
Mapping of Some Further Alkylation-Initiated Pathways to Polyheterocyclic Compounds from Indigo and Indirubin

 , , , and
, , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
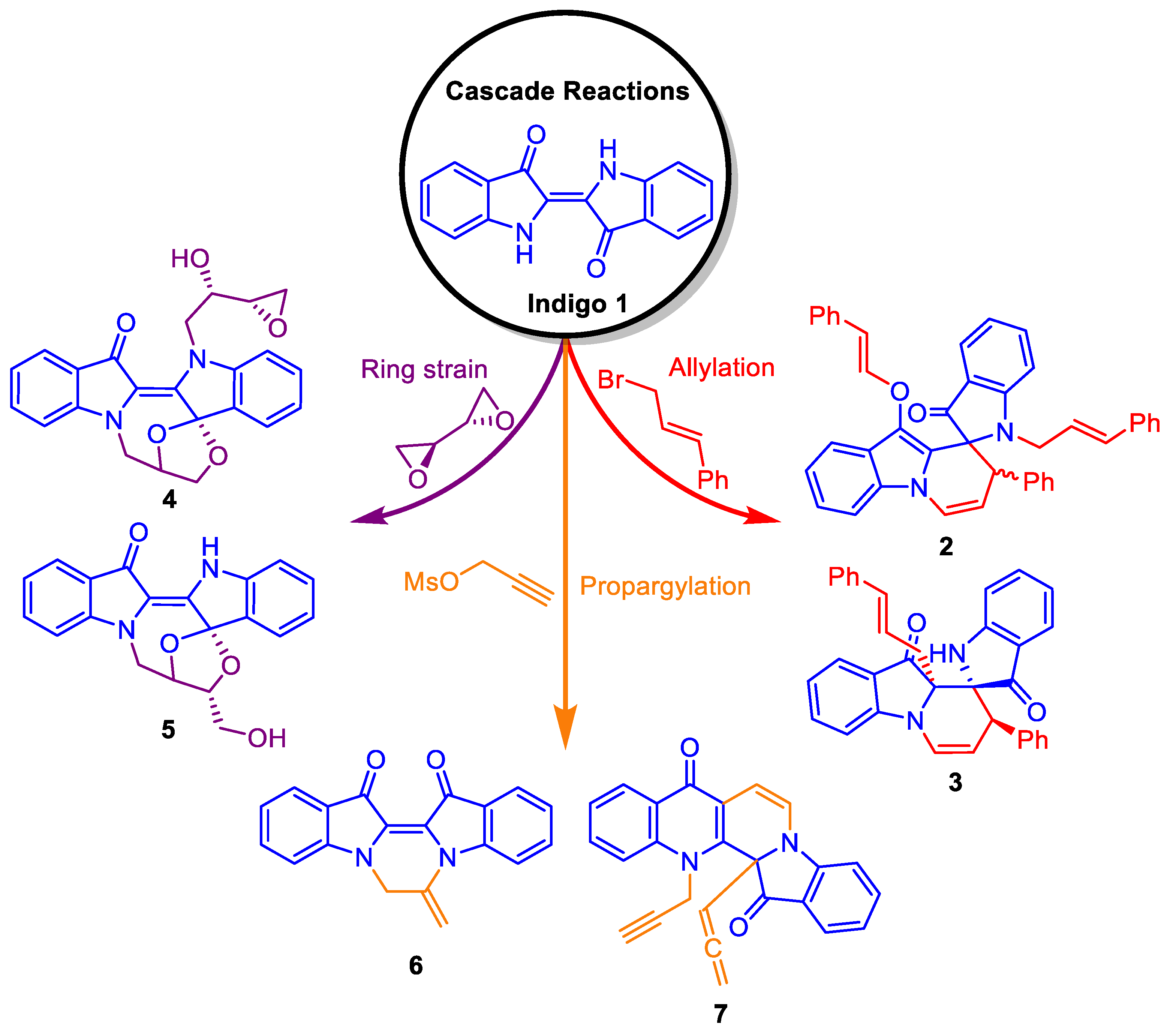
1. Introduction
2. Results
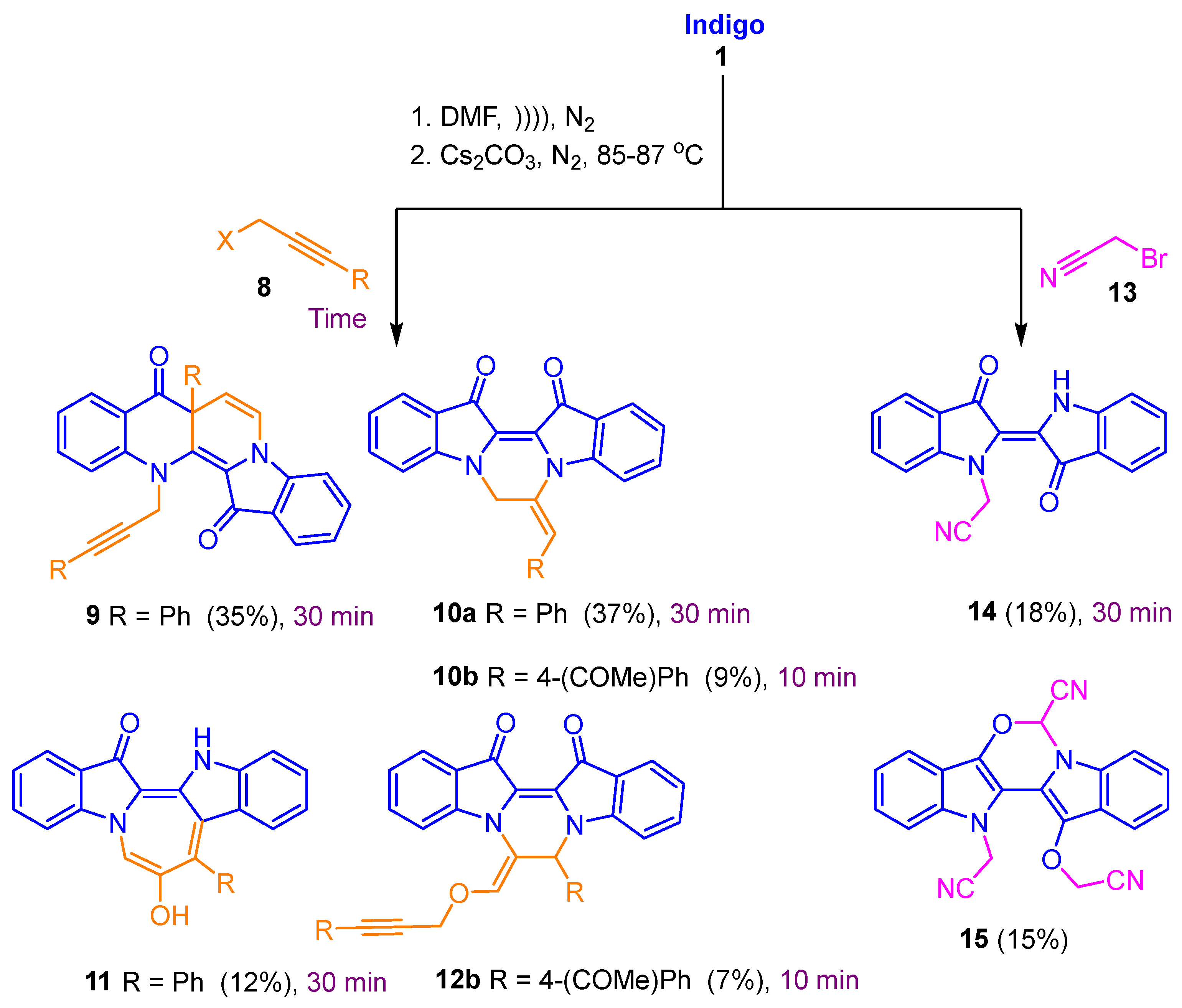
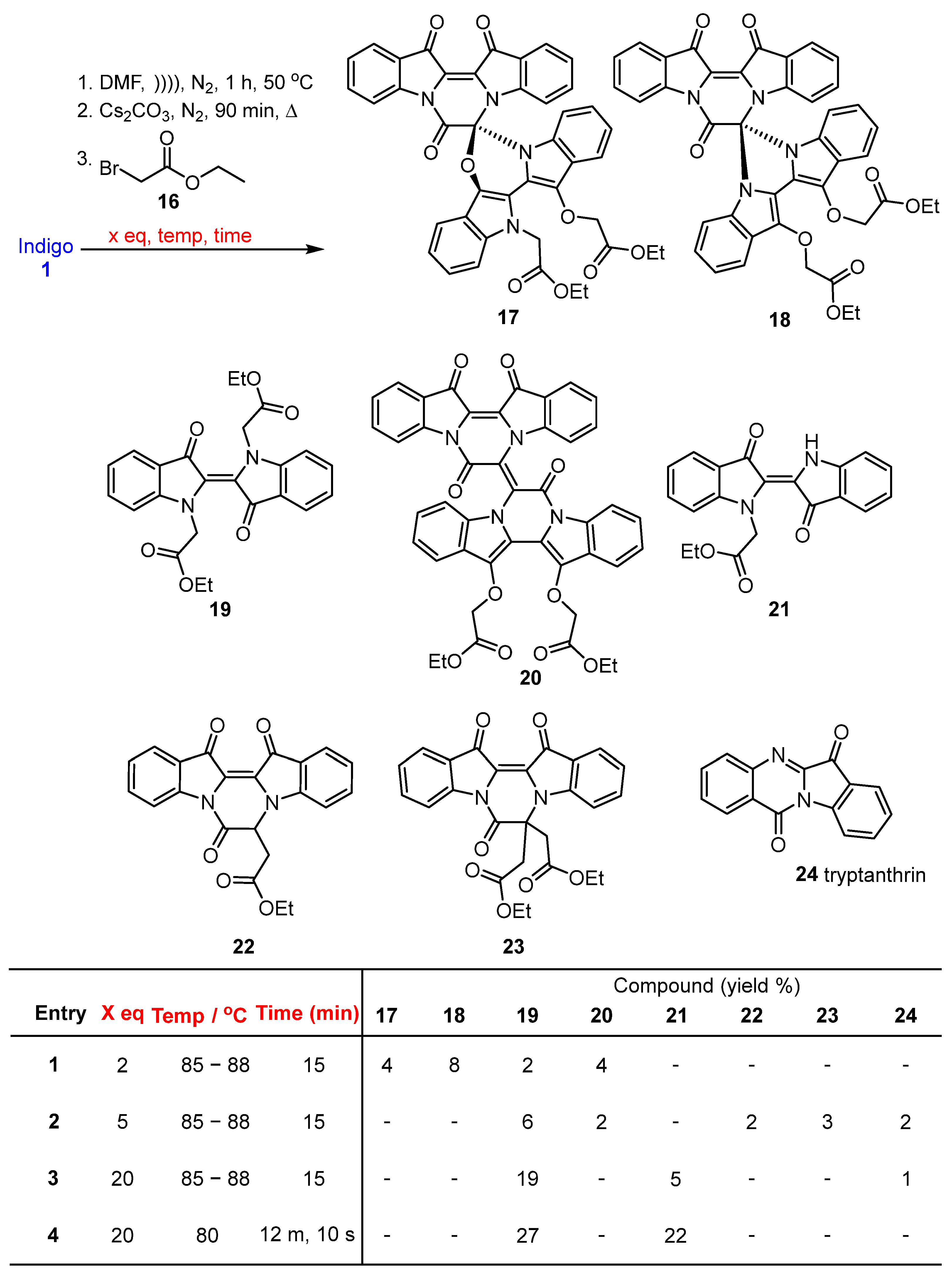
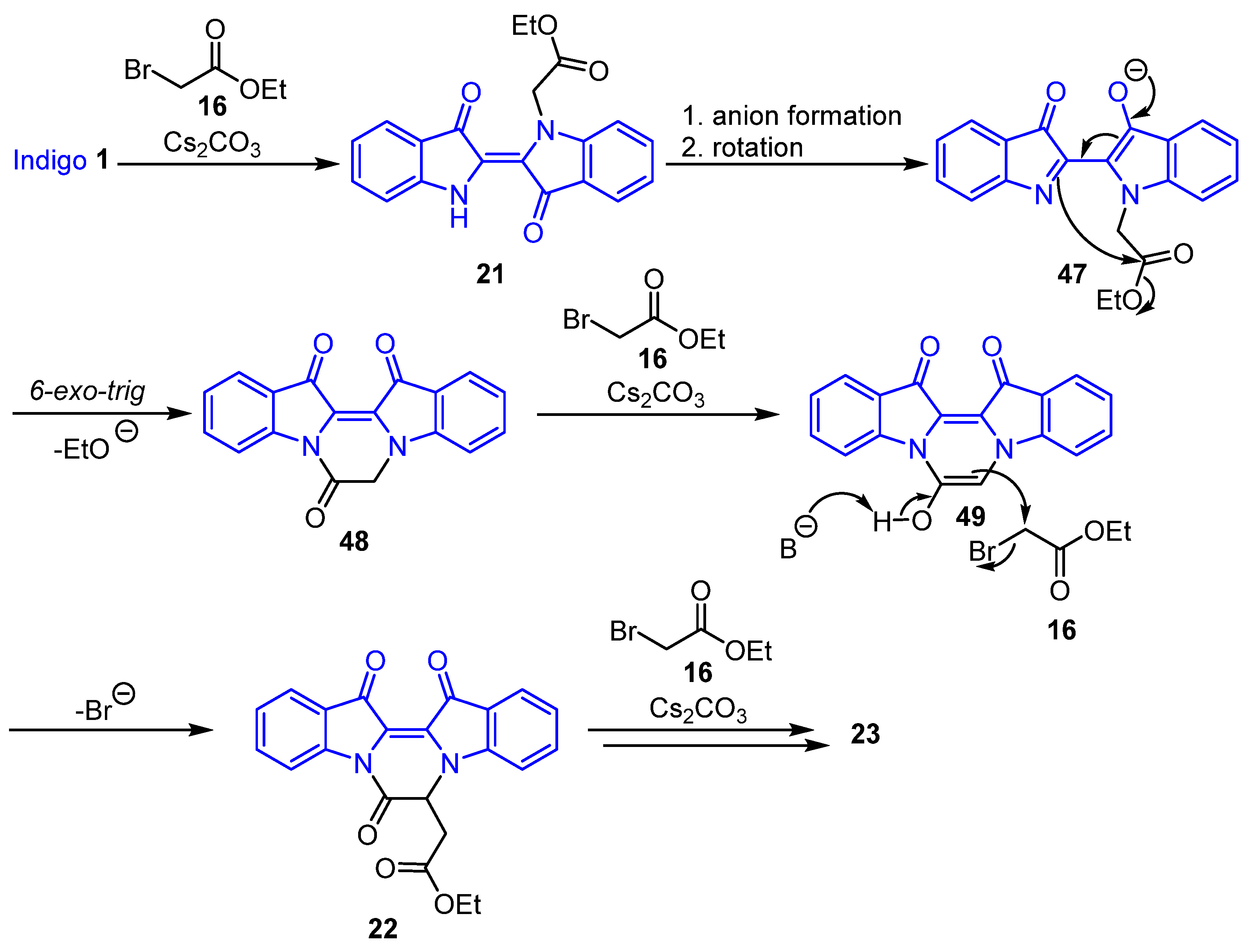
2.1. Reaction of Indigo with Ethyl Bromoacetate
2.2. Investigation of the Effect of Reaction Conditions on Product Outcomes for the Indigo and Ethyl Bromoacetate Reaction
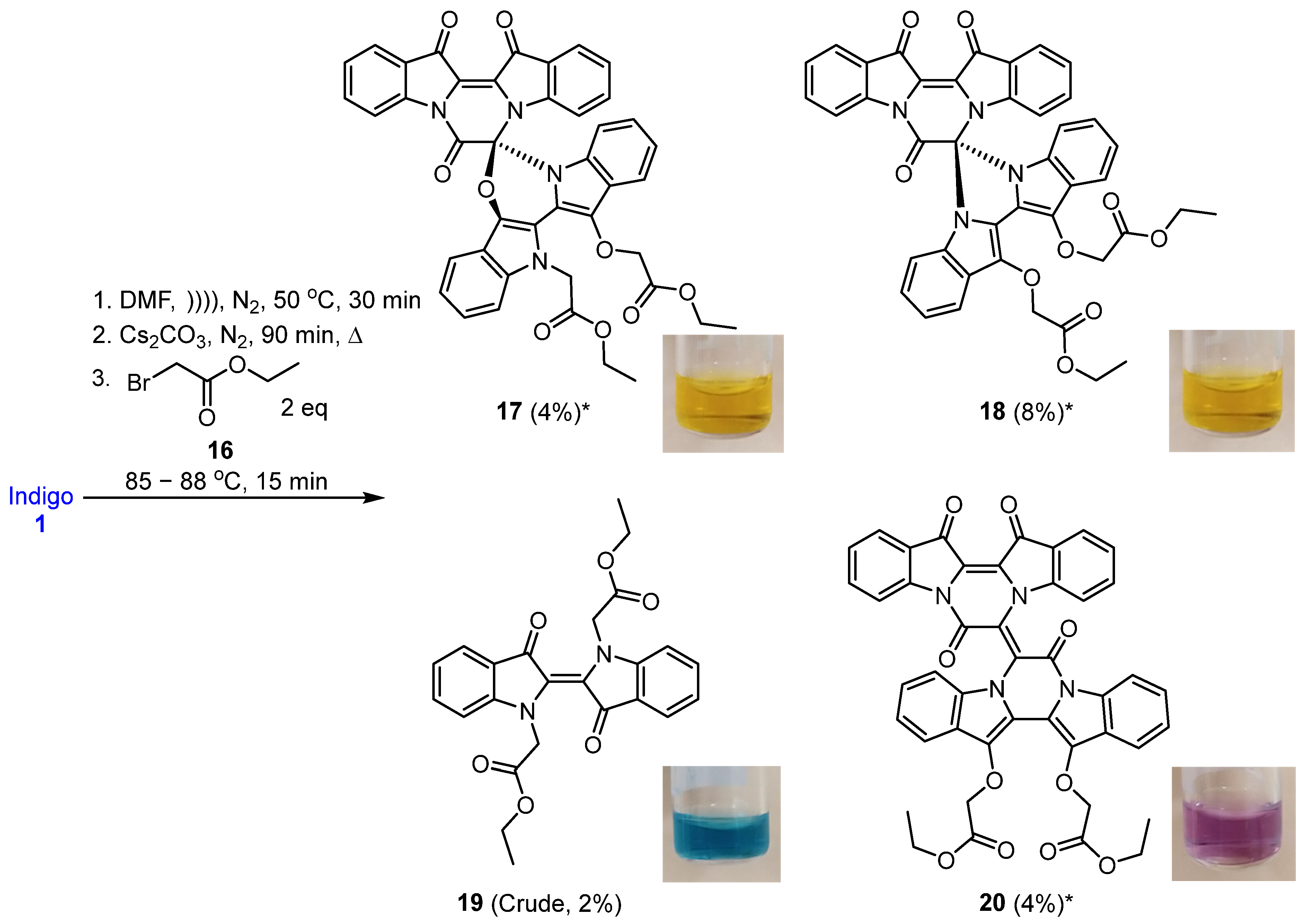
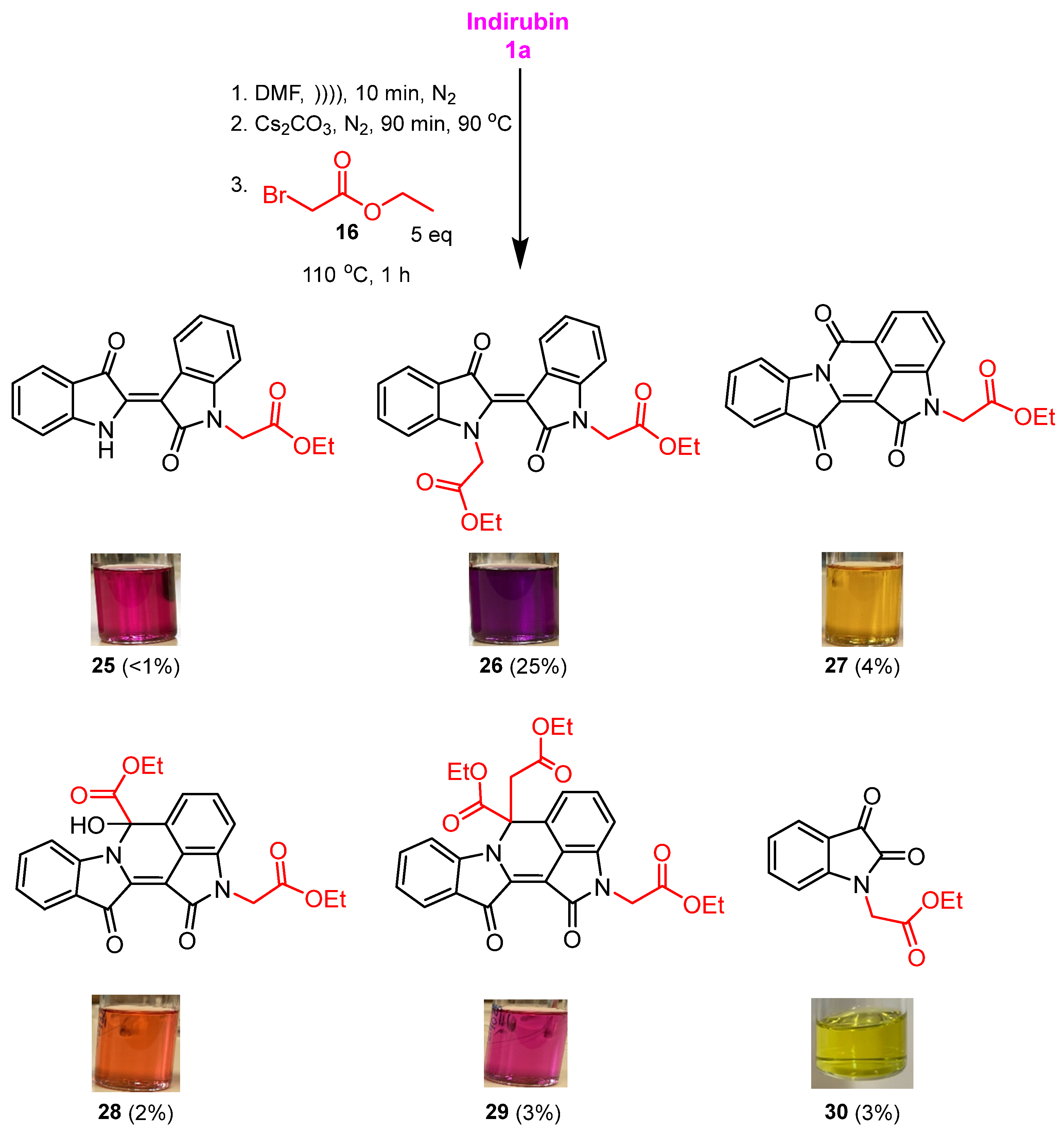
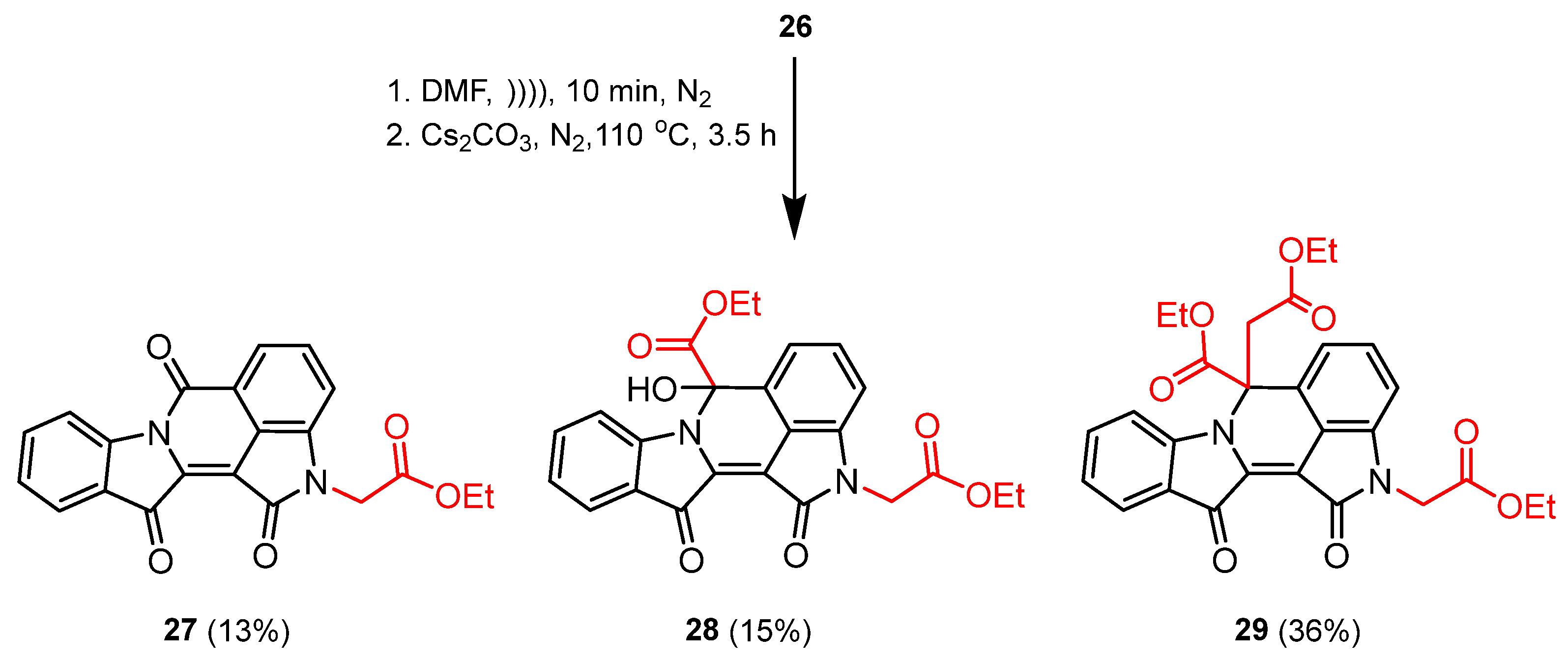
2.3. Reaction of Indirubin with Ethyl Bromoacetate
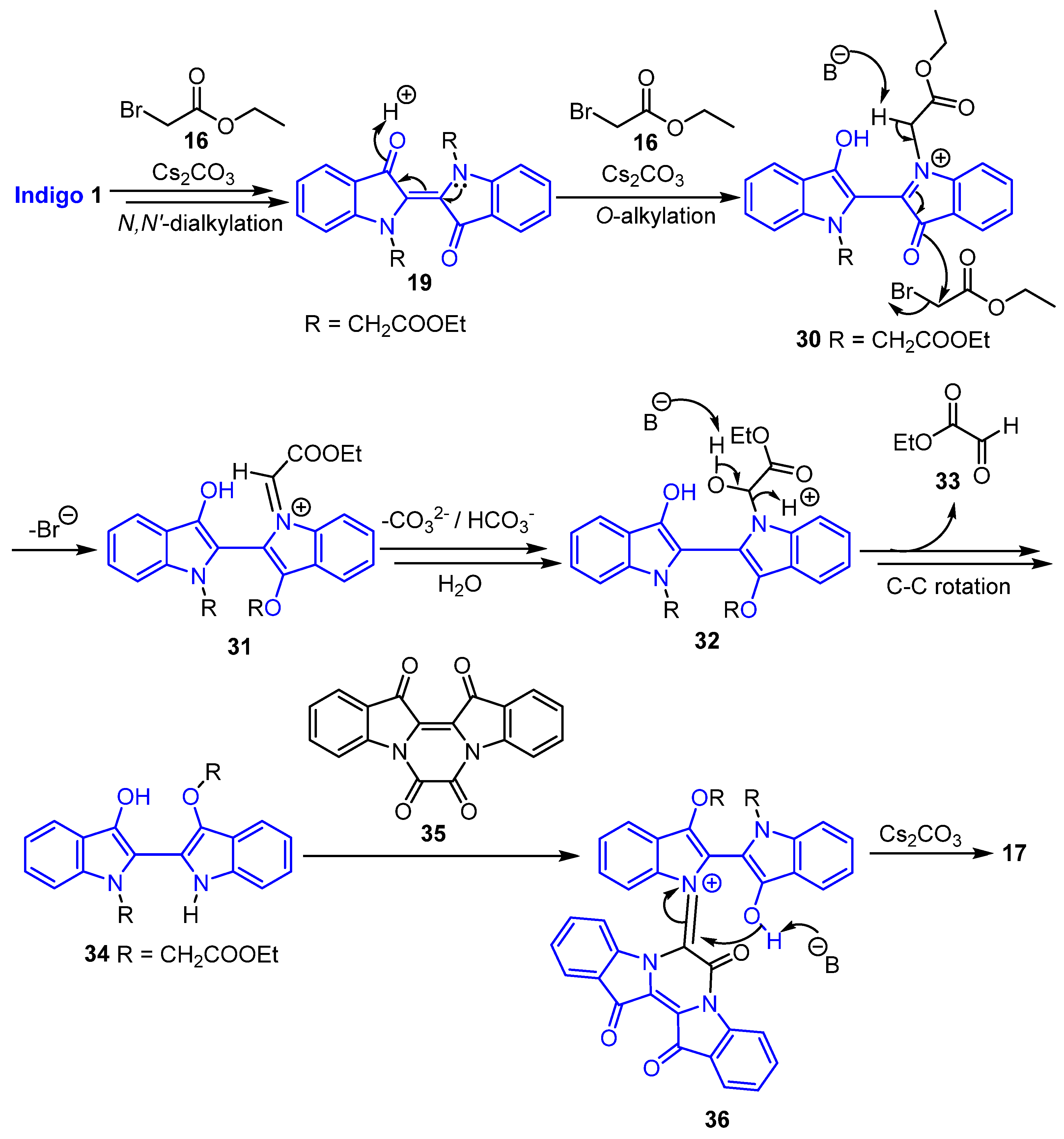
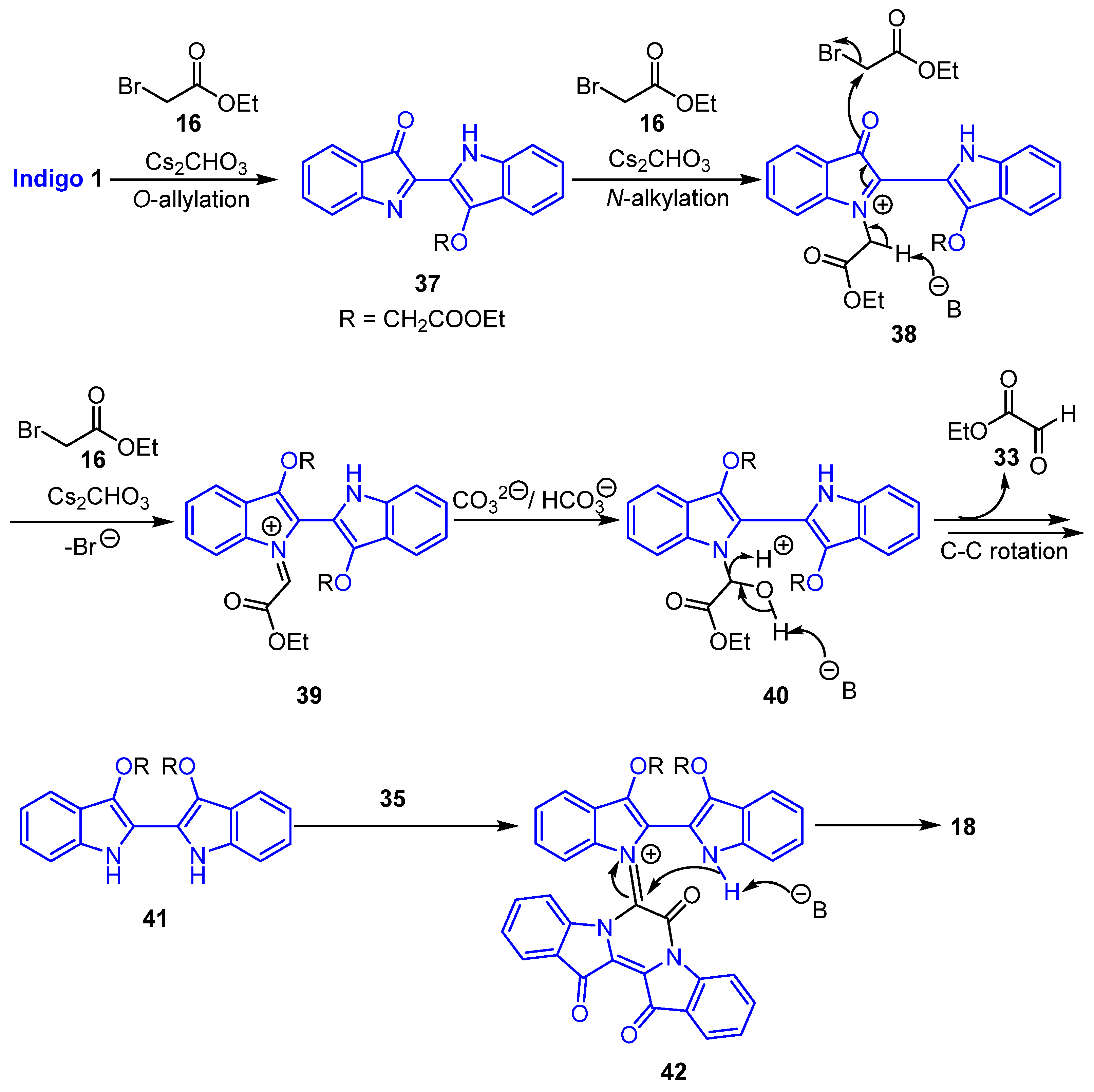
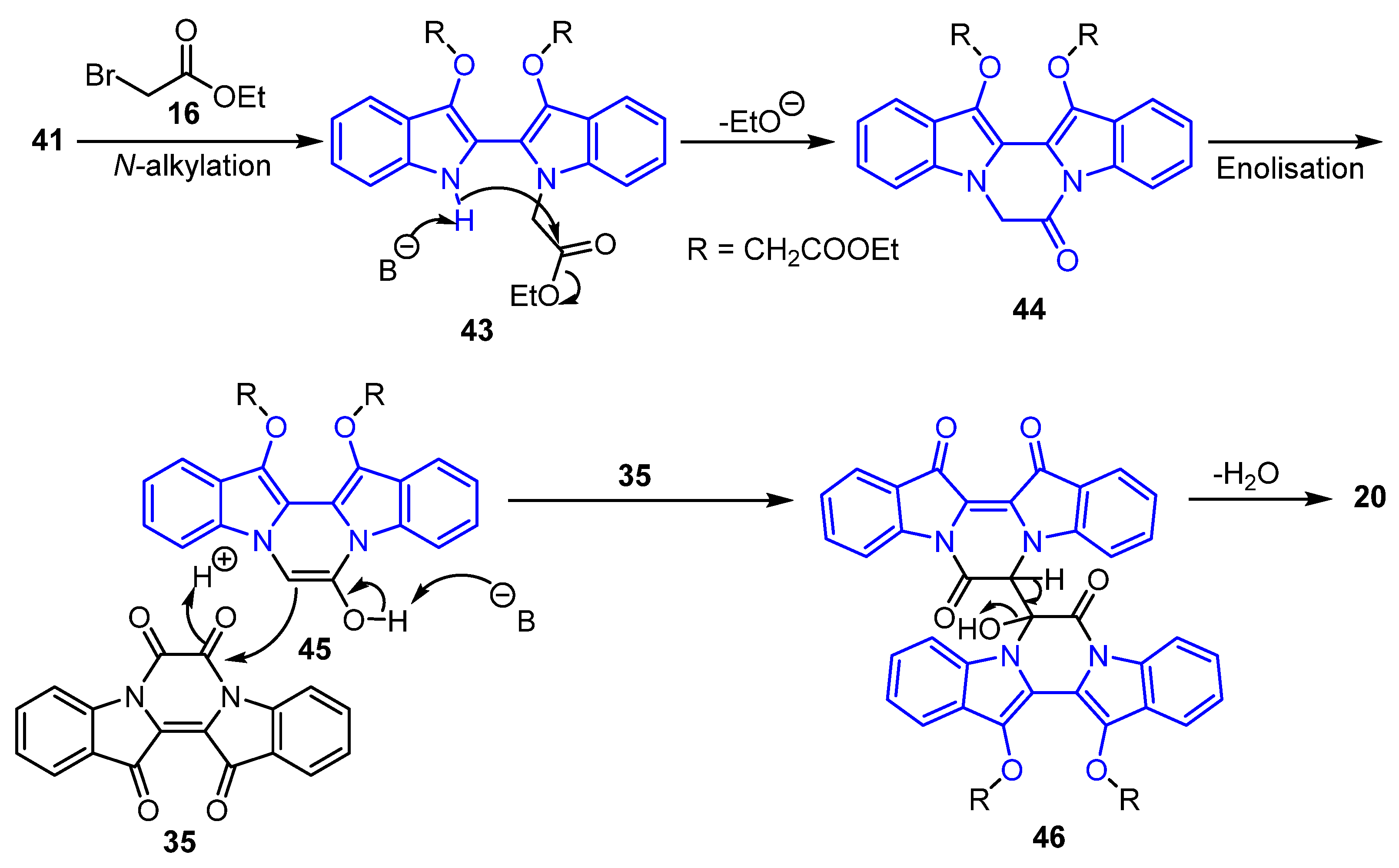
2.4. Mechanistic Discussion of the Indigo-Ethyl Bromoacetate Reactions
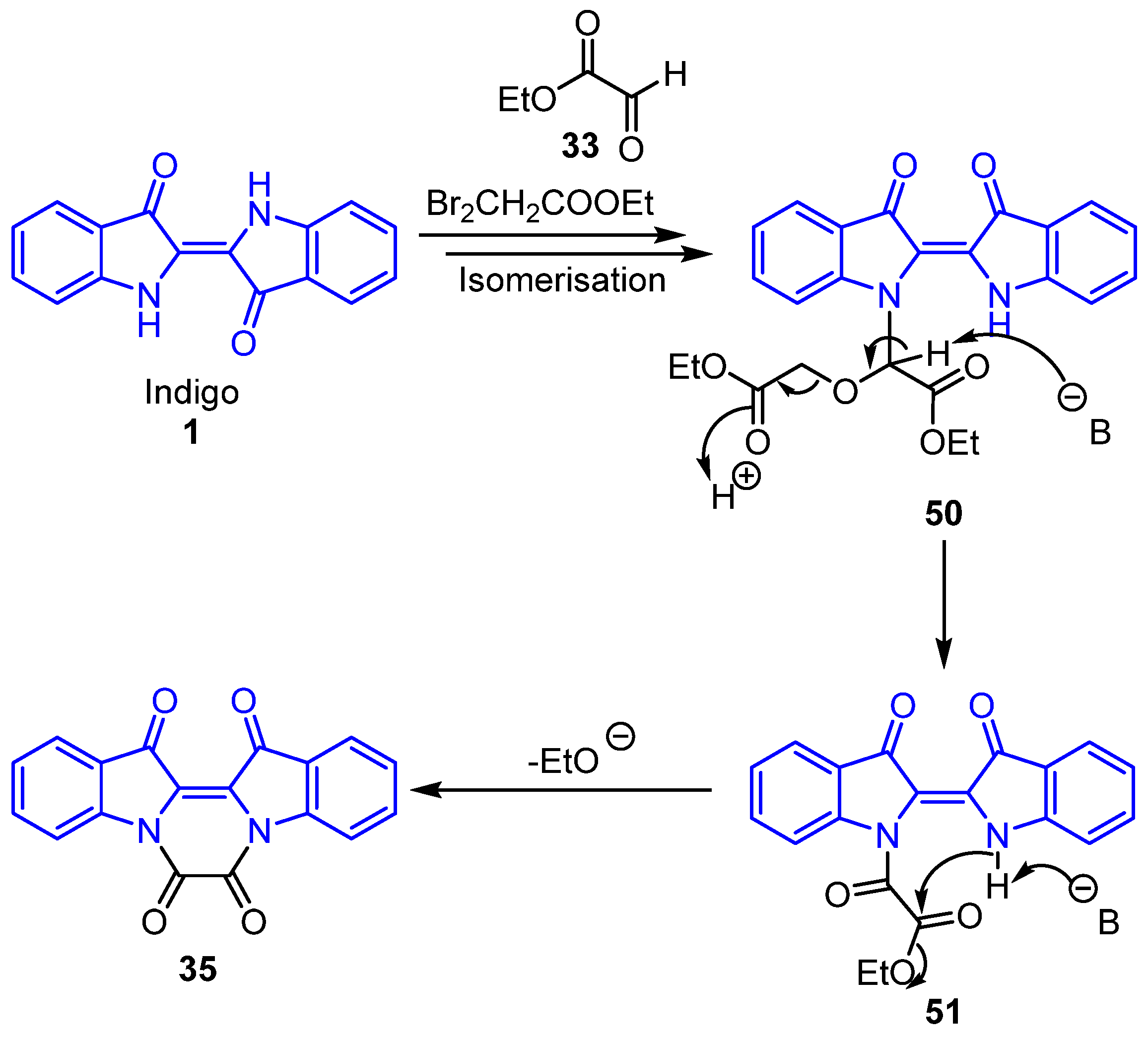
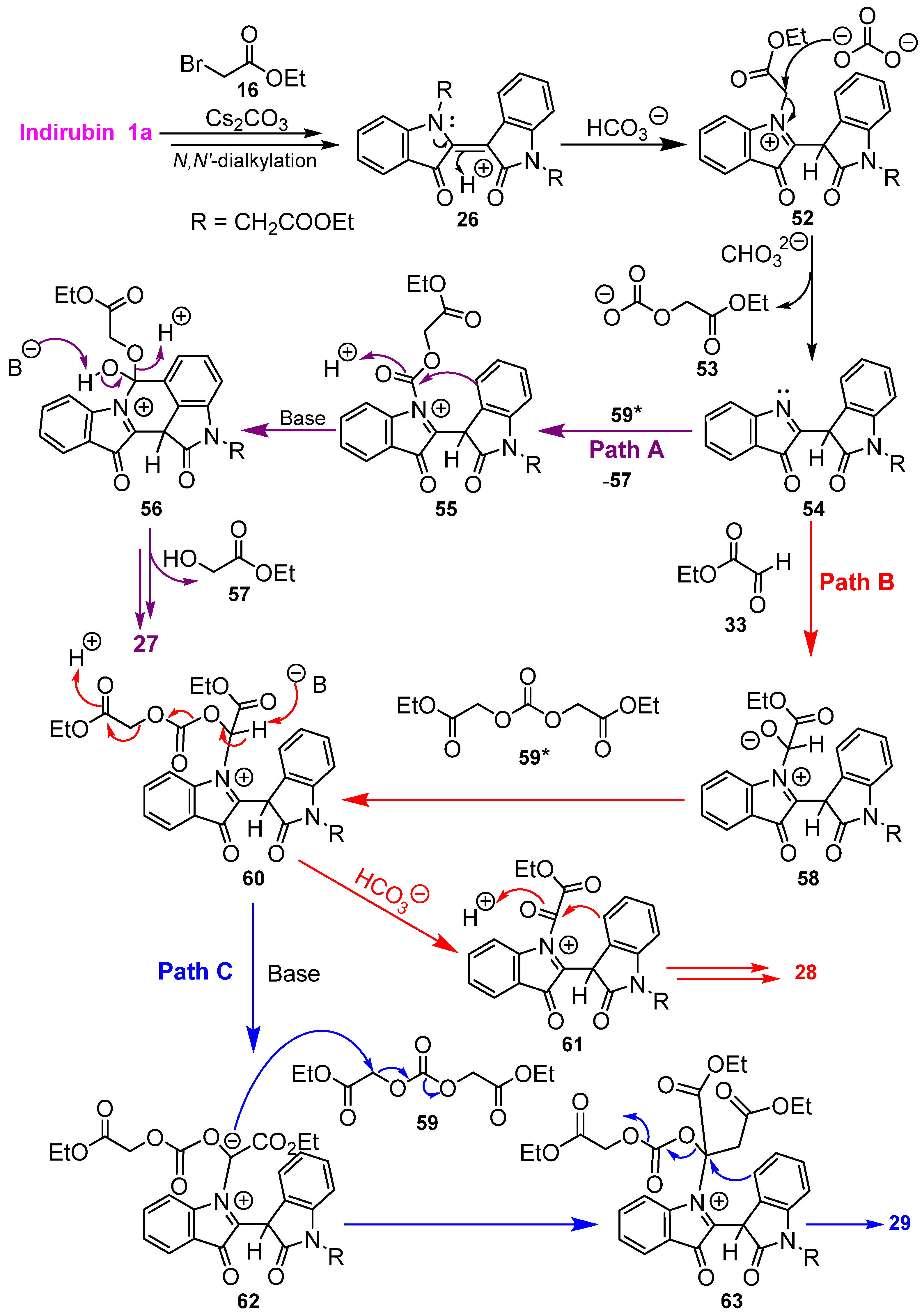
2.5. Mechanistic Discussion of the Indirubin-Ethyl Bromoacetate Reaction
3. Materials and Methods
3.1. Chemistry
3.2. Reactions of Indigo with Ethyl Bromoacetate
3.2.1. Method 1
3.2.2. Method 2
3.2.3. Method 3
3.3. Reactions of Indirubin with Ethyl Bromoacetate
3.3.1. Alkylation Reaction of Indirubin 1a with Ethyl Bromoacetate 16
3.3.2. Cascade Reaction of Indirubin 1a with Ethyl Bromoacetate 16
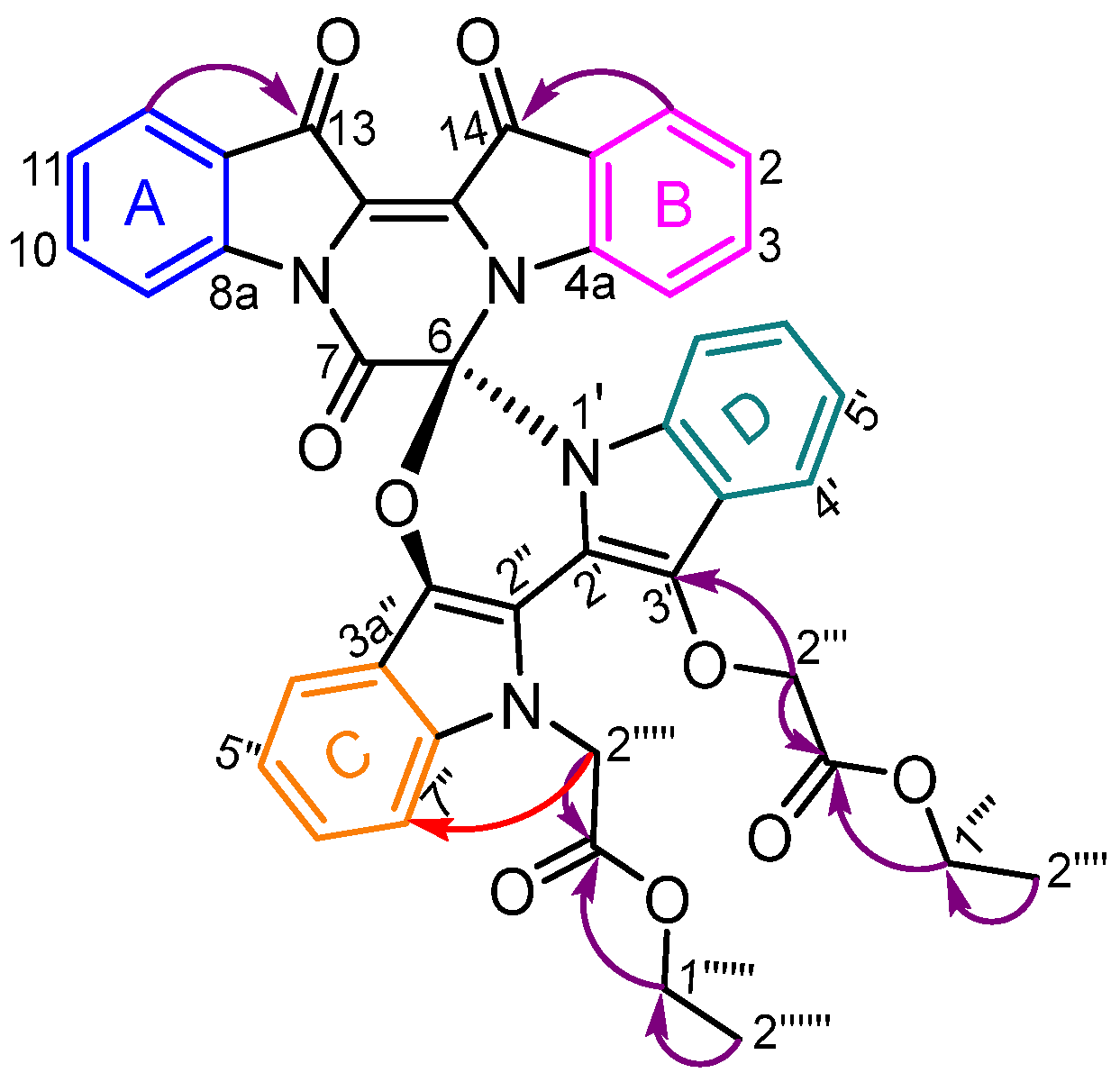
