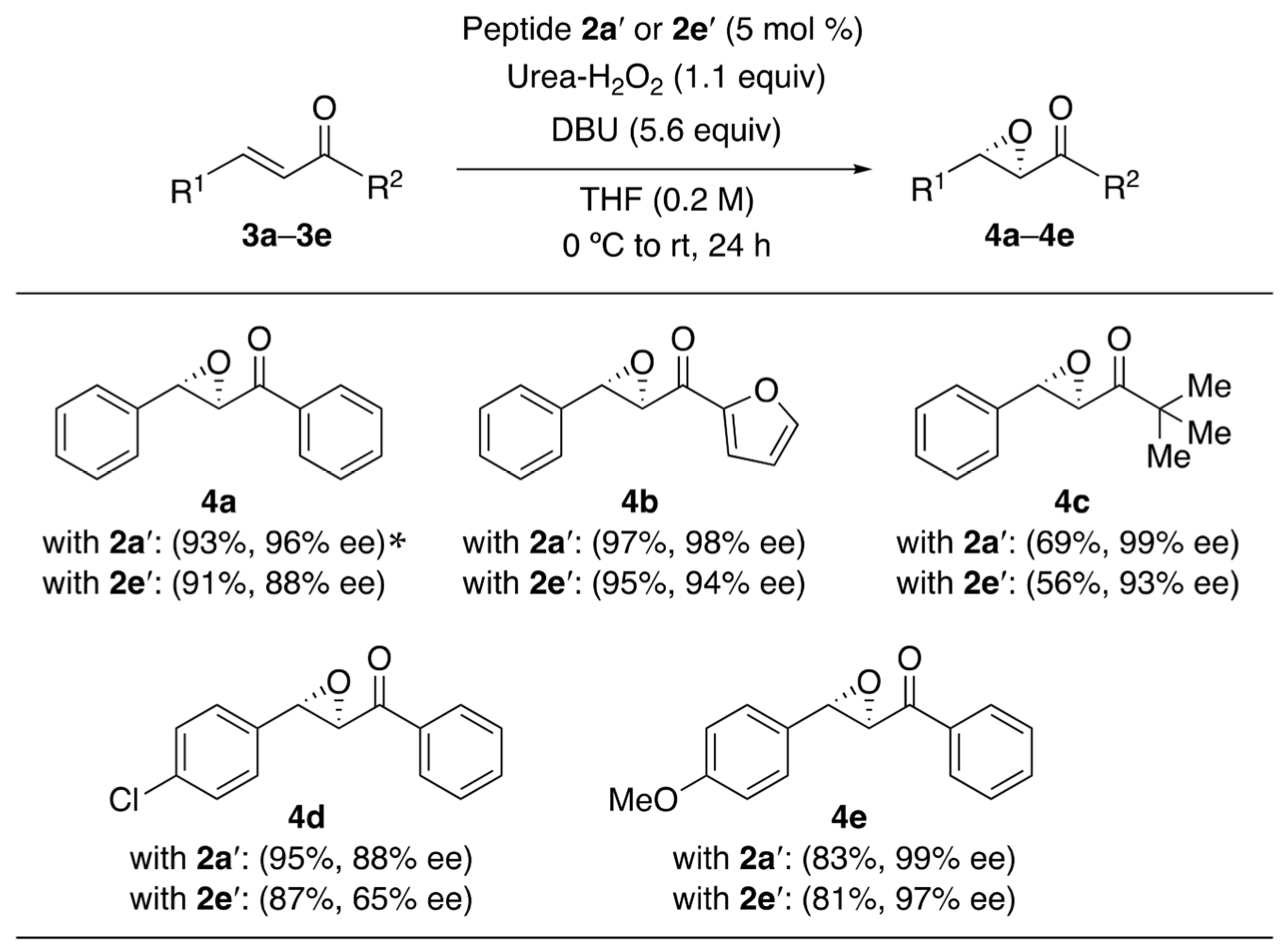
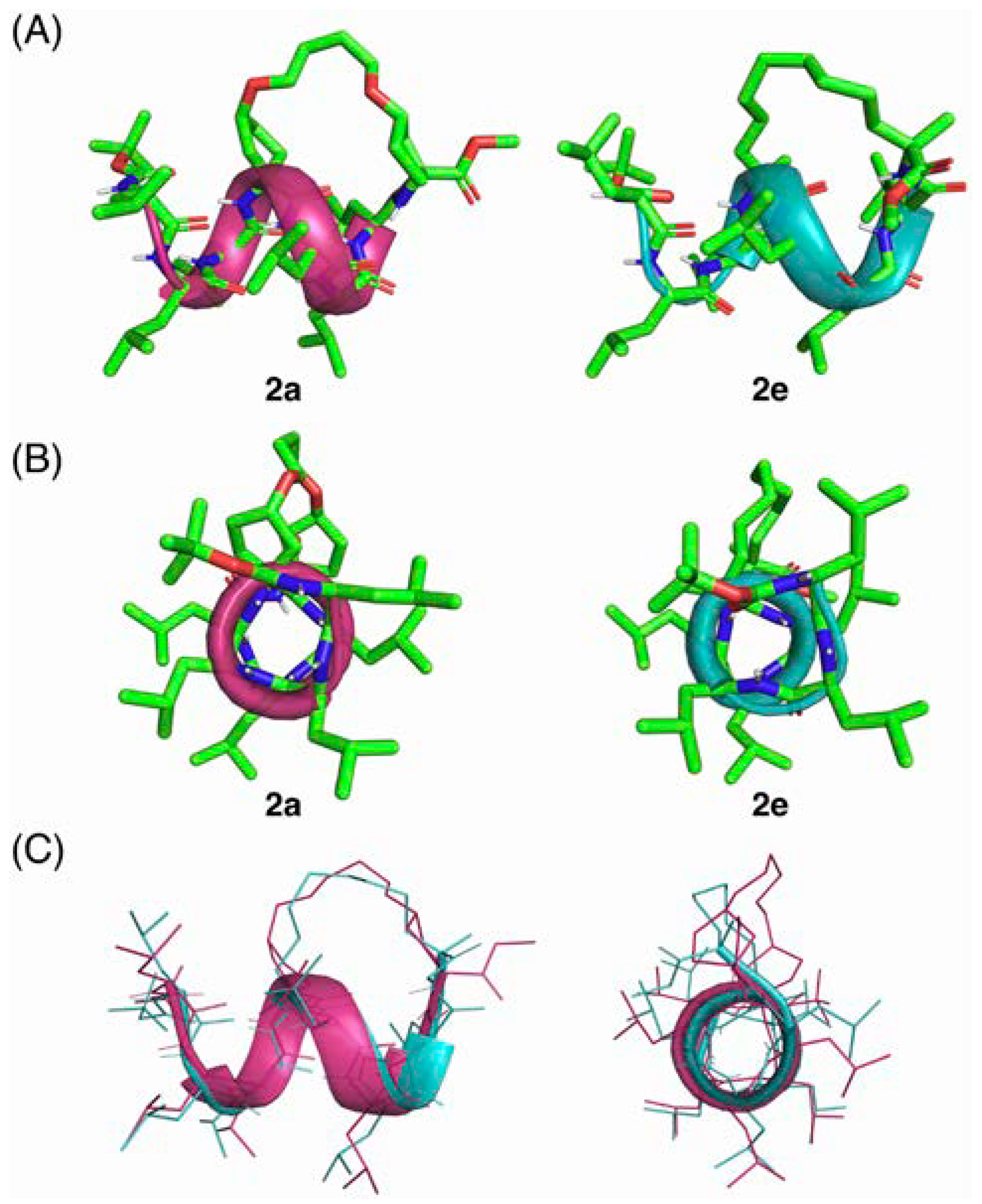
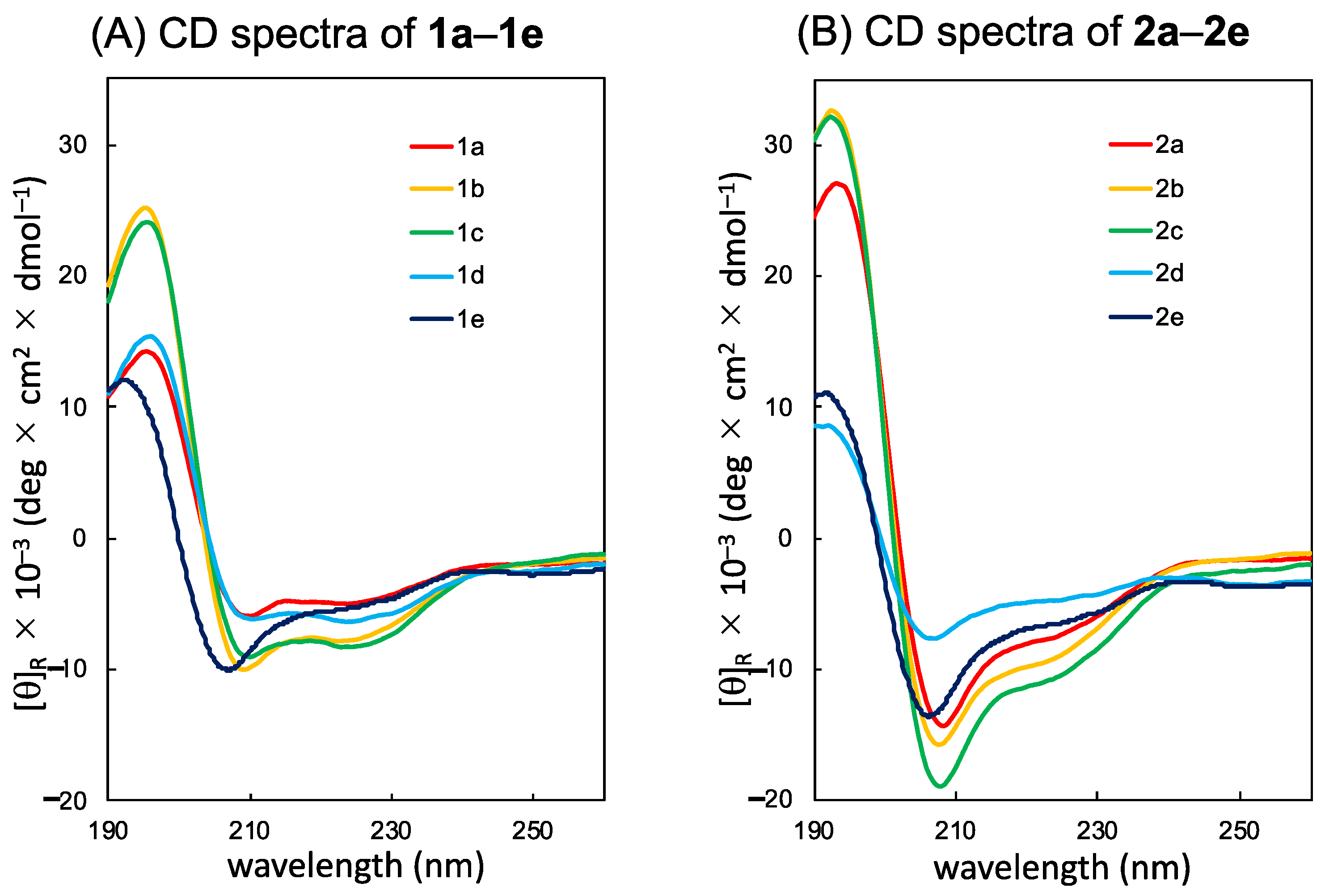
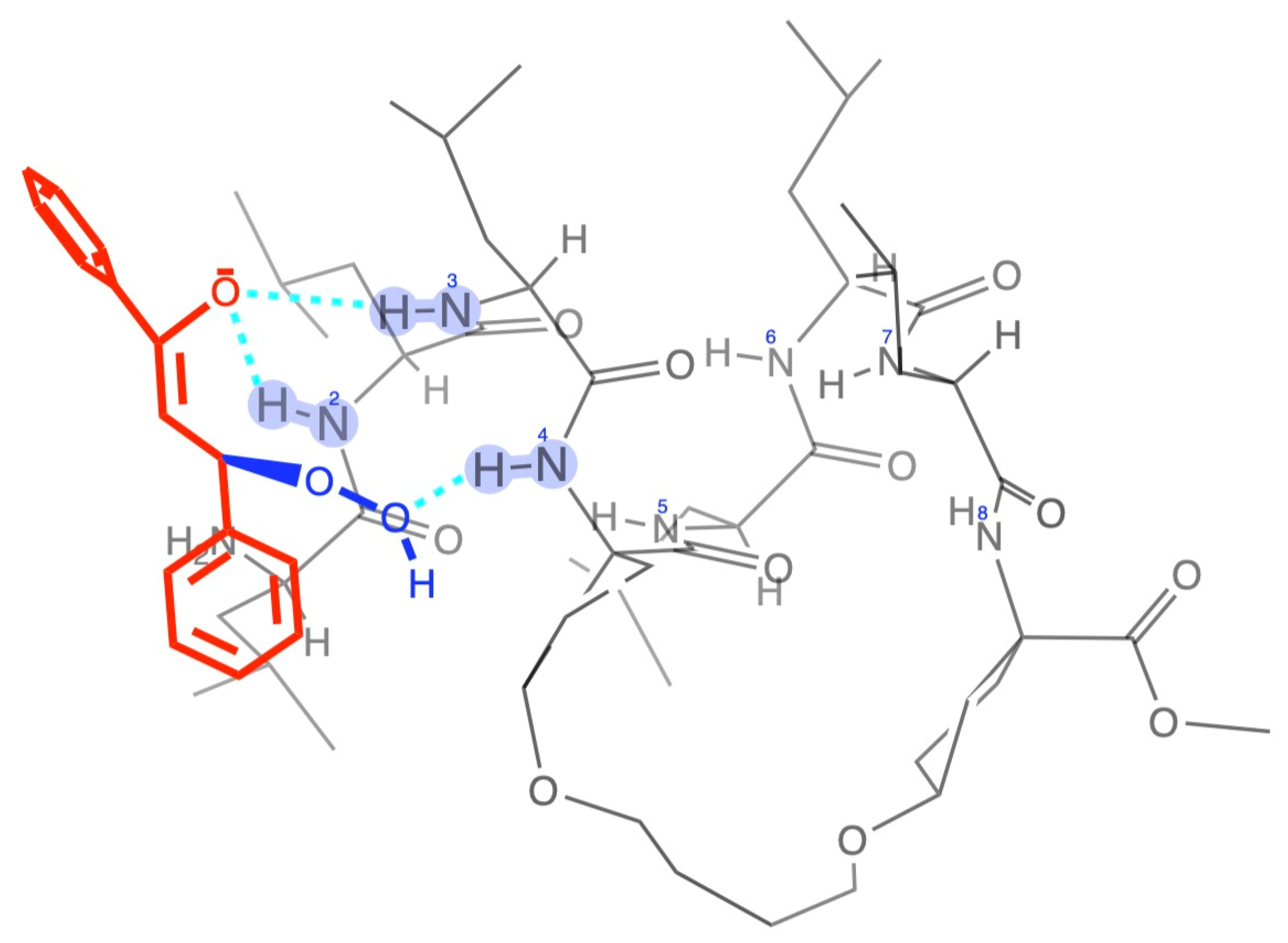
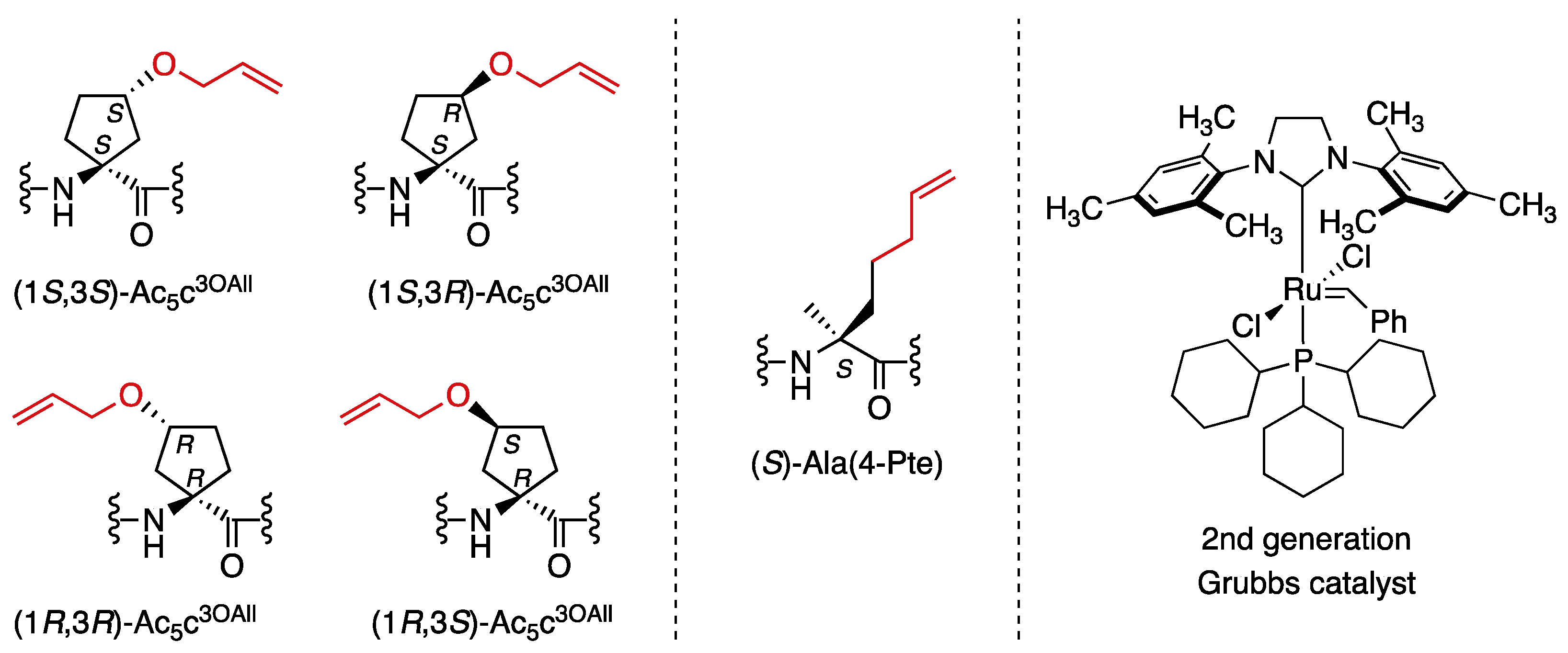
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure and Method
3.2. General Procedure for Peptide-Catalyzed Asymmetric Epoxidation
3.3. Synthesis of Stapled Peptides 2a–2e
3.4. Synthesis of Unstapled Peptide Catalysts 1a′–1e′ and Stapled Peptide Catalysts 2a′–2e′
3.5. Synthesis Outlined in Figure 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gellman, S.H. Foldamers: A manifesto. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar] [CrossRef]
- Goodman, C.M.; Choi, S.; Shandler, S.; DeGrado, W.F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [Google Scholar] [CrossRef]
- Martinek, T.A.; Fülöp, F. Peptidic foldamers: Ramping up diversity. Chem. Soc. Rev. 2012, 41, 687–702. [Google Scholar] [CrossRef]
- Girvin, Z.C.; Gellman, S.H. Foldamer catalysis. J. Am. Chem. Soc. 2020, 142, 17211–17223. [Google Scholar] [CrossRef]
- Juliá, S.; Masana, J.; Vega, J.C. “Synthetic enzymes”. Highly stereoselective epoxidation of chalcone in a triphasic toluene-water-poly[(S)-alanine] system. Angew. Chem. Int. Ed. Engl. 1980, 19, 929–931. [Google Scholar] [CrossRef]
- Juliá, S.; Guixer, J.; Masana, J.; Rocas, J.; Colonna, S.; Annuziata, R.; Molinari, H. Synthetic enzymes. Part 2. Catalytic asymmetric epoxidation by means of polyamino-acids in a triphase system. J. Chem. Soc. Perkin Trans. 1982, 1, 1317–1324. [Google Scholar] [CrossRef]
- Bentley, P.A.; Bergeron, S.; Cappi, M.W.; Hibbs, D.E.; Hursthouse, M.B.; Nugent, T.C.; Pulido, R.; Roberts, S.M.; Wu, L.E. Asymmetric epoxidation of enones employing polymeric α-amino acids in non-aqueous media. Chem. Commun. 1997, 1997, 739–740. [Google Scholar] [CrossRef]
