
Theoretical Study on Vibrationally Resolved Electronic Spectra of Chiral Nanographenes


Abstract
1. Introduction
2. Results and Discussion
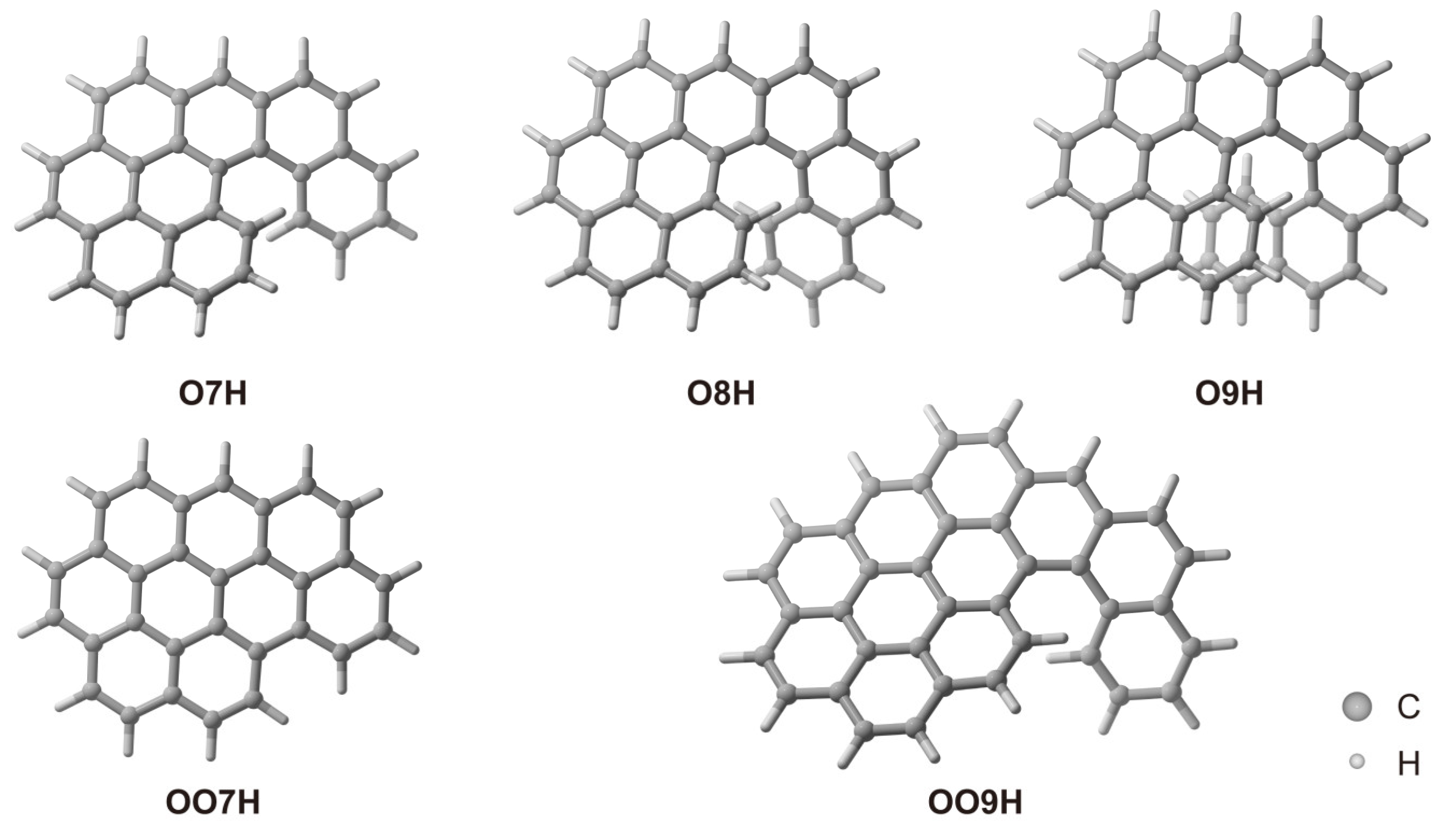
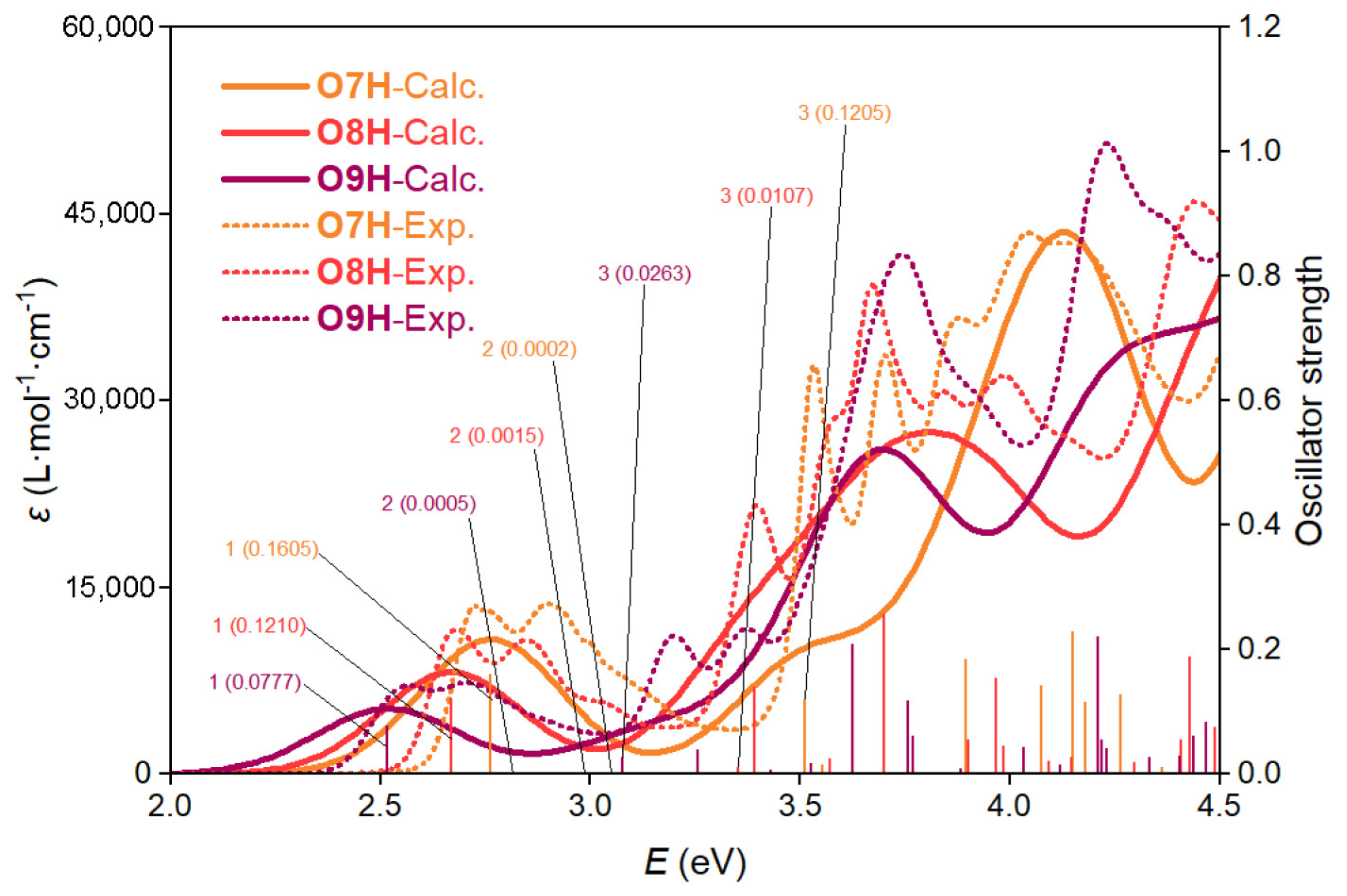
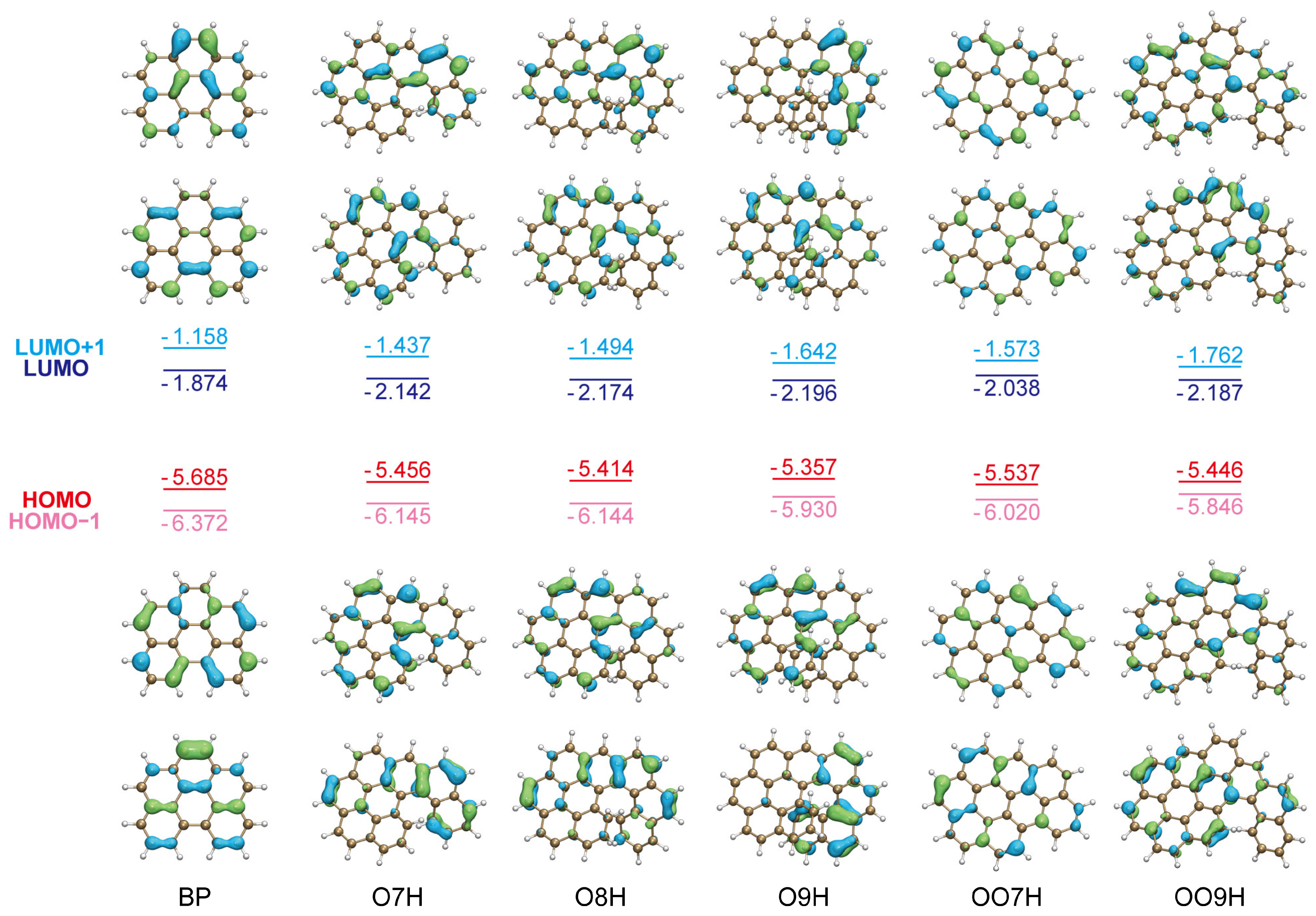
2.1. Pure Electronic Transition of O7H, O8H, and O9H
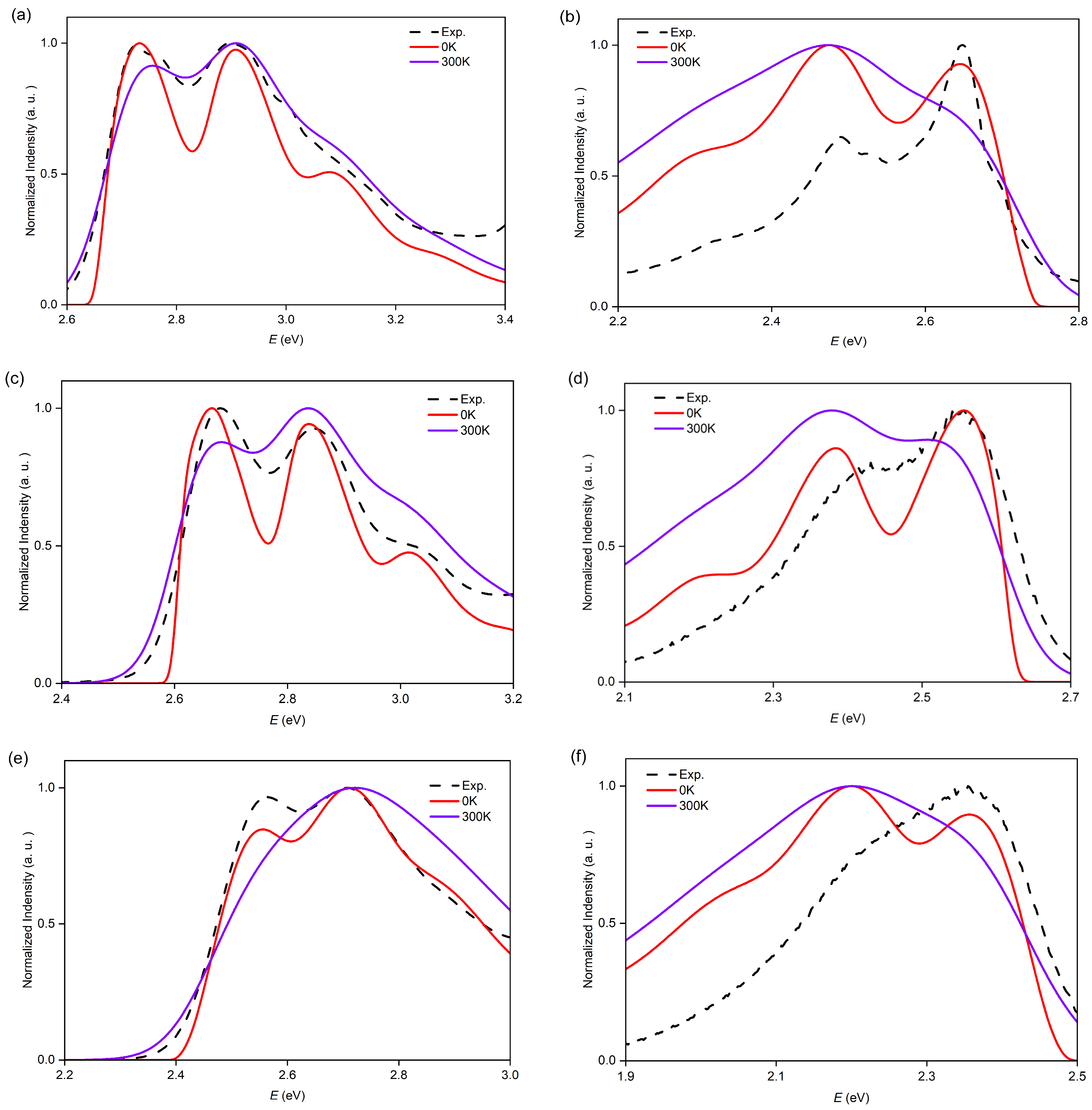
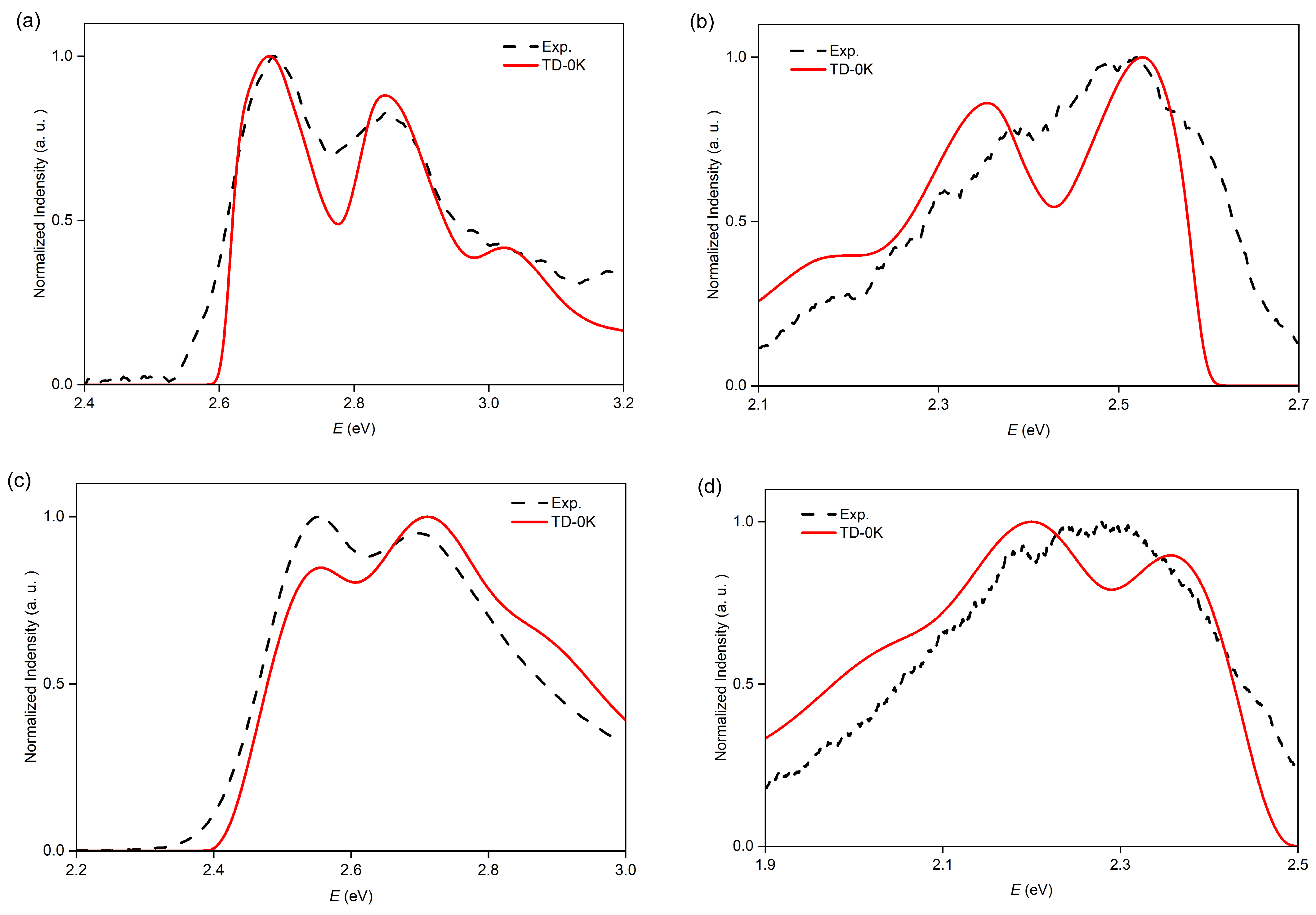
2.2. Vibrationally Resolved Electronic Transition of O7H, O8H, and O9H
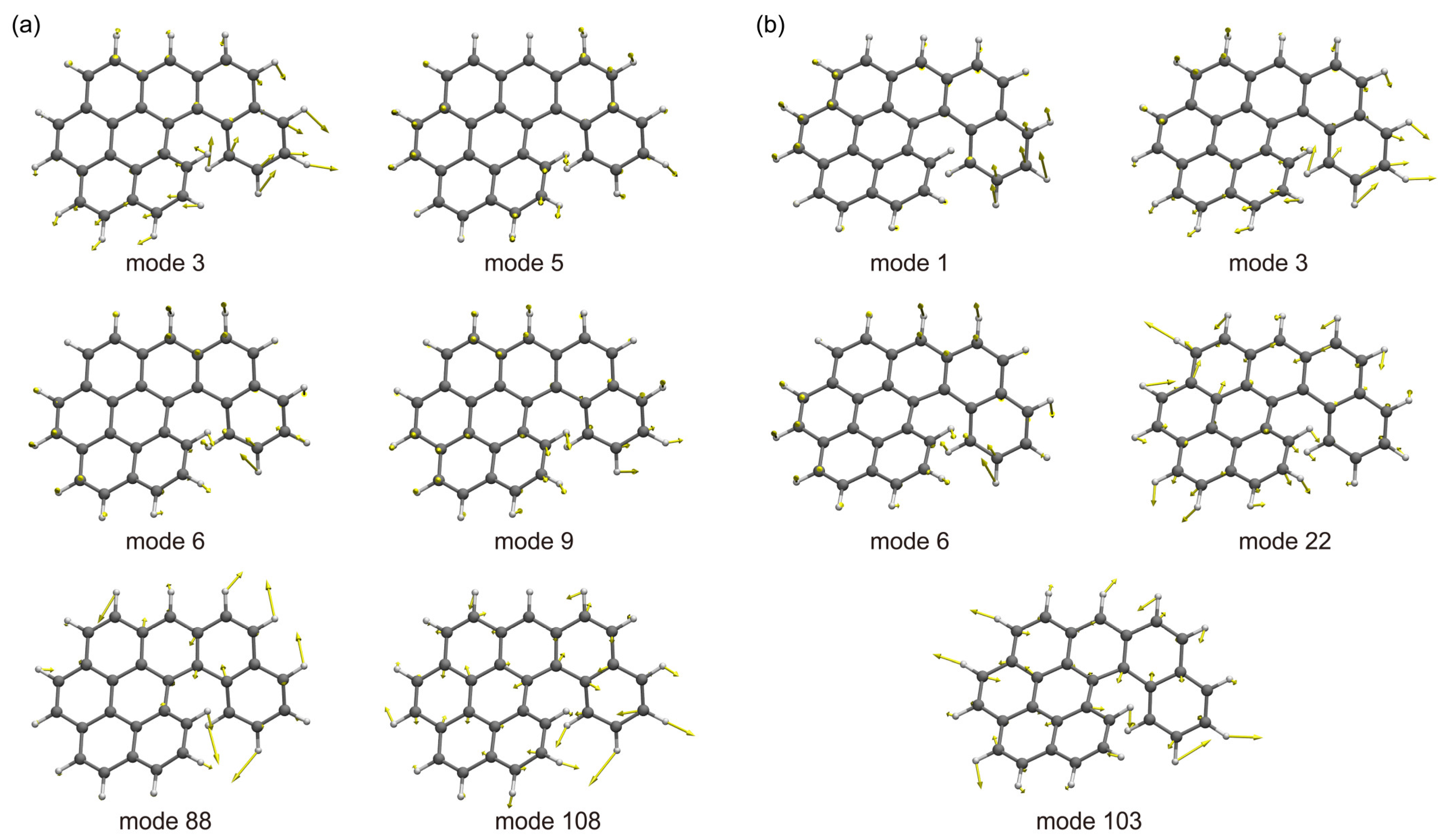
2.3. TI Spectrum and Assignment of the Main Vibronic Bands
2.4. Electronic Transition of OO7H and OO9H
2.5. Vibrationally Resolved ECD and CPL of O8H and O9H
3. Methods
Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Z.; Fu, S.; Liu, X.; Narita, A.; Samorì, P.; Bonn, M.; Wang, H.I. Small Size, Big Impact: Recent Progress in Bottom-Up Synthesized Nanographenes for Optoelectronic and Energy Applications. Adv. Sci. 2022, 9, 2106055. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, Y.; Chandresh, A.; Pati, P.B.; Monnier, V.; Heinke, L.; Odobel, F.; Diring, S.; Haldar, R.; Wöll, C. Nanographene-Based Metal-Organic Framework Thin Films: Optimized Packing and Efficient Electron-Hole Separation Yielding Efficient Photodetector. Adv. Funct. Mater. 2024, 34, 2308847. [Google Scholar] [CrossRef]
- Castro-Fernández, S.; Cruz, C.M.; Mariz, I.F.A.; Márquez, I.R.; Jiménez, V.G.; Palomino-Ruiz, L.; Cuerva, J.M.; Maçôas, E.; Campaña, A.G. Two-Photon Absorption Enhancement by the Inclusion of a Tropone Ring in Distorted Nanographene Ribbons. Angew. Chem. Int. Ed. 2020, 59, 7139–7145. