Activated Iron-Porous Carbon Nanomaterials as Adsorbents for Methylene Blue and Congo Red

 , , , , , and
, , , , , and 
Abstract
1. Introduction
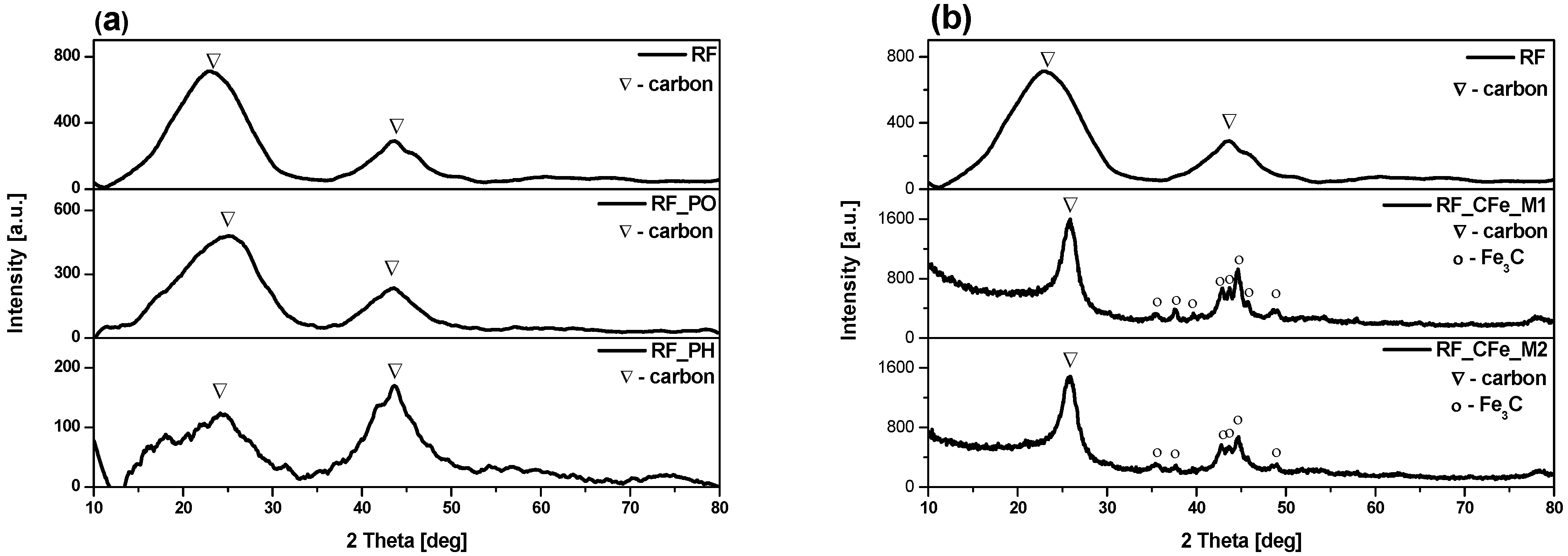
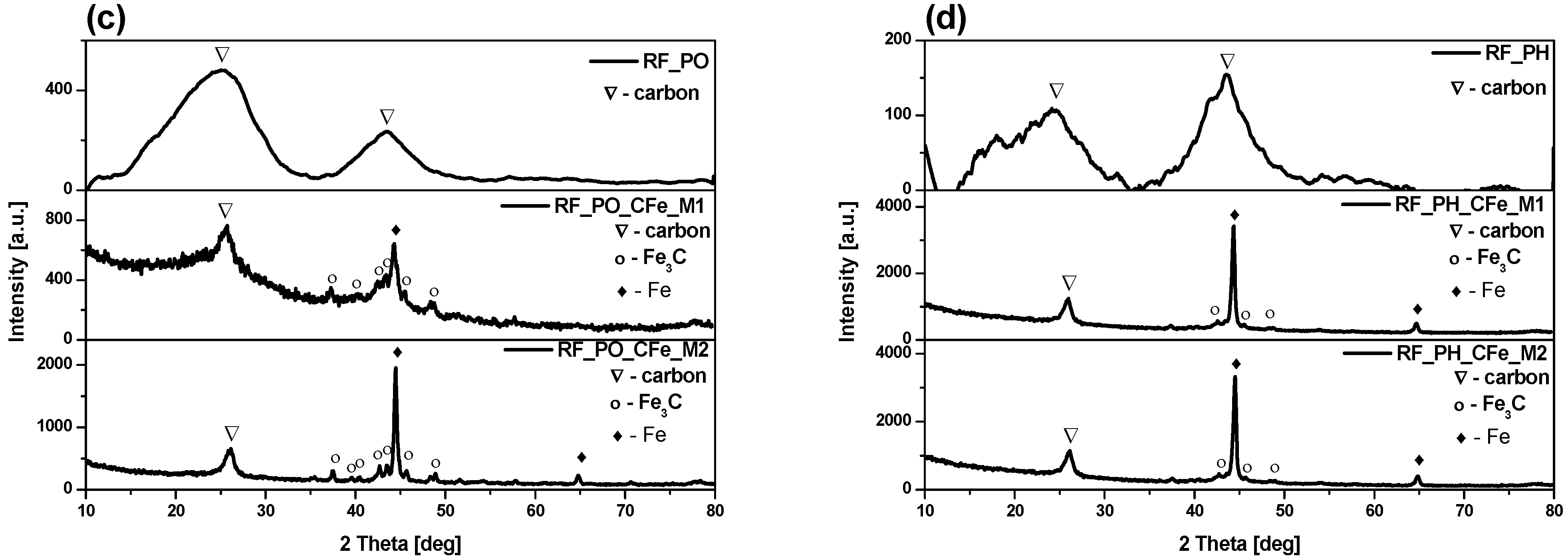
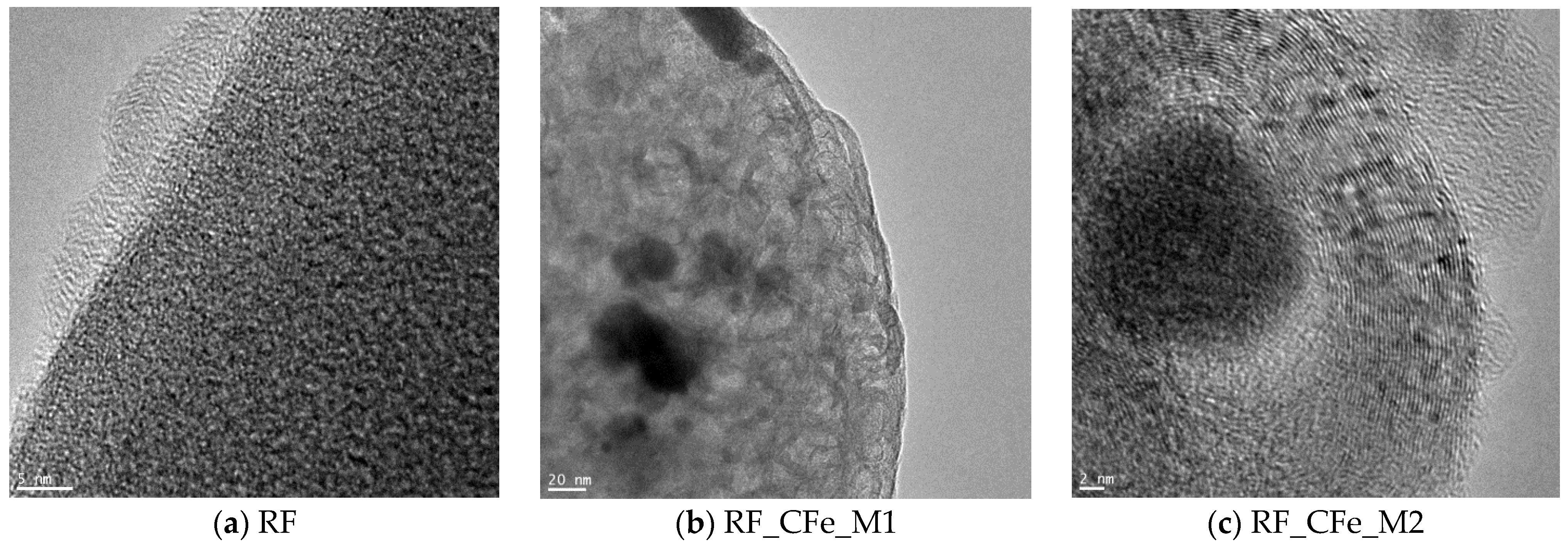
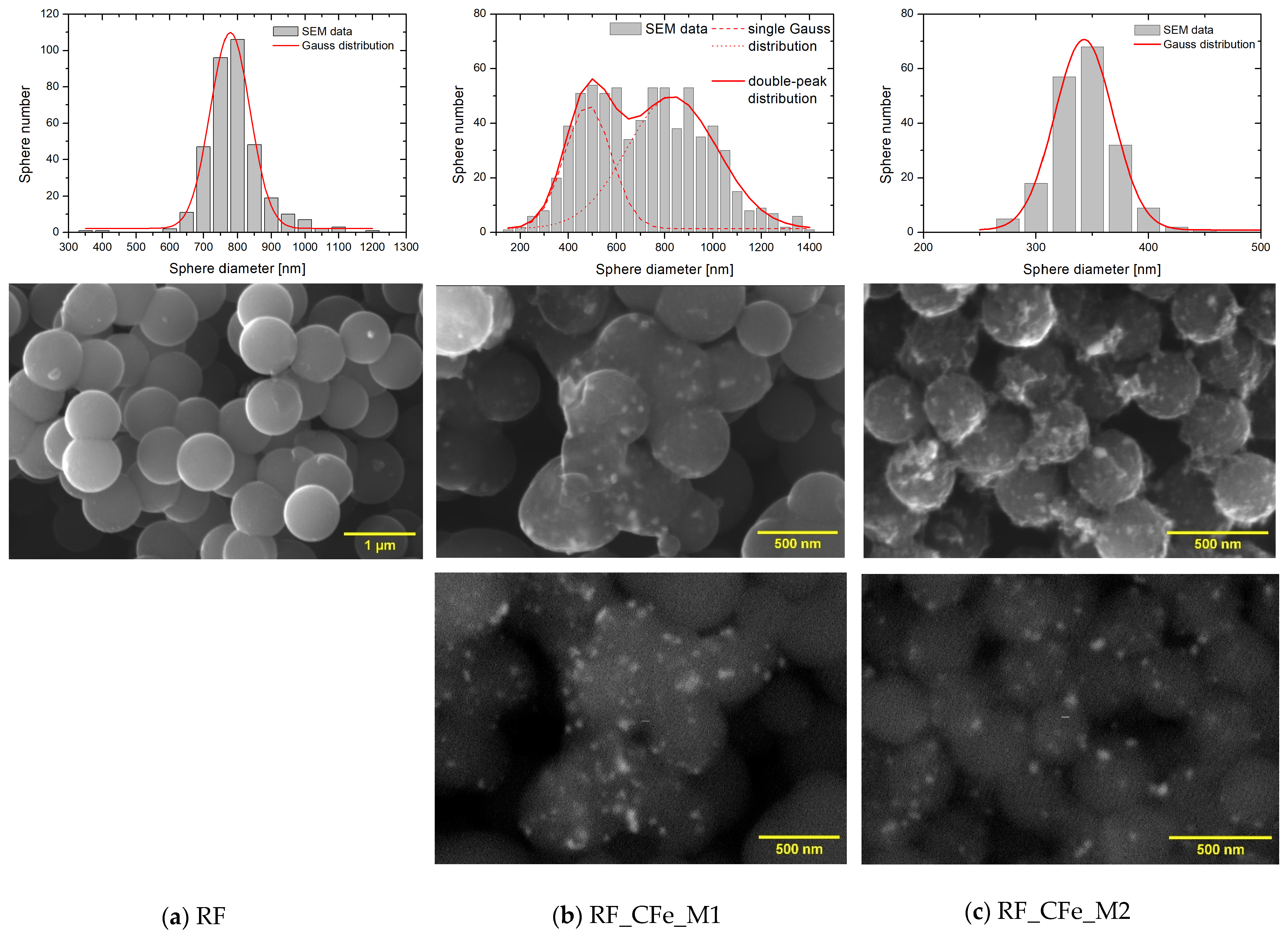
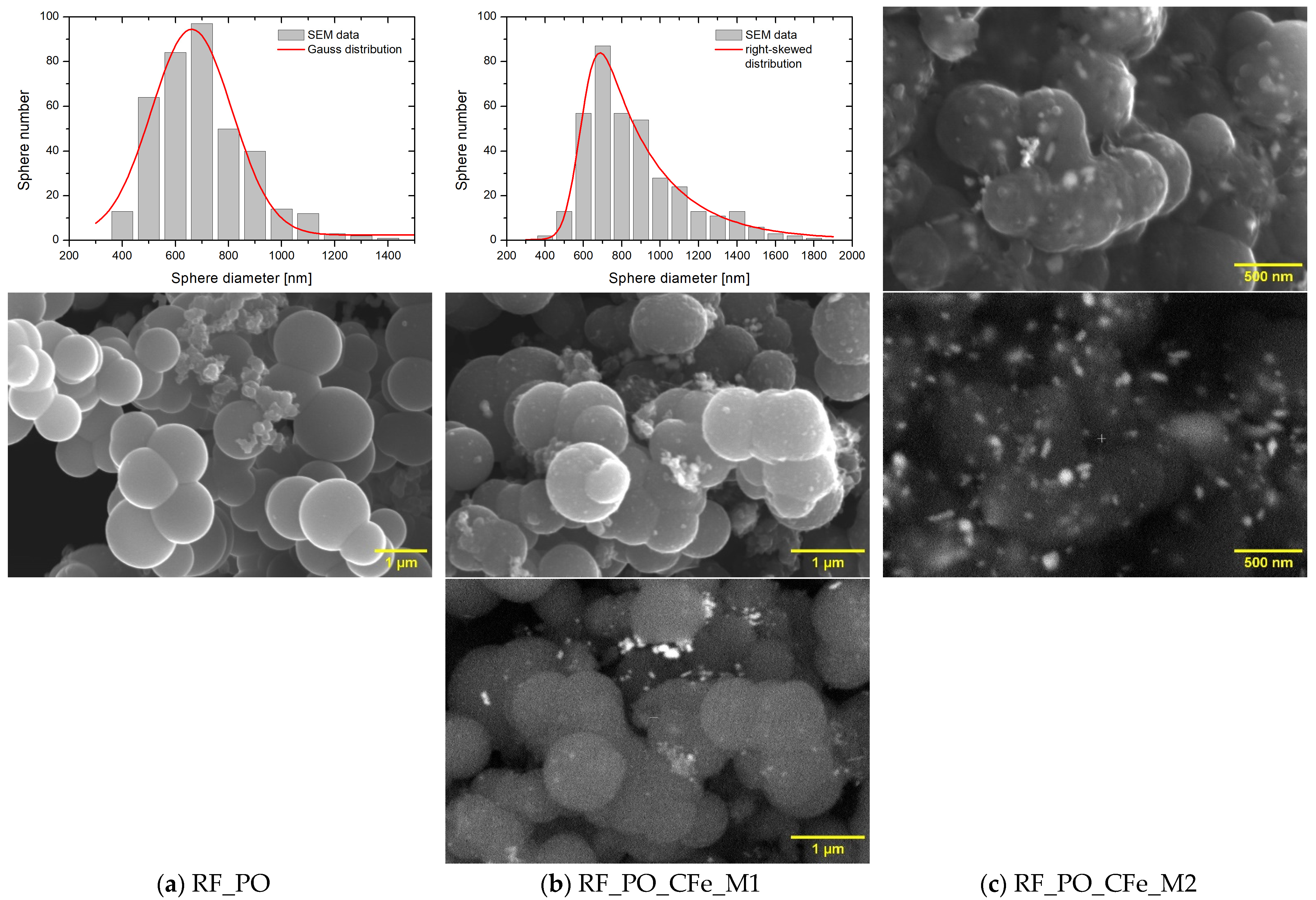
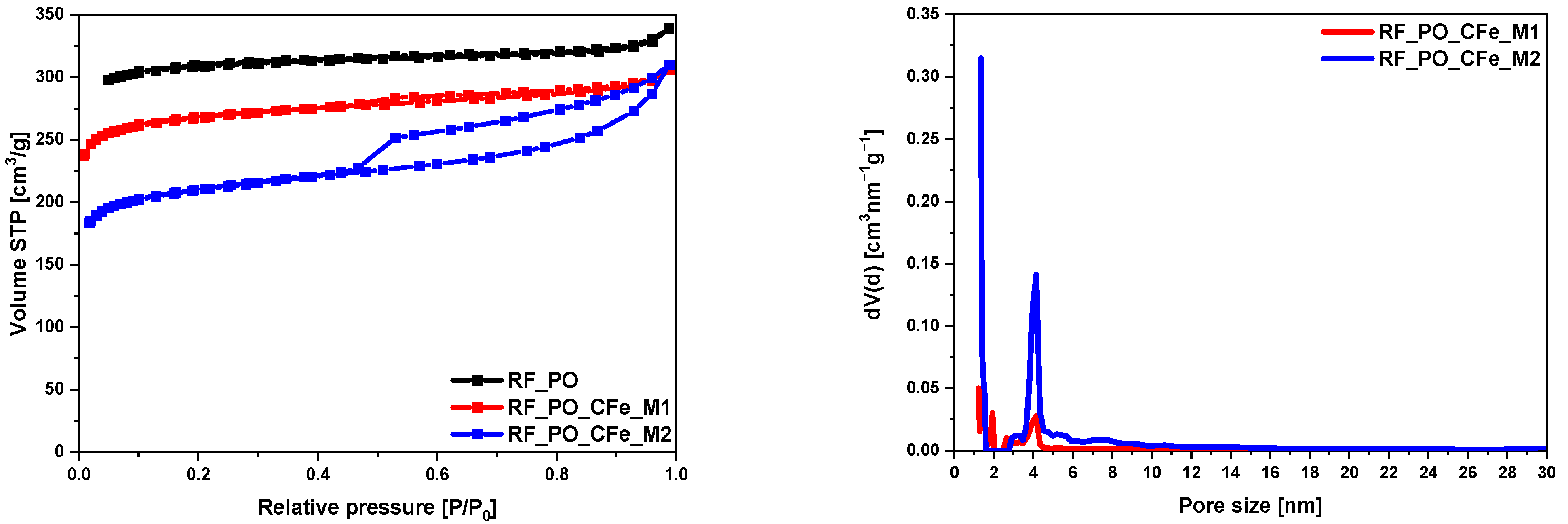
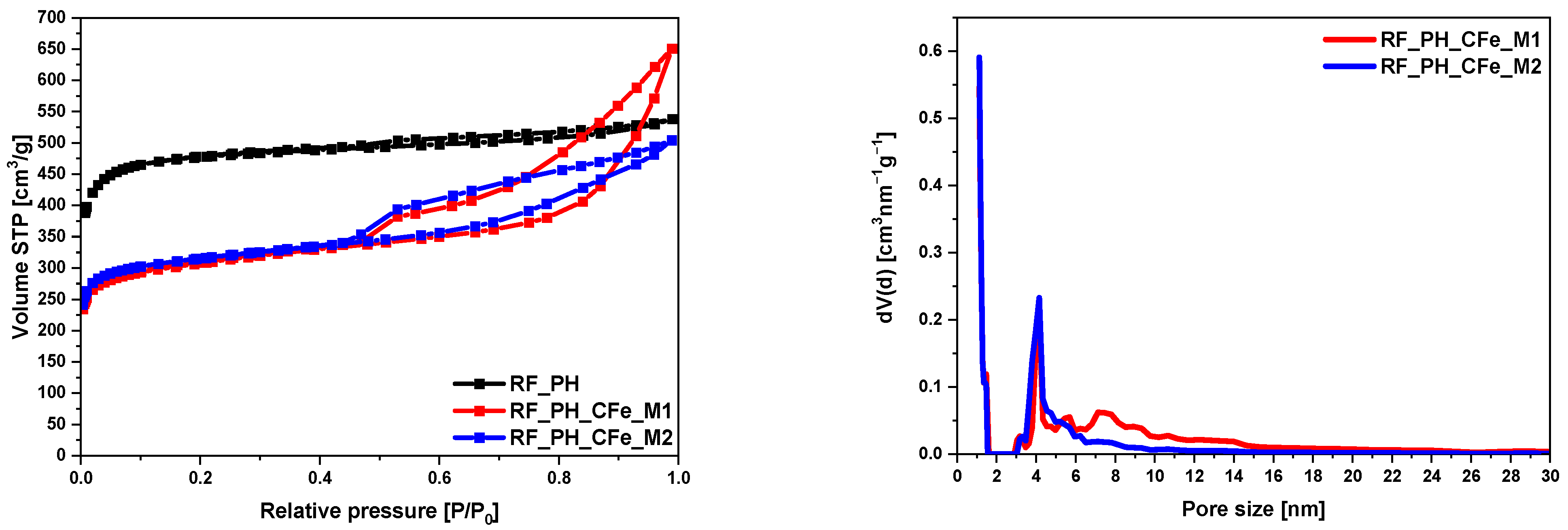
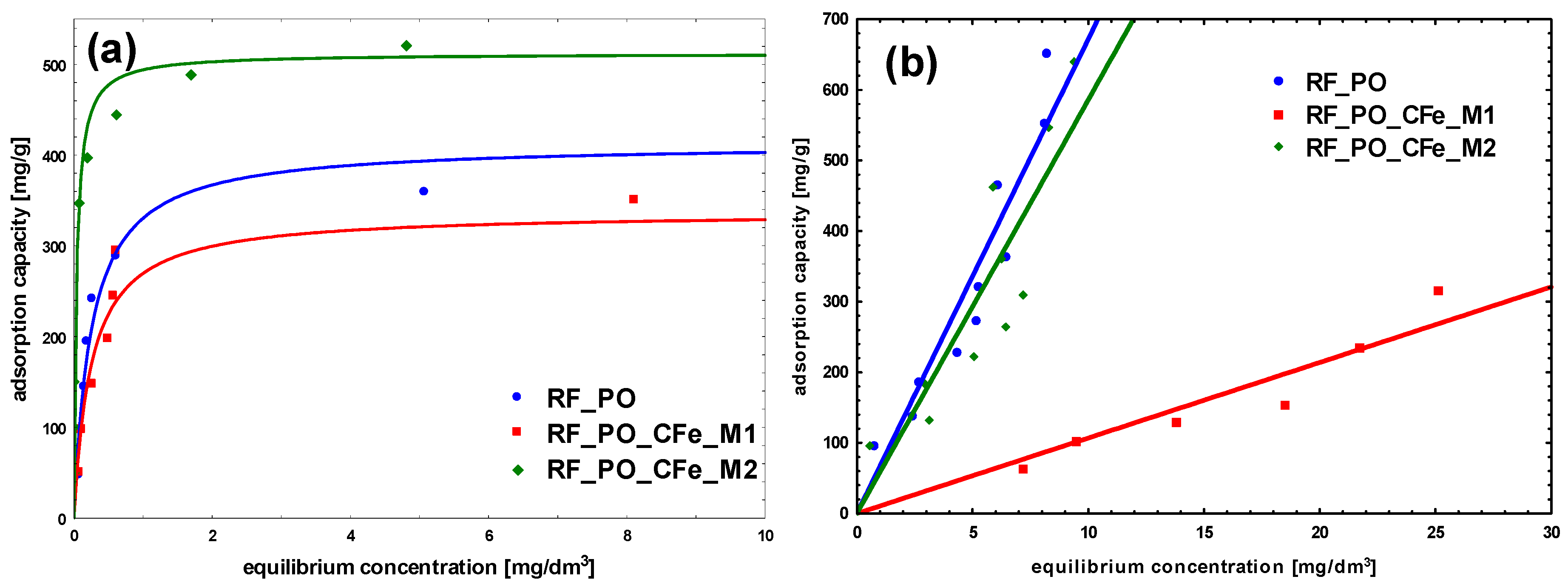
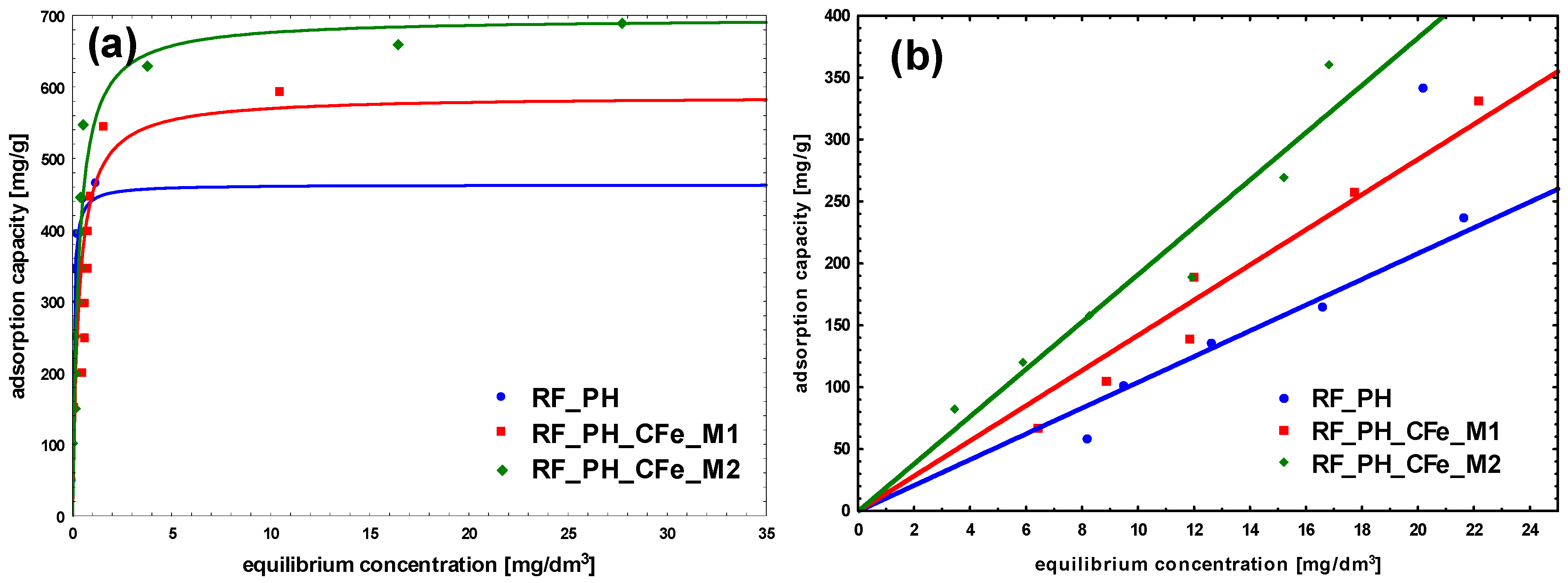
2. Results and Discussion
3. Materials and Methods
3.1. Materials Preparation
3.2. Materials Characterization
3.3. Dye Solution Preparation
3.4. Adsorption Experiments
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sen, T.K.; Afroze, S.; Ang, H.M. Equilibrium, Kinetics and Mechanism of Removal of Methylene Blue from Aqueous Solution by Adsorption onto Pine Cone Biomass of Pinus radiata. Water Air Soil. Pollut. 2011, 218, 499–515. [Google Scholar] [CrossRef]
- Yagub, M.T.; Sen, T.K.; Afroze, S.; Ang, H.M. Dye and its removal from aqueous solution by adsorption: A review. Adv. Colloid Interface Sci. 2014, 209, 172–184. [Google Scholar] [CrossRef]
- Kadhim, R.J.; Al-Ani, F.H.; Al-shaeli, M.; Alsalhy, Q.F.; Figoli, A. Removal of Dyes Using Graphene Oxide (GO) Mixed Matrix Membranes. Membranes 2020, 10, 366. [Google Scholar] [CrossRef]
- Januário, E.F.D.; Beluci, N.L.; Vidovix, T.B.; Vieira, M.F.; Bergamasco, R.; Vieira, A.M.S. Functionalization of membrane surface by layer-by-layer self-assembly method for dyes removal. Process Saf. Environ. Prot. 2020, 134, 140–148. [Google Scholar] [CrossRef]