3.3.3. Mechanistic Investigation of N,N′-Dialkylated Indirubin 26
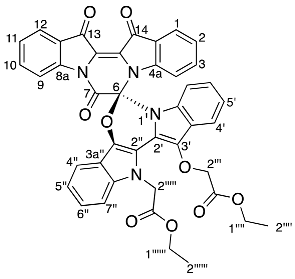
- Ethyl (R)-2-(13′-(2-ethoxy-2-oxoethoxy)-7,13,14-trioxo-13,14-dihydro-7H,12′H-spiro[pyrazino[1,2-a:4,3-a′]diindole-6,6′-[1,3]oxazino[3,4-a:5,6-b′]diindol]-12′-yl)acetate 17
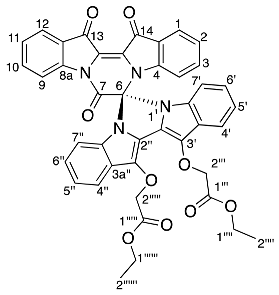
- Diethyl 2,2′-((7′,13′,14′-trioxo-13′,14′-dihydro-7′H-spiro[imidazo[1,5-a:3,4-a′]diindole-6,6′-pyrazino[1,2-a:4,3-a′]diindole]-12,13-diyl)bis(oxy))diacetate 18
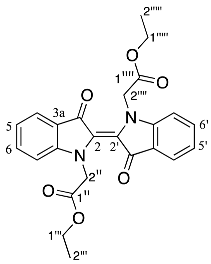
- Diethyl (E)-2,2′-(3,3′-dioxo-[2,2′-biindolinylidene]-1,1′-diyl) diacetate 19

- Diethyl (E)-2,2′-((7,7′,13′,14′-tetraoxo-13′,14′-dihydro-7H,7′H-[6,6′-bipyrazino[1,2-a:4,3-a′]diindolylidene]-13,14-diyl)bis(oxy)) diacetate 20
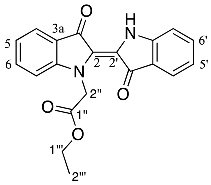
- Ethyl (E)-2-(3,3′-dioxo-[2,2′-biindolinylidene]-1-yl)acetate 21
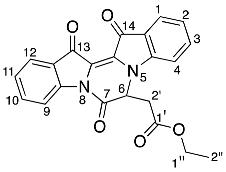
- Ethyl 2-(7,13,14-trioxo-6,7,13,14-tetrahydropyrazino[1,2-a:4,3-a′]diindol-6-yl) acetate 22
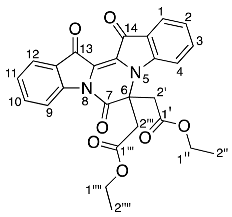
- Diethyl 2,2′-(7,13,14-trioxo-6,7,13,14-tetrahydropyrazino[1,2-a:4,3-a′]diindole-6,6-diyl) diacetate 23
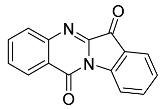
- Tryptanthrin 24

- Ethyl (Z)-2-(2′,3-dioxo-[2,3′-biindolinylidene]-1′-yl)acetate 25
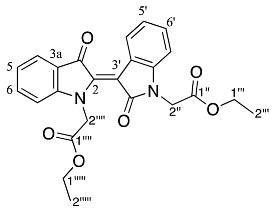
- Diethyl (Z)-2,2′-(2′,3-dioxo-[2,3′-biindolinylidene]-1,1′-diyl) diacetate 26
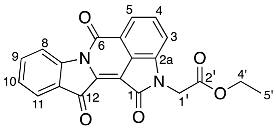