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Oda, S.; Yoshiura, K.; Nakajima, K.; Yasuda, N.; Hatakeyama, T. One-Shot Multiple Borylation toward BN-Doped Nanographenes. J. Am. Chem. Soc. 2018, 140, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-A.; Sato, Y.; Segawa, Y.; Nishihara, T.; Sugimoto, N.; Scott, L.T.; Higashiyama, T.; Itami, K. A Water-Soluble Warped Nanographene: Synthesis and Applications for Photoinduced Cell Death. Angew. Chem. Int. Ed. 2018, 57, 2874–2878. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Q.; Zhao, H.; Yang, Q.; Goudappagouda; Gelléri, M.; Ritz, S.; Ng, D.; Koynov, K.; Parekh, S.H.; et al. Intrinsic Burst-Blinking Nanographenes for Super-Resolution Bioimaging. J. Am. Chem. Soc. 2024, 146, 5195–5203. [Google Scholar] [CrossRef]
- Song, S.; Pinar Solé, A.; Matěj, A.; Li, G.; Stetsovych, O.; Soler, D.; Yang, H.; Telychko, M.; Li, J.; Kumar, M.; et al. Highly entangled polyradical nanographene with coexisting strong correlation and topological frustration. Nat. Chem. 2024, 16, 938–944. [Google Scholar] [CrossRef]
- Borissov, A.; Maurya, Y.K.; Moshniaha, L.; Wong, W.-S.; Żyła-Karwowska, M.; Stępień, M. Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds. Chem. Rev. 2022, 122, 565–788. [Google Scholar] [CrossRef]
- Anderson, H.V.; Gois, N.D.; Chalifoux, W.A. New advances in chiral nanographene chemistry. Org. Chem. Front. 2023, 10, 4167–4197. [Google Scholar] [CrossRef]
- Jassas, R.S.; Mughal, E.A.; Sadiq, A.; Alsantali, L.I.; Al-Rooqi, M.M.; Naeem, N.; Moussa, Z.; Ahmed, S.A. Scholl reaction as a powerful tool for the synthesis of nanographenes: A systematic review. RSC Adv. 2021, 11, 32158–32202. [Google Scholar] [CrossRef]
- Muzammil, E.M.; Halilovic, D.; Stuparu, M.C. Synthesis of corannulene-based nanographenes. Commun. Chem. 2019, 2, 58. [Google Scholar] [CrossRef]
- Li, R.; Wang, D.; Li, S.; An, P. Construction of hexabenzocoronene-based chiral nanographenes. Beilstein J. Org. Chem. 2023, 19, 736–751. [Google Scholar] [CrossRef] [PubMed]