- Wei, Y.; Cheng, X.; Ding, A.; Xu, J. Magnesium Silicate Polymer as a Coagulant for Reactive Dye Removal from Wastewater: Considering the Intrinsic pH in Magnesium Silicate Polymer and Coagulation Behavior. ACS Omega 2020, 5, 26094–26100. [Google Scholar] [CrossRef]
- Hu, E.; Shang, S.; Chiu, K.-L. Removal of Reactive Dyes in Textile Effluents by Catalytic Ozonation Pursuing on-Site Effluent Recycling. Molecules 2019, 24, 2755. [Google Scholar] [CrossRef]
- Rajagopal, S.; Paramasivam, B.; Muniyasamy, K. Photocatalytic removal of cationic and anionic dyes in the textile wastewater by H2O2 assisted TiO2 and micro-cellulose composites. Sep. Purif. Technol. 2020, 252, 117444. [Google Scholar] [CrossRef]
- Wong, S.; Ghafar, N.A.; Ngadi, N.; Razmi, F.A.; Inuwa, I.M.; Mat, R.; Amin, N.A.S. Effective removal of anionic textile dyes using adsorbent synthesized from coffee waste. Sci. Rep. 2020, 10, 2928. [Google Scholar] [CrossRef]
- Soltani, A.; Faramarzi, M.; Parsa, S.A.M. A review on adsorbent parameters for removal of dye products from industrial wastewater. Water Qual. Res. J. 2021, 56, 181–193. [Google Scholar] [CrossRef]
- Geng, Z.; Lin, Y.; Yu, X.; Shen, Q.; Ma, L.; Li, Z.; Pan, N.; Wang, X. Highly efficient dye adsorption and removal: A functional hybrid of reduced graphene oxide–Fe3O4 nanoparticles as an easily regenerative adsorbent. J. Mater. Chem. 2012, 22, 3527–3535. [Google Scholar] [CrossRef]
- Khurana, I.; Saxena, A.; Bharti; Khurana, J.M.; Rai, P.K. Removal of Dyes Using Graphene-Based Composites: A Review. Water Air Soil. Pollut. 2021, 228, 180. [Google Scholar] [CrossRef]
- Jinendra, U.; Bilehal, D.; Nagabhushana, B.M.; Kumar, A.P. Adsorptive removal of Rhodamine B dye from aqueous solution by using graphene–based nickel nanocomposite. Heliyon 2021, 7, e06851. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Mahanpoor, K.; Moradi, O. Removal of dye molecules from aqueous solution by carbon nanotubes and carbon nanotube functional groups: Critical review. RSC Adv. 2017, 7, 47083–47090. [Google Scholar] [CrossRef]