- Porter, M.J.; Roberts, S.M.; Skidmore, J. Polyamino acids as catalysts in asymmetric synthesis. Bioorg. Med. Chem. 1999, 7, 2145–2156. [Google Scholar] [CrossRef]
- Takagi, R.; Manabe, T.; Shiraki, A.; Yoneshige, A.; Hiraga, Y.; Kojima, S.; Ohkata, K. The Juliá-Colonna asymmetric epoxidation reaction of chalcone catalyzed by length defined oligo-L-leucine: Importance of the N-terminal functional group and helical structure of the catalyst in the asymmetric induction. Bull. Chem. Soc. Jpn. 2000, 73, 2115–2121. [Google Scholar] [CrossRef]
- Takagi, R.; Shiraki, A.; Manabe, T.; Kojima, S.; Ohkata, K. The Juliá-Colonna type asymmetric epoxidation reaction catalyzed by soluble oligo-L-leucines containing an α-aminoisobutyric acid residue: Importance of helical structure of the catalyst on asymmetric induction. Chem. Lett. 2000, 29, 366–367. [Google Scholar] [CrossRef]
- Kelly, D.R.; Bui, T.T.T.; Caroff, E.; Drake, A.F.; Roberts, S.M. Structure and catalytic activity of some soluble polyethylene glycol–peptide conjugates. Tetrahedron Lett. 2004, 45, 3885–3888. [Google Scholar] [CrossRef]
- Carrea, G.; Colonna, S.; Kelly, D.R.; Lazcano, A.; Ottolina, G.; Roberts, S.M. Polyamino acids as synthetic enzymes: Mechanism, applications and relevance to prebiotic catalysis. Trends Biotechnol. 2005, 23, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.R.; Roberts, S.M. Oligopeptides as catalysts for asymmetric epoxidation. Biopolymers 2006, 84, 74–89. [Google Scholar] [CrossRef]
- Nagano, M.; Doi, M.; Kurihara, M.; Suemune, H.; Tanaka, M. Stabilized α-helix-catalyzed enantioselective epoxidation of α,β-unsaturated ketones. Org. Lett. 2010, 12, 3564–3566. [Google Scholar] [CrossRef]
