
Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Kinetics of Excited States of Photocatalysts
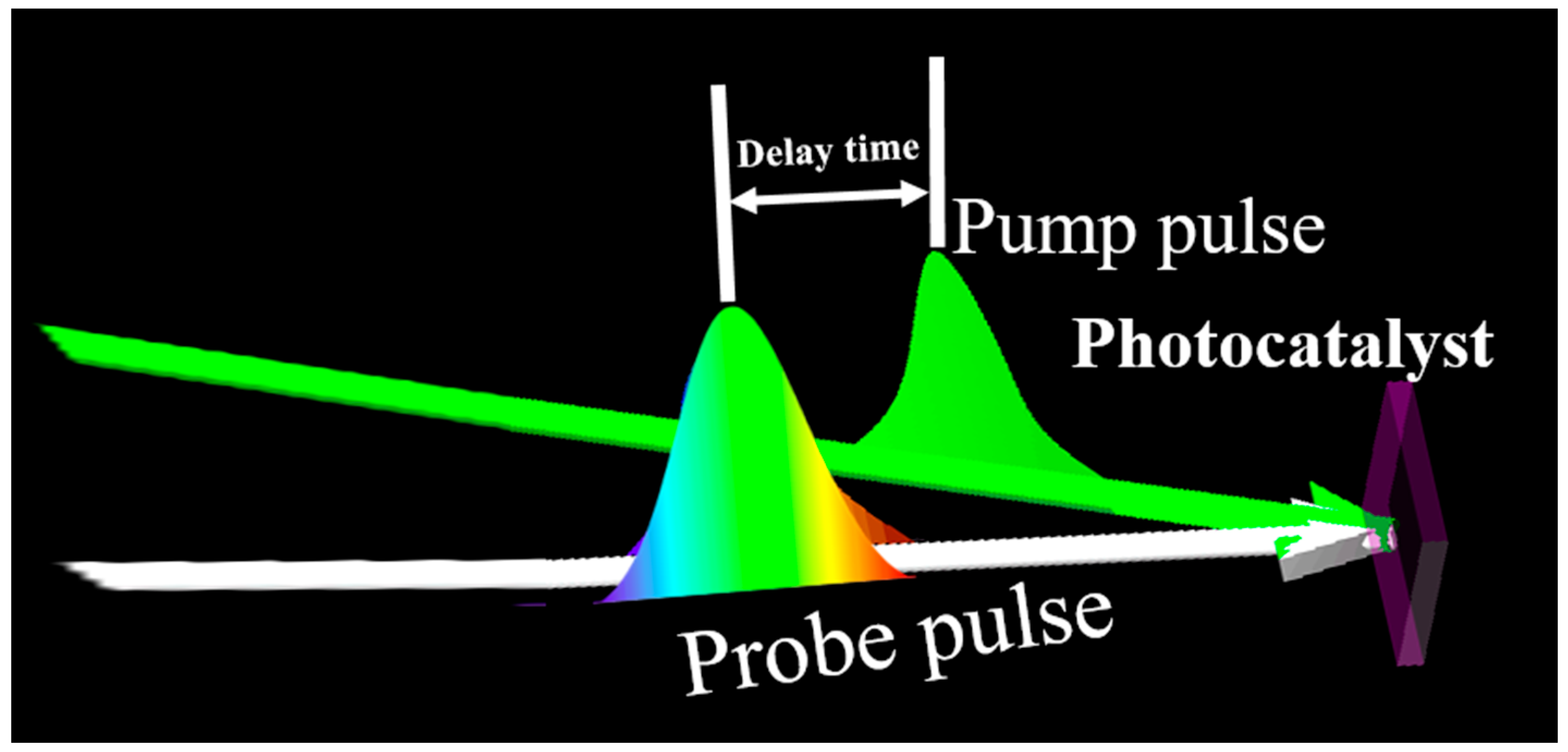
2.1. The Principle of Transient Absorption
2.2. Ultrafast Dynamic Process