- Ai, L.; Zhang, C.; Liao, F.; Wang, Y.; Li, M.; Meng, L.; Jiang, J. Removal of methylene blue from aqueous solution with magnetite loaded multi-wall carbon nanotube: Kinetic, isotherm and mechanism analysis. J. Hazard. Mater. 2011, 198, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhao, Y.; Liu, P.; Men, Y.-L.; Pan, Y.-X. Boosting the adsorption and removal of dye from water by COOH-functionalized carbon nanotubes. Green. Chem. Eng. 2023, 4, 88–98. [Google Scholar] [CrossRef]
- Jurkiewicz, M.; Pełech, R. Adsorption of 1,2-Dichlorobenzene from the Aqueous Phase onto Activated Carbons and Modified Carbon Nanotubes. Int. J. Mol. Sci. 2021, 22, 13152. [Google Scholar] [CrossRef]
- Kundu, S.; Chowdhury, I.H.; Naskar, M.K. Hierarchical Porous Carbon Nanospheres for Efficient Removal of Toxic Organic Water Contaminants of Phenol and Methylene Blue. J. Chem. Eng. Data 2018, 63, 559–573. [Google Scholar] [CrossRef]
- Manippady, S.R.; Singh, A.; Basavaraja, B.M.; Samal, A.K.; Srivastava, S.; Saxena, M. Iron–Carbon Hybrid Magnetic Nanosheets for Adsorption-Removal of Organic Dyes and 4-Nitrophenol from Aqueous Solution. ACS Appl. Nano Mater. 2020, 3, 1571–1582. [Google Scholar] [CrossRef]
- Nizam, N.U.M.; Hanafiah, M.M.; Mahmoudi, E.; Halim, A.A.; Mohammad, A.W. The removal of anionic and cationic dyes from an aqueous solution using biomass-based activated carbon. Sci. Rep. 2021, 11, 8623. [Google Scholar] [CrossRef]