- Demizu, Y.; Yamagata, N.; Nagoya, S.; Sato, Y.; Doi, M.; Tanaka, M.; Nagasawa, K.; Okuda, H.; Kurihara, M. Enantioselective epoxidation of α,β-unsaturated ketones catalyzed by stapled helical L-Leu-based peptides. Tetrahedron 2011, 67, 6155–6165. [Google Scholar] [CrossRef]
- Yamagata, N.; Demizu, Y.; Sato, Y.; Doi, M.; Tanaka, M.; Nagasawa, K.; Okuda, H.; Kurihara, M. Design of a stabilized short helical peptide and its application to catalytic enantioselective epoxidation of (E)-chalcone. Tetrahedron Lett. 2011, 52, 798–801. [Google Scholar] [CrossRef]
- Akagawa, K.; Hirata, T.; Kudo, K. Asymmetric epoxidation of enones by peptide-based catalyst: A strategy inverting Juliá–Colonna stereoselectivity. Synlett 2016, 27, 1217–1222. [Google Scholar] [CrossRef]
- Akagawa, K.; Suzuki, R.; Kudo, K. Development of a peptide-based primary aminocatalyst with a helical structure. Asian J. Org. Chem. 2014, 3, 514–522. [Google Scholar] [CrossRef]
- Ueda, A.; Umeno, T.; Doi, M.; Akagawa, K.; Kudo, K.; Tanaka, M. Helical-peptide-catalyzed enantioselective Michael addition reactions and their mechanistic insights. J. Org. Chem. 2016, 81, 6343–6356. [Google Scholar] [CrossRef]
- Ueda, A.; Higuchi, M.; Umeno, T.; Tanaka, M. Enantioselective synthesis of 2,4,5-trisubstituted tetrahydropyrans via peptide-catalyzed Michael addition followed by Kishi’s reductive cyclization. Heterocycles 2019, 99, 989–1002. [Google Scholar] [CrossRef]
- Umeno, T.; Ueda, A.; Doi, M.; Kato, T.; Oba, M.; Tanaka, M. Helical foldamer-catalyzed enantioselective 1,4-addition reaction of dialkyl malonates to cyclic enones. Tetrahedron Lett. 2019, 60, 151301. [Google Scholar] [CrossRef]
- Sato, K.; Umeno, T.; Ueda, A.; Kato, T.; Doi, M.; Tanaka, M. Asymmetric 1,4-addition reactions catalyzed by N-terminal thiourea-modified helical L-Leu peptide with cyclic amino acids. Chem. Eur. J. 2021, 27, 11216–11220. [Google Scholar] [CrossRef] [PubMed]