- Ethyl 2-(1,6,12-trioxo-6,12-dihydroindolo[1,2-b]pyrrolo[4,3,2-de]isoquinolin-2(1H)-yl) acetate 27
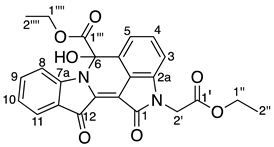
- Ethyl 2-(2-ethoxy-2-oxoethyl)-6-hydroxy-1,12-dioxo-1,2,6,12-tetrahydroindolo[1,2-b]pyrrolo[4,3,2-de]isoquinoline-6-carboxylate 28
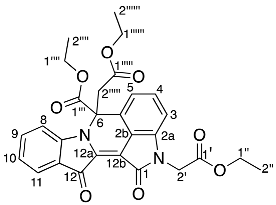
- Diethyl 2,2′-(6-(ethoxycarbonyl)-1,12-dioxo-6,12-dihydroindolo[1,2-b]pyrrolo[4,3,2-de]isoquinoline-2,6(1H)-diyl) diacetate 29
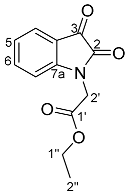
- Ethyl 2-(2,3-dioxo-1H-indol-1-yl)acetate 30
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, K.-Y. Pigments, A review of recent progress in the synthesis of bio-indigoids and their biologically assisted end-use applications. Dye. Pigment. 2020, 181, 108570. [Google Scholar] [CrossRef]
- Ma, L.; Sun, T.; Liu, Y.; Zhao, Y.; Liu, X.; Li, Y.; Chen, X.; Cao, L.; Kang, Q.; Guo, J. Enzymatic synthesis of indigo derivatives by tuning P450 BM3 peroxygenases. Synth. Syst. Biotechnol. 2023, 8, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Hecht, S. A blueprint for transforming indigos to photoresponsive molecular tools. Chem. Eur. J. 2023, 29, e202300981. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Bora, S.R.; Bishen, S.M.; Mishra, H.; Kalita, D.J.; Wahab, A. Photophysics of a monoannulated indigo: Intra-and intermolecular charge transfer. J. Phys. Chem. A 2024, 128, 2565–2573. [Google Scholar] [CrossRef]