- Moosavi, S.; Lai, C.W.; Gan, S.; Zamiri, G.; Akbarzadeh Pivehzhani, O.; Johan, M.R. Application of efficient magnetic particles and activated carbon for dye removal from wastewater. ACS Omega 2020, 5, 20684–20697. [Google Scholar] [CrossRef]
- Kheddo, A.; Rhyman, L.; Elzagheid, M.I.; Jeetah, P.; Ramasami, P. Adsorption of synthetic dyed wastewater using activated carbon from rice husk. SN Appl. Sci. 2020, 2, 2170. [Google Scholar] [CrossRef]
- Sultana, M.; Rownok, M.H.; Sabrin, M.; Rahaman, M.R.; Nur Alam, S.M. A review on experimental chemically modified activated carbon to enhance dye and heavy metals adsorption. Clean. Eng. Technol. 2022, 6, 100382. [Google Scholar] [CrossRef]
- Foroutan, R.; Peighambardoust, S.J.; Peighambardoust, S.H.; Pateiro, M.; Lorenzo, J.M. Adsorption of Crystal Violet Dye Using Activated Carbon of Lemon Wood and Activated Carbon/Fe3O4 Magnetic Nanocomposite from Aqueous Solutions: A Kinetic, Equilibrium and Thermodynamic Study. Molecules 2021, 26, 2241. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, J.; Jia, D.M. Facile synthesis of shell-core structured Fe3O4@ACS as recyclable magnetic adsorbent for methylene blue removal. J. Dispers. Sci. Technol. 2019, 40, 1736–1743. [Google Scholar] [CrossRef]
- Jiang, W.; Zhang, L.; Guo, X.; Yang, M.; Lu, Y.; Wang, Y.; Zheng, Y.; Wei, G. Adsorption of cationic dye from water using an iron oxide/activated carbon magnetic composites prepared from sugarcane bagasse by microwave method. Environ. Technol. 2021, 42, 337–350. [Google Scholar] [CrossRef]
- Siyasukh, A.; Chimupala, Y.; Tonanon, N. Preparation of magnetic hierarchical porous carbon spheres with graphitic features for high methyl orange adsorption capacity. Carbon 2018, 134, 207–221. [Google Scholar] [CrossRef]