- Tamaribuchi, K.; Tian, J.; Akagawa, K.; Kudo, K. Enantioselective nitro-Michael addition catalyzed by N-terminal guanidinylated helical peptide. Adv. Synth. Catal. 2022, 364, 82–86. [Google Scholar] [CrossRef]
- Tian, J.; Tamaribuchi, K.; Yoshikawa, I.; Kudo, K. Kinetic resolution of a planar-chiral [2.2]paracyclophane via Michael addition to nitroolefins catalyzed by N-terminal guanidinylated helical peptide. Eur. J. Org. Chem. 2024, 27, e202400117. [Google Scholar] [CrossRef]
- Rossi, P.; Felluga, F.; Tecilla, P.; Formaggio, F.; Crisma, M.; Toniolo, C.; Scrimin, P. A bimetallic helical heptapeptide as a transphosphorylation catalyst in water. J. Am. Chem. Soc. 1999, 121, 6948–6949. [Google Scholar] [CrossRef]
- Scarso, A.; Scheffer, U.; Göbel, M.; Broxterman, Q.B.; Kaptein, B.; Formaggio, F.; Toniolo, C.; Scrimin, P. A peptide template as an allosteric supramolecular catalyst for the cleavage of phosphate esters. Proc. Natl. Acad. Sci. USA 2002, 99, 5144–5149. [Google Scholar] [CrossRef]
- Miller, S.J.; Copeland, G.T.; Papaioannou, N.; Horstmann, T.E.; Ruel, E.M. Kinetic resolution of alcohols catalyzed by tripeptides containing the N-alkylimidazole substructure. J. Am. Chem. Soc. 1998, 120, 1629–1630. [Google Scholar] [CrossRef]
- Horstmann, T.E.; Guerin, D.J.; Miller, S.J. Asymmetric conjugate addition of azide to α,β-unsaturated carbonyl compounds catalyzed by simple peptides. Angew. Chem. Int. Ed. 2000, 39, 3635–3638. [Google Scholar] [CrossRef]
- Akagawa, K.; Sakai, N.; Kudo, K. Histidine-containing peptide catalysts developed by a facile library screening method. Angew. Chem. Int. Ed. 2015, 54, 1822–1826. [Google Scholar] [CrossRef]