- Kaplan, G.; Seferoğlu, Z.; Berdnikova, D.V. Photochromic derivatives of indigo: Historical overview of development, challenges and applications. Beilstein J. Org. Chem. 2024, 20, 228–242. [Google Scholar] [CrossRef]
- Reva, I.; Lapinski, L.; Nowak, M.J. Stability of monomeric indigo in the electronic ground state: An experimental matrix isolation infrared and theoretical study. Dyes. Pigment. 2024, 222, 111827. [Google Scholar] [CrossRef]
- Xu, H.; Chakraborty, R.; Adak, A.K.; Das, A.; Yang, B.; Meier, D.; Riss, A.; Reichert, J.; Narasimhan, S.; Barth, J.V.; et al. On-surface isomerization of indigo within 1D coordination polymers. Angew. Chem. Int. Ed. 2024, 136, e202319162. [Google Scholar] [CrossRef]
- Shakoori, A.; Bremner, J.B.; Willis, A.C.; Haritakun, R.; Keller, P.A. Rapid cascade synthesis of poly-heterocyclic architectures from indigo. J. Org. Chem. 2013, 78, 7639–7647. [Google Scholar] [CrossRef]
- Shakoori, A.; Bremner, J.B.; Abdel-Hamid, M.K.; Willis, A.C.; Haritakun, R.; Keller, P.A. Further exploration of the heterocyclic diversity accessible from the allylation chemistry of indigo. Beilstein J. Org. Chem. 2015, 11, 481–492. [Google Scholar] [CrossRef]
- Butler, N.M.; Hendra, R.; Bremner, J.B.; Willis, A.C.; Lucantoni, L.; Avery, V.M.; Keller, P.A. Cascade reactions of indigo with oxiranes and aziridines: Efficient access to dihydropyrazinodiindoles and spiro-oxazocinodiindoles. Org. Biomol. Chem. 2018, 16, 6006–6016. [Google Scholar] [CrossRef]