- Pełech, I.; Lewinska, S.; Arciszewska, M.; Khaliq, A.; Ślawska-Waniewska, A.; Sibera, D.; Staciwa, P.; Narkiewicz, U. Iron–Carbon Nanospheres as Promising Material for Magnetic Assisted Adsorption and Separation of Impurities from a Liquid Phase. Materials 2024, 17, 2111. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Rodriguez, C.E.; Tovar-Martinez, E.; Reyes-Reyes, M.; Chazaro-Ruiz, L.F.; López-Sandoval, R. Synthesis of hollow carbon spheres by chemical activation of carbon nanoparticles for their use in electrochemical capacitor. Carbon. Trends 2022, 9, 100220. [Google Scholar] [CrossRef]
- Mansuri, I.; Farzana, R.; Rajarao, R.; Sahajwalla, V. Carbon Dissolution Using Waste Biomass—A Sustainable Approach for Iron-Carbon Alloy Production. Metals 2018, 8, 290. [Google Scholar] [CrossRef]
- Juhl, A.C.; Schneider, A.; Ufer, B.; Brezesinski, T.; Janek, J.; Fröba, M. Mesoporous hollow carbon spheres for lithium–sulfur batteries: Distribution of sulfur and electrochemical performance. Beilstein J. Nanotechnol. 2016, 7, 1229–1240. [Google Scholar] [CrossRef]
- Krstic, S.; Kragovic, M.; Pagnacco, M.; Dodevski, V.; Kaluđerovic, B.; Momcilovic, M.; Ristovic, I.; Stojmenovic, M. Hydrothermal Synthesized and Alkaline Activated Carbons Prepared from Glucose and Fructose—Detailed Characterization and Testing in Heavy Metals and Methylene Blue Removal. Minerals 2018, 8, 246. [Google Scholar] [CrossRef]
- Pełech, I.; Sibera, D.; Staciwa, P.; Sobczuk, K.S.; Narkiewicz, U. Influence of Potassium-Based Activation on Adsorptive Properties of Carbon Spheres Modified with Iron(III) Citrate. Materials 2023, 16, 5227. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wei, C.; Jin, X.; Akhtar, R.; Zhang, W. Carbon spheres as lubricant additives for improving tribological performance of polyetheretherketone. J. Mater. Sci. 2019, 54, 5127–5135. [Google Scholar] [CrossRef]