- Peris, G.; Jakobsche, C.E.; Miller, S.J. Aspartate-catalyzed asymmetric epoxidation reactions. J. Am. Chem. Soc. 2007, 129, 8710–8711. [Google Scholar] [CrossRef]
- Lichtor, P.A.; Miller, S.J. Combinatorial evolution of site- and enantioselective catalysts for polyene epoxidation. Nat. Chem. 2012, 4, 990–995. [Google Scholar] [CrossRef]
- Toniolo, C.; Polese, A.; Formaggio, F.; Crisma, M.; Kamphuis, J. Circular dichroism spectrum of a peptide 310-helix. J. Am. Chem. Soc. 1996, 118, 2744–2745. [Google Scholar] [CrossRef]
- Tanaka, M. Design and synthesis of chiral α,α-disubstituted amino acids and conformational study of their oligopeptides. Chem. Pharm. Bull. 2007, 55, 349–358. [Google Scholar] [CrossRef]
- Kato, T.; Oba, M.; Nishida, K.; Tanaka, M. Cell-penetrating helical peptides having L-arginines and five-membered ring α,α-disubstituted α-amino acids. Bioconjugate Chem. 2014, 25, 1761–1768. [Google Scholar] [CrossRef]
- Crisma, M.; Toniolo, C. Helical screw-sense preferences of peptides based on chiral, Cα-tetrasubstituted α-amino acids. Biopolymers 2015, 104, 46–64. [Google Scholar] [CrossRef]
- Umeno, T.; Ueda, A.; Oba, M.; Doi, M.; Hirata, T.; Suemune, H.; Tanaka, M. Helical structures of L-Leu-based peptides having chiral six-membered ring amino acids. Tetrahedron 2016, 72, 3124–3131. [Google Scholar] [CrossRef]
- Koba, Y.; Ueda, A.; Oba, M.; Doi, M.; Demizu, Y.; Kurihara, M.; Tanaka, M. Helical L-Leu-based peptides having chiral five-membered carbocyclic ring amino acids with an ethylene acetal moiety. ChemistrySelect 2017, 2, 8108–8114. [Google Scholar] [CrossRef]
- Kato, T.; Oba, M.; Nishida, K.; Tanaka, M. Cell-penetrating peptides using cyclic α,α-disubstituted α-amino acids with basic functional groups. ACS Biomater. Sci. Eng. 2018, 4, 1368–1376. [Google Scholar] [CrossRef]