- Narita, A.; Wang, X.-Y.; Feng, X.; Müllen, K. New advances in nanographene chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-García, P.; Fernández-García, J.M.; Fernández, I.; Perles, J.; Martín, N. Helically Arranged Chiral Molecular Nanographenes. J. Am. Chem. Soc. 2021, 143, 11864–11870. [Google Scholar] [CrossRef]
- Evans, P.J.; Ouyang, J.; Favereau, L.; Crassous, J.; Fernández, I.; Perles, J.; Martín, N. Synthesis of a Helical Bilayer Nanographene. Angew. Chem. Int. Ed. 2018, 57, 6774–6779. [Google Scholar] [CrossRef]
- Yamamoto, K.; Saitho, Y.; Iwaki, D.; Ooka, T. [7.7]Circulene, a Molecule Shaped Like a Figure of Eight. Angew. Chem. Int. Ed. Engl. 1991, 30, 1173–1174. [Google Scholar] [CrossRef]
- Pun, S.H.; Wang, Y.; Chu, M.; Chan, C.K.; Li, Y.; Liu, Z.; Miao, Q. Synthesis, Structures, and Properties of Heptabenzo[7]circulene and Octabenzo[8]circulene. J. Am. Chem. Soc. 2019, 141, 9680–9686. [Google Scholar] [CrossRef]
- Pun, S.H.; Miao, Q. Toward Negatively Curved Carbons. Acc. Chem. Res. 2018, 51, 1630–1642. [Google Scholar] [CrossRef]
- Chueng, K.Y.; Chan, C.K.; Liu, Z.; Qian, M. A Twisted Nanographene Consisting of 96 Carbon Atoms. Angew. Chem. Int. Ed. 2017, 56, 9003–9007. [Google Scholar] [CrossRef]
- Dong, Y.; Zhang, Z.; Hashikawa, Y.; Meng, H.; Bai, F.; Itami, K.; Chaolumen. A Double Twisted Nanographene with a Contorted Pyrene Core. Angew. Chem. Int. Ed. 2024, 136, e202406927. [Google Scholar] [CrossRef]
- Li, J.-K.; Chen, X.-Y.; Zhao, W.-L.; Guo, Y.-L.; Zhang, Y.; Wang, X.-C.; Sue, A.C.-H.; Cao, X.-Y.; Li, M.; Chen, C.-F.; et al. Synthesis of Highly Luminescent Chiral Nanographene. Angew. Chem. Int. Ed. 2023, 62, e202215367. [Google Scholar] [CrossRef]