- Zhao, M.; Song, H. Catalytic Graphitization of Phenolic Resin. J. Mater. Sci. Technol. 2011, 27, 266–270. [Google Scholar] [CrossRef]
- Hunter, R.D.; Ramírez-Rico, J.; Schnepp, Z. Iron-Catalyzed Graphitization for the Synthesis of Nanostructured Graphitic Carbons. J. Mater. Chem. A 2022, 10, 4489–4516. [Google Scholar] [CrossRef]
- Barnakov, C.N.; Khokhlova, G.P.; Popova, A.N.; Sozinov, S.A.; Ismagilov, Z.R. XRD characterization of the structure of graphites and carbon materials obtained by the low-temperature graphitization of coal tar pitch. Eurasian Chem. J. 2015, 17, 87–93. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Provisional). Pure Appl. Chem. 1982, 54, 2201–2218. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, T.; Chen, X.; He, Y.; Liang, X. Model construction of micro-pores in shale: A case study of Silurian Longmaxi Formation shale in Dianqianbei area, SW China. Pet. Explor. Dev. 2018, 45, 412–421. [Google Scholar] [CrossRef]
- Lv, M.; Xie, W.; Sun, S.; Wu, G.; Zheng, L.; Chu, S.; Gao, C.; Bao, J. Activated-carbon-supported K–Co–Mo catalysts for synthesis of higher alcohols from syngas. Catal. Sci. Technol. 2015, 5, 2925–2934. [Google Scholar] [CrossRef]
- Sobczuk, K.S.; Pełech, I.; Narkiewicz, U.; Staciwa, P.; Sibera, D.; Moszyński, D. The influence of the synthesis pH on the morphology and adsorption properties of carbon spheres. Appl. Surf. Sci. 2023, 608, 155196. [Google Scholar] [CrossRef]