- McCosker, P.M.; Butler, N.M.; Shakoori, A.; Perry, M.J.; Mullen, J.W.; Willis, A.C.; Clark, T.; Bremner, J.B.; Guldi, D.M.; Keller, P.A.; et al. The cascade reactions of indigo with propargyl substrates for heterocyclic and photophysical diversity. Chem. Eur. J. 2021, 27, 3708–3721. [Google Scholar] [CrossRef]
- Sele, A.M.; Bremner, J.B.; Willis, A.C.; Haritakun, R.; Griffith, R.; Keller, P.A. A cascade synthetic route to new bioactive spiroindolinepyrido [1, 2-a] indolediones from indirubin. Tetrahedron 2015, 71, 8357–8367. [Google Scholar] [CrossRef]
- Kaur, R.; Manjal, S.K.; Rawal, R.K.; Kumar, K.J. B. Recent synthetic and medicinal perspectives of tryptanthrin. Bioorg. Med. Chem. 2017, 25, 4533–4552. [Google Scholar] [CrossRef]
- Pinheiro, D.; Pineiro, M.; Pina, J.; Brandão, P.; Galvão, A.M.; Seixas de Melo, J.S. Tryptanthrin from indigo: Synthesis, excited state deactivation routes and efficient singlet oxygen sensitization. Dye. Pigment. 2020, 175, 108125. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Bonasera, A.; Hristov, L.; Garmshausen, Y.; Schmidt, B.M.; Jacquemin, D.; Hecht, S. N,N′-disubstituted indigos as readily available red-light photoswitches with tunable thermal half-lives. J. Am. Chem. Soc. 2017, 139, 15205–15211. [Google Scholar] [CrossRef]