- Gan, F.; Shen, C.; Cui, W.; Qiu, H. [1,4]Diazocine-Embedded Electron-Rich Nanographenes with Cooperatively Dynamic Skeletons. J. Am. Chem. Soc. 2023, 145, 5952–5959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, J.; Tao, Y.; Gan, F.; Lin, G.; Liang, J.; Shen, C.; Zhang, Y.; Qiu, H. Facile fabrication of recyclable robustnoncovalent porous crystals from low-symmetry helicene derivative. Nat. Commun. 2024, 15, 5469. [Google Scholar] [CrossRef] [PubMed]
- Berezhnaia, V.; Roy, M.; Vanthuyne, N.; Villa, M.; Naubron, J.-V.; Rodriguez, J.; Coquerel, Y.; Gingras, M. Chiral Nanographene Propeller Embedding Six Enantiomerically Stable [5]Helicene Units. J. Am. Chem. Soc. 2017, 139, 18508–18511. [Google Scholar] [CrossRef]
- Shen, Y.-J.; Yao, N.-T.; Diao, L.-N.; Yang, Y.; Chen, X.-L.; Gong, H.-Y. A π-Extended Pentadecabenzo[9]helicene. Angew. Chem. Int. Ed. 2023, 62, e202300840. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Asako, S.; Hu, Y.; Ju, C.W.; Liu, T.; Rondin, L.; Schollmeyer, D.; Lauret, J.S.; Müllen, K.; Narita, A. Negatively Curved Nanographene with Heptagonal and [5]Helicene Units. J. Am. Chem. Soc. 2020, 142, 14814–14819. [Google Scholar] [CrossRef]
- Qiu, Z.; Ju, C.W.; Frederic, L.; Hu, Y.; Schollmeyer, D.; Pieters, G.; Müllen, K.; Narita, A. Amplification of Dissymmetry Factors in π-Extended [7]- and [9]Helicenes. J. Am. Chem. Soc. 2020, 143, 4661–4667. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, C.; Luo, Z.; Yin, Z.; Shi, H.; Zhu, Y.; W, J. Double π-Extended Undecabenzo[7]helicene. Angew. Chem. Int. Ed. 2021, 60, 7796–7801. [Google Scholar] [CrossRef]
- Sánchez-Carnerero, E.M.; Agarrabeitia, A.R.; Moreno, F.; Maroto, B.L.; Muller, G.; Ortiz, M.J.; de la Moya, S. Circularly polarized luminescence from simple organic molecules. Chem. Eur. J. 2015, 21, 13488–13500. [Google Scholar] [CrossRef]
- Bai, W.; Liang, M.; Xuan, T.; Gong, T.; Bian, L.; Li, H.; Xie, R.-J. Ligand Engineering Enables Efficient Pure Red Tin-Based Perovskite Light-Emitting Diodes. Angew. Chem. Int. Ed. 2023, 62, e202312728. [Google Scholar] [CrossRef]