- Maneerung, T.; Liew, J.; Dai, Y.; Kawi, S.; Chong, C.; Wang, C.-H. Activated carbon derived from carbon residue from biomass gasification and its application for dye adsorption: Kinetics, isotherms and thermodynamic studies. Bioresour. Technol. 2016, 200, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ji, T.; Mu, L.; Shi, Y.; Wang, H.; Zhu, J. Pore size dependent molecular adsorption of cationic dye in biomass derived hierarchically porous carbon. J. Environ. Manag. 2017, 196, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Ighalo, J.O.; Iwuozor, K.O.; Igwegbe, C.A.; Adeniyi, A.G. Verification of pore size effect on aqueous-phase adsorption kinetics: A case study of methylene blue. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127119. [Google Scholar] [CrossRef]
- Khan, I.; Saeed, K.; Zekker, I.; Zhang, B.; Hendi, A.H.; Ahmad, A.; Ahmad, S.; Zada, N.; Ahmad, H.; Shah, L.A.; et al. Review on Methylene Blue: Its Properties, Uses, Toxicity and Photodegradation. Water 2022, 14, 242. [Google Scholar] [CrossRef]
- Pełech, I.; Staciwa, P.; Sibera, D.; Kusiak-Nejman, E.; Morawski, A.W.; Kapica-Kozar, J.; Narkiewicz, U. The Effect of the Modification of Carbon Spheres with ZnCl2 on the Adsorption Properties towards CO2. Molecules 2022, 27, 1387. [Google Scholar] [CrossRef]
- El-Bery, H.M.; Saleh, M.; El-Gendy, R.A.; Saleh, M.R.; Thabet, S.M. High adsorption capacity of phenol and methylene blue using activated carbon derived from lignocellulosic agriculture wastes. Sci. Rep. 2022, 12, 5499. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SBET [m2/g] | TPV [cm3/g] | Vm (<2 nm) [cm3/g] | Vmeso [cm3/g] | |
|---|---|---|---|---|
| RF | 455 | 0.26 | 0.22 | 0.04 |
| RF_CFe_M1 | 400 | 0.33 | 0.13 | 0.20 |
| RF_CFe_M2 | 439 | 0.39 | 0.14 | 0.25 |
| RF_PO | 942 | 0.53 | 0.44 | 0.07 |
| RF_PO_CFe_M1 | 826 | 0.47 | 0.37 | 0.10 |
| RF_PO_CFe_M2 | 656 | 0.48 | 0.27 | 0.21 |
| RF_PH | 1473 | 0.83 | 0.67 | 0.16 |