- Oba, M.; Nakajima, S.; Misao, K.; Yokoo, H.; Tanaka, M. Effect of helicity and hydrophobicity on cell-penetrating ability of arginine-rich peptides. Bioorg. Med. Chem. 2023, 91, 117409. [Google Scholar] [CrossRef]
- Oba, M.; Shibuya, M.; Yamaberi, Y.; Yokoo, H.; Uchida, S.; Ueda, A.; Tanaka, M. An amphipathic structure of a dipropylglycine-containing helical peptide with sufficient length enables safe and effective Intracellular siRNA delivery. Chem. Pharm. Bull. 2023, 71, 250–256. [Google Scholar] [CrossRef]
- Schafmeister, C.E.; Po, J.; Verdine, G.L. An All-Hydrocarbon Cross-Linking System for Enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 2000, 122, 5891–5892. [Google Scholar] [CrossRef]
- Ueda, A.; Higuchi, M.; Sato, K.; Umeno, T.; Tanaka, M. Design and synthesis of helical N-terminal L-Prolyl oligopeptides possessing hydrocarbon stapling. Molecules 2020, 25, 4667. [Google Scholar] [CrossRef]
- Makura, Y.; Ueda, A.; Kato, T.; Iyoshi, A.; Higuchi, M.; Doi, M.; Tanaka, M. X-ray crystallographic structure of alpha-helical peptide stabilized by hydrocarbon stapling at i,i + 1 positions. Int. J. Mol. Sci. 2021, 22, 5364. [Google Scholar] [CrossRef]
- Oba, M.; Kunitake, M.; Kato, T.; Ueda, A.; Tanaka, M. Enhanced and prolonged cell-penetrating abilities of arginine-rich peptides by introducing cyclic alpha,alpha-disubstituted alpha-amino acids with stapling. Bioconjugate Chem. 2017, 28, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Makura, Y.; Kakazu, S.; Kato, T.; Umeno, T.; Hirayama, K.; Doi, M.; Oba, M.; Tanaka, M. E-Selective ring-closing metathesis in alpha-helical stapled peptides using carbocyclic alpha,alpha-disubstituted alpha-amino acids. Org. Lett. 2022, 24, 1049–1054. [Google Scholar] [CrossRef]