- Kihara, Y.; Tani, S.; Higashi, Y.; Teramoto, T.; Nagasawa, Y. Ultrafast excited state dynamics of forward and reverse trans-cis photoisomerization of red-light-absorbing indigo derivatives. J. Phys. Chem. B 2022, 126, 3539–3550. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Z.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L.; Chen, Y. Anticancer potential of indirubins in medicinal chemistry: Biological activity, structural modification, and structure-activity relationship. J. Med. Chem. 2021, 223, 113652. [Google Scholar] [CrossRef]
- Jautelat, R.; Brumby, T.; Schäfer, M.; Briem, H.; Eisenbrand, G.; Schwahn, S.; Krüger, M.; Lücking, U.; Prien, O.; Siemeister, G. From the insoluble dye indirubin towards highly active, soluble CDK2-inhibitors. ChemBioChem 2005, 6, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Eisenbrand, G.; Hippe, F.; Jakobs, S.; Muehlbeyer, S. Molecular mechanisms of indirubin and its derivatives: Novel anticancer molecules with their origin in traditional Chinese phytomedicine. J. Cancer Res. Clin. Oncol. 2004, 130, 627–635. [Google Scholar] [CrossRef]
- Alex, D.; Lam, I.K.; Lin, Z.; Lee, S.M. Y. Indirubin shows anti-angiogenic activity in an in vivo zebrafish model and an in vitro HUVEC model. J. Ethnopharmacol. 2010, 131, 242–247. [Google Scholar] [CrossRef]
- Meijer, L.; Shearer, J.; Bettayeb, K.; Ferandin, Y. Diversity of the Intracellular Mechanisms Underlying the Anti-Tumor Properties of Indirubins. Int. Congr. Ser. 2007, 1304, 60–74. [Google Scholar]