- Niu, W.; Fu, Y.; Deng, Q.; Qiu, Z.-L.; Liu, F.; Popov, A.A.; Komber, H.; Ma, J.; Feng, X. Enhancing Chiroptical Responses in Helical Nanographenes via Geometric Engineering of Double [7]Helicenes. Angew. Chem. Int. Ed. 2024, 63, e202319874. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shen, C.; Zhang, G.; Gan, F.; Ding, Y.; Qiu, H. Transformation of Crowded Oligoarylene into Perylene-Cored Chiral Nanographene by Sequential Oxidative Cyclization and 1,2-Phenyl Migration. Angew. Chem. Int. Ed. 2022, 61, e202115979. [Google Scholar] [CrossRef]
- Barroso, J.; Cabellos, J.L.; Pan, S.; Murillo, F.; Zarate, X.; Fernandez-Herrera, M.A.; Merino, G. Revisiting the Racemization Mechanism of Helicenes. Chem. Commun. 2017, 54, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Luo, W.; Zhang, H.; Liu, P.; Shi, Y.; An, P. Cycl[2,2,4]azine-embedded non-alternant nanographenes containing fused antiaromatic azepine ring. Chem. Sci. 2024, 15, 1511–1519. [Google Scholar] [CrossRef]
- Minkin, V.I.; Starikov, A.G.; Starikova, A.A.; Gapurenko, O.A.; Minyaev, R.M.; Boldyrev, A.I. Electronic Structure and Magnetic Properties of the Triangular Nanographenes with Radical Substituents: A DFT Study. Phys. Chem. Chem. Phys. 2020, 22, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Escayola, S.; Poater, A.; Muñoz-Castro, A.; Solà, M. An Unprecedented π-Electronic Circuit Involving an Odd Number of Carbon Atoms in a Grossly Warped Non-Planar Nanographene. Chem. Commun. 2021, 57, 3087–3090. [Google Scholar] [CrossRef]
- Guido, C.A.; Zinna, F.; Pescitelli, G. CPL calculations of [7]helicenes with alleged exceptional emission dissymmetry values. J. Mater. Chem. C 2023, 11, 10474–10482. [Google Scholar] [CrossRef]