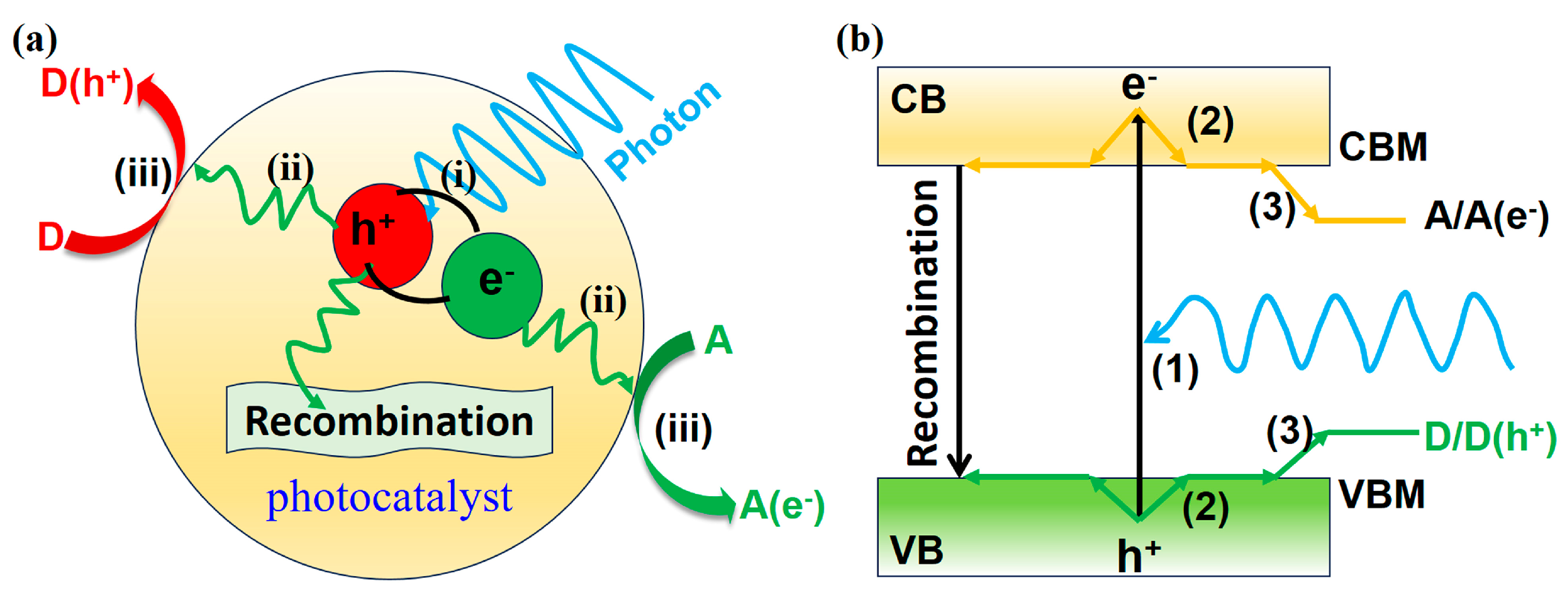
2.3. Kinetic Process of Photocatalyst
3. Application of Transient Absorption Spectroscopy in Photocatalysis
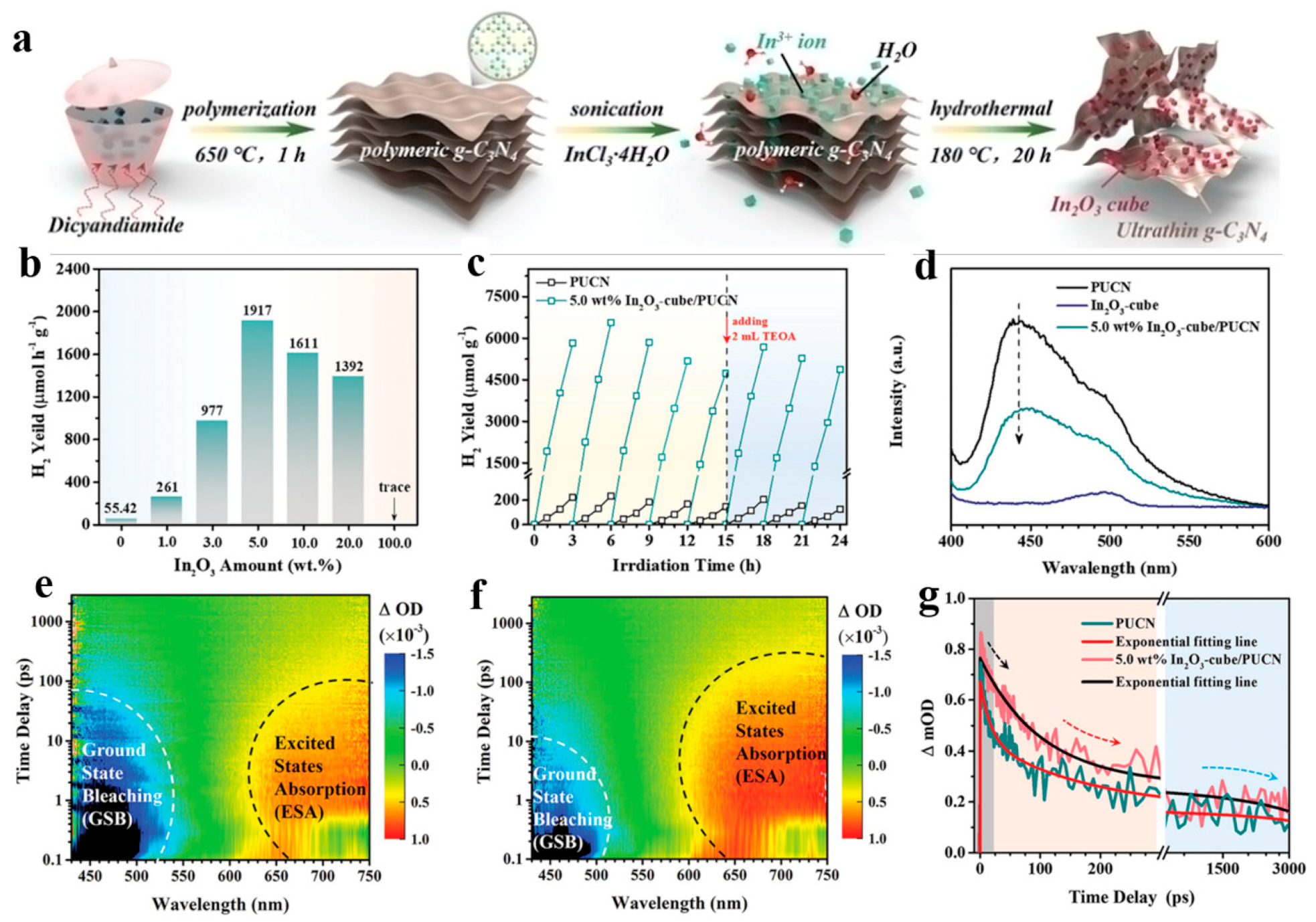