- Zhu, J.; Cai, Y.; Kong, M.; Li, Y.; Zhu, L.; Zhang, J.; Yu, Z.; Xu, S.; Hong, L.; Chen, C.; et al. Design, Synthesis, and Biological Evaluation for First GPX4 and CDK Dual Inhibitors. J. Med. Chem. 2024, 67, 2758–2776. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Chen, X.; Yin, F.; Li, S.; Zhang, Y.; Luo, H.; Luo, Z.; Cui, N.; Chen, Y.; Li, X.; et al. Indirubin derivatives as bifunctional molecules inducing DNA damage and targeting PARP for the treatment of cancer. Eur. J. Med. Chem. 2023, 261, 115843. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-W.; He, J.-B.; You, Y.-H.; Zhang, Y.-M.; Zhang, S.-L. Spectroelectrochemistry of solid indirubin and its sulfonated form. Electrochim. Acta 2011, 56, 1219–1226. [Google Scholar] [CrossRef]
- Nobre, D.C.; Delgado-Pinar, E.; Cunha, C.; Galvao, A.M.; Seixas de Melo, J.S. Indirubin: Nature finding efficient light-activated protective molecular mechanisms. Dye. Pigment. 2023, 212, 111116. [Google Scholar] [CrossRef]
- Görner, H.; Pouliquen, J.; Kossanyi, J. Trans to cis photoisomerization of N,N′-disubstituted indigo dyes via excited singlet states; a laser flash photolysis and steady state irradiation study. Can. J. Chem. 1987, 65, 708–717. [Google Scholar] [CrossRef]