- Yang, Z.; Gai, X.; Zou, Y.; Jiang, Y. The Physical Mechanism of Linear and Nonlinear Optical Properties of Nanographene-Induced Chiral Inversion. Molecules 2024, 29, 1053. [Google Scholar] [CrossRef]
- Liu, Y.; Cerezo, J.; Mazzeo, G.; Lin, N.; Zhao, X.; Longhi, G.; Abbate, S.; Santoro, F. Vibronic Coupling Explains the Different Shape of Electronic Circular Dichroism and of Circularly Polarized Luminescence Spectra of Hexahelicenes. J. Chem. Theory Comput. 2016, 12, 2799–2819. [Google Scholar] [CrossRef]
- Bhattacharyya, P.; Rai, D.K.; Shukla, A. Pariser–Parr–Pople Model Based Configuration-Interaction Study of Linear Optical Absorption in Lower-Symmetry Polycyclic Aromatic Hydrocarbon Molecules. J. Phys. Chem. C 2020, 124, 14297–14305. [Google Scholar] [CrossRef]
- Weber, I.; Lee, Y.-P. Fluorescence Excitation and Dispersed Fluorescence Spectra of the First Electronic Excited (S1) State of peri-Hexabenzocoronene (C42H18) Isolated in Solid para-Hydrogen. J. Phys. Chem. A 2024, 128, 4984–4991. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Y.; Zhao, X.; Chen, S.; Li, Q.; Wang, M.; Yang, C. Vibrationally resolved electronic circular dichroism and circularly polarized luminescence spectra of a boron-fused double helicene: A theoretical study. Spectrochim. Acta Part A 2020, 231, 118132. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Q.; Sun, J.; Wang, L.; He, D.; Wang, M.; Yang, C. Insights for vibronic effects on spectral shapes of electronic circular dichroism and circularly polarized luminescence of aza[7]helicene. Spectrochim. Acta Part A 2020, 239, 118475. [Google Scholar] [CrossRef] [PubMed]
- Mori, T. Significance of Vibronic Coupling that Shapes Circularly Polarized Luminescence of Double Helicenes. Angew. Chem. Int. Ed. 2024, 63, e202319702. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, G.; Ding, Y.; Yang, N.; Gan, F.; Crassous, J.; Qiu, H. Oxidative cyclo-rearrangement of helicenes into chiral nanographenes. Nat. Commun. 2021, 12, 2786. [Google Scholar] [CrossRef]