| RF_PH_CFe_M1 | 991 | 0.78 | 0.39 | 0.39 |
| RF_PH_CFe_M2 | 974 | 1.01 | 0.37 | 0.64 |
| Methylene Blue | Congo Red | ||||
|---|---|---|---|---|---|
| am, mg/g | b, dm3/g | R2 | H, dm3/g | R2 | |
| RF | 12 | 0.04 | 0.99 | 61.5 | 0.98 |
| RF_CFe_M1 | 117 | 0.67 | 0.94 | 86.1 | 0.98 |
| RF_CFe_M2 | 178 | 4.30 | 0.91 | 171.0 | 0.98 |
| RF_PO | 413 | 4.0 | 0.92 | 67.2 | 0.98 |
| RF_PO_CFe_M1 | 337 | 4.0 | 0.91 | 10.7 | 0.98 |
| RF_PO_CFe_M2 | 492 | 31.5 | 0.98 | 58.6 | 0.95 |
| RF_PH | 463 | 20.3 | 0.97 | 10.4 | 0.99 |
| RF_PH_CFe_M1 | 587 | 3.3 | 0.90 | 14.2 | 0.99 |
| RF_PH_CFe_M2 | 696 | 3.4 | 0.95 | 19.1 | 0.99 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sibera, D.; Pełech, I.; Staciwa, P.; Pełech, R.; Ekiert, E.; Kayalar, G.Y.; Narkiewicz, U. Activated Iron-Porous Carbon Nanomaterials as Adsorbents for Methylene Blue and Congo Red. Molecules 2024, 29, 4090. https://doi.org/10.3390/molecules29174090
Sibera D, Pełech I, Staciwa P, Pełech R, Ekiert E, Kayalar GY, Narkiewicz U. Activated Iron-Porous Carbon Nanomaterials as Adsorbents for Methylene Blue and Congo Red. Molecules. 2024; 29(17):4090. https://doi.org/10.3390/molecules29174090
Chicago/Turabian StyleSibera, Daniel, Iwona Pełech, Piotr Staciwa, Robert Pełech, Ewa Ekiert, Gulsen Yagmur Kayalar, and Urszula Narkiewicz. 2024. "Activated Iron-Porous Carbon Nanomaterials as Adsorbents for Methylene Blue and Congo Red" Molecules 29, no. 17: 4090. https://doi.org/10.3390/molecules29174090
APA StyleSibera, D., Pełech, I., Staciwa, P., Pełech, R., Ekiert, E., Kayalar, G. Y., & Narkiewicz, U. (2024). Activated Iron-Porous Carbon Nanomaterials as Adsorbents for Methylene Blue and Congo Red. Molecules, 29(17), 4090. https://doi.org/10.3390/molecules29174090