- Wang, C.; Yan, J.; Du, M.; Burlison, J.A.; Li, C.; Sun, Y.; Zhao, D.; Liu, J. One step synthesis of indirubins by reductive coupling of isatins with KBH4. Tetrahedron 2017, 73, 2780–2785. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Ramadan, E.S.; Hamid, H.A.; Hagar, M. Microwave irradiation for enhancing the regioselective synthesis of 6H-indolo [2,3-b] quinoxalines. J. Chem. Res. 2005, 2005, 229–232. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, S.; McCosker, P.M.; Willis, A.C.; Pyne, S.G.; Richardson, C.; Bremner, J.B.; Keller, P.A. Mapping of Some Further Alkylation-Initiated Pathways to Polyheterocyclic Compounds from Indigo and Indirubin. Molecules 2024, 29, 4242. https://doi.org/10.3390/molecules29174242
Ali S, McCosker PM, Willis AC, Pyne SG, Richardson C, Bremner JB, Keller PA. Mapping of Some Further Alkylation-Initiated Pathways to Polyheterocyclic Compounds from Indigo and Indirubin. Molecules. 2024; 29(17):4242. https://doi.org/10.3390/molecules29174242
Chicago/Turabian StyleAli, Sarfaraz, Patrick M. McCosker, Anthony C. Willis, Stephen G. Pyne, Christopher Richardson, John B. Bremner, and Paul A. Keller. 2024. "Mapping of Some Further Alkylation-Initiated Pathways to Polyheterocyclic Compounds from Indigo and Indirubin" Molecules 29, no. 17: 4242. https://doi.org/10.3390/molecules29174242
APA StyleAli, S., McCosker, P. M., Willis, A. C., Pyne, S. G., Richardson, C., Bremner, J. B., & Keller, P. A. (2024). Mapping of Some Further Alkylation-Initiated Pathways to Polyheterocyclic Compounds from Indigo and Indirubin. Molecules, 29(17), 4242. https://doi.org/10.3390/molecules29174242