- Qiu, S.; Valdivia, A.C.; Zhuang, W.; Hung, F.F.; Che, C.M.; Casado, J.; Liu, J. Nonalternant Nanographenes Containing N- Centered Cyclopenta[ef]heptalene and Aza[7]Helicene Units. J. Am. Chem. Soc. 2024, 146, 16161–16172. [Google Scholar] [CrossRef]
- Avila Ferrer, F.J.; Cerezo, J.; Soto, J.; Improta, R.; Santoro, F. First-principle computation of absorption and fluorescence spectra in solution accounting for vibronic structure, temperature effects and solvent inhomogenous broadening. Comput. Theo. Chem. 2014, 1040, 328–337. [Google Scholar] [CrossRef]
- Santoro, F.; Lami, A.; Improta, R.; Bloino, J.; Barone, V. Effective method for the computation of optical spectra of large molecules at finite temperature including the Duschinsky and Herzberg-Teller effect: The Q(x) band of porphyrin as a case study. J. Chem. Phys. 2008, 128, 224311. [Google Scholar] [CrossRef]
- Santoro, F.; Improta, R.; Lami, A.; Bloino, J.; Barone, V. Effective method to compute Franck-Condon integrals for optical spectra of large molecules in solution. J. Chem. Phys. 2007, 126, 084509. [Google Scholar] [CrossRef]
- Santoro, F.; Lami, A.; Improta, R.; Barone, V. Effective method to compute vibrationally resolved optical spectra of large molecules at finite temperature in the gas phase and in solution. J. Chem. Phys. 2007, 126, 184102. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian, version 09, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Cerezo, J.; Santoro, F. FCclasses3: Vibrationally-resolved spectra simulated at the edge of the harmonic approximation. J. Comput. Chem. 2023, 44, 626–643. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 2020, 165, 461–467. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absorption | Emission | Stokes Shift (nm) | |||||