- Verdine, G.L.; Hilinski, G.J. Chapter one—Stapled peptides for intracellular drug targets. In Methods in Enzymology; Wittrup, K.D., Verdine, G.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 503, pp. 3–33. [Google Scholar]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N. Hydrocarbon stapled peptides as modulators of biological function. ACS Chem. Biol. 2015, 10, 1362–1375. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, C.; Benedetti, E. The polypeptide 310-helix. Trends Biochem. Sci. 1991, 16, 350–353. [Google Scholar] [CrossRef]
- Toniolo, C.; Crisma, M.; Formaggio, F.; Peggion, C.; Broxterman, Q.B.; Kaptein, B. Molecular spacers for physicochemical investigations based on novel helical and extended peptide structures. Biopolymers 2004, 76, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.R.; Roberts, S.M. The mechanism of polyleucine catalysed asymmetric epoxidation. Chem. Commun. 2004, 2004, 2018–2020. [Google Scholar] [CrossRef] [PubMed]
- Bougauchi, M.; Watanabe, S.; Arai, T.; Sasaki, H.; Shibasaki, M. Catalytic asymmetric epoxidation of α,β-unsaturated ketones promoted by lanthanoid complexes. J. Am. Chem. Soc. 1997, 119, 2329–2330. [Google Scholar] [CrossRef]
- Choudary, B.M.; Kantam, M.L.; Ranganath, K.V.S.; Mahendar, K.; Sreedhar, B. Bifunctional nanocrystalline MgO for chiral epoxy ketones via Claisen−Schmidt condensation−asymmetric epoxidation reactions. J. Am. Chem. Soc. 2004, 126, 3396–3397. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Ohshima, T.; Yamaguchi, K.; Shibasaki, M. Catalytic asymmetric epoxidation of enones using La−BINOL−triphenylarsine oxide complex: Structural determination of the asymmetric catalyst. J. Am. Chem. Soc. 2001, 123, 2725–2726. [Google Scholar] [CrossRef] [PubMed]