|---|---|---|---|---|---|---|---|
| E (eV) | λ (nm) | f | E (eV) | λ (nm) | f | ||
| O7H | 2.76 | 449 | 0.1605 | 2.22 | 558 | 0.1302 | 109 |
| O8H | 2.67 | 465 | 0.1210 | 2.22 | 559 | 0.1263 | 94 |
| O9H | 2.52 | 493 | 0.0777 | 1.89 | 657 | 0.0495 | 164 |
| OO7H | 3.03 | 409 | 0.1212 | 2.80 | 443 | 0.1883 | 34 |
| OO9H | 2.60 | 477 | 0.0824 | 1.93 | 641 | 0.0554 | 164 |
| O7H | O8H | O9H | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| S0 Mode | ω0 (cm−1) | S1 Mode | ω1 (cm−1) | S0 Mode | ω0 (cm−1) | S1 Mode | ω1 (cm−1) | S0 Mode | ω0 (cm−1) | S1 Mode | ω1 (cm−1) |
| 3 | 87 | 1 | 44 | 1 | 39 | 1 | 34 | 3 | 56 | 3 | 64 |
| 5 | 114 | 3 | 88 | 3 | 65 | 3 | 64 | 7 | 112 | 7 | 110 |
| 6 | 152 | 6 | 156 | 4 | 86 | 5 | 99 | 9 | 168 | 10 | 182 |
| 9 | 217 | 22 | 443 | 20 | 400 | 13 | 270 | 13 | 244 | 15 | 281 |
| 88 | 1315 | 103 | 1502 | 99 | 1292 | 108 | 1422 | 138 | 1591 | 122 | 1423 |
| 108 | 1592 | 108 | 1427 | 136 | 1562 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Feng, X.; Yu, W.; Shen, C. Theoretical Study on Vibrationally Resolved Electronic Spectra of Chiral Nanographenes. Molecules 2024, 29, 3999. https://doi.org/10.3390/molecules29173999
Ma Y, Feng X, Yu W, Shen C. Theoretical Study on Vibrationally Resolved Electronic Spectra of Chiral Nanographenes. Molecules. 2024; 29(17):3999. https://doi.org/10.3390/molecules29173999
Chicago/Turabian StyleMa, Yijian, Xian Feng, Wenxiong Yu, and Chengshuo Shen. 2024. "Theoretical Study on Vibrationally Resolved Electronic Spectra of Chiral Nanographenes" Molecules 29, no. 17: 3999. https://doi.org/10.3390/molecules29173999
APA StyleMa, Y., Feng, X., Yu, W., & Shen, C. (2024). Theoretical Study on Vibrationally Resolved Electronic Spectra of Chiral Nanographenes. Molecules, 29(17), 3999. https://doi.org/10.3390/molecules29173999