- Lygo, B.; Wainwright, P.G. Phase-transfer catalysed asymmetric epoxidation of enones using N-anthracenylmethyl-substituted Cinchona alkaloids. Tetrahedron 1999, 55, 6289–6300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
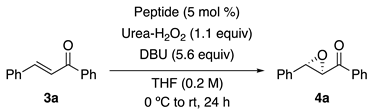 | |||
| Entry | Peptide | Conv (%) 1 | Ee (%) 2 |
| 1 | 1a′ | >99 | 97 |
| 2 | 2a′ | >99 | 97 |
| 3 | 1b′ | >99 | 98 |
| 4 | 2b′ | >99 | 98 |
| 5 | 1c′ | >99 | 97 |
| 6 | 2c′ | >99 | 98 |
| 7 | 1d′ | >99 | 98 |
| 8 | 2d′ | >99 | 97 |
| 9 | 1e′ | >99 | 89 |
| 10 | 2e′ | 92 | 90 |
| 11 | none | 71 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyoshi, A.; Ueda, A.; Umeno, T.; Kato, T.; Hirayama, K.; Doi, M.; Tanaka, M. Conformational Analysis and Organocatalytic Activity of Helical Stapled Peptides Containing α-Carbocyclic α,α-Disubstituted α-Amino Acids. Molecules 2024, 29, 4340. https://doi.org/10.3390/molecules29184340
Iyoshi A, Ueda A, Umeno T, Kato T, Hirayama K, Doi M, Tanaka M. Conformational Analysis and Organocatalytic Activity of Helical Stapled Peptides Containing α-Carbocyclic α,α-Disubstituted α-Amino Acids. Molecules. 2024; 29(18):4340. https://doi.org/10.3390/molecules29184340
Chicago/Turabian StyleIyoshi, Akihiro, Atsushi Ueda, Tomohiro Umeno, Takuma Kato, Kazuhiro Hirayama, Mitsunobu Doi, and Masakazu Tanaka. 2024. "Conformational Analysis and Organocatalytic Activity of Helical Stapled Peptides Containing α-Carbocyclic α,α-Disubstituted α-Amino Acids" Molecules 29, no. 18: 4340. https://doi.org/10.3390/molecules29184340
APA StyleIyoshi, A., Ueda, A., Umeno, T., Kato, T., Hirayama, K., Doi, M., & Tanaka, M. (2024). Conformational Analysis and Organocatalytic Activity of Helical Stapled Peptides Containing α-Carbocyclic α,α-Disubstituted α-Amino Acids. Molecules, 29(18), 4340. https://doi.org/10.3390/molecules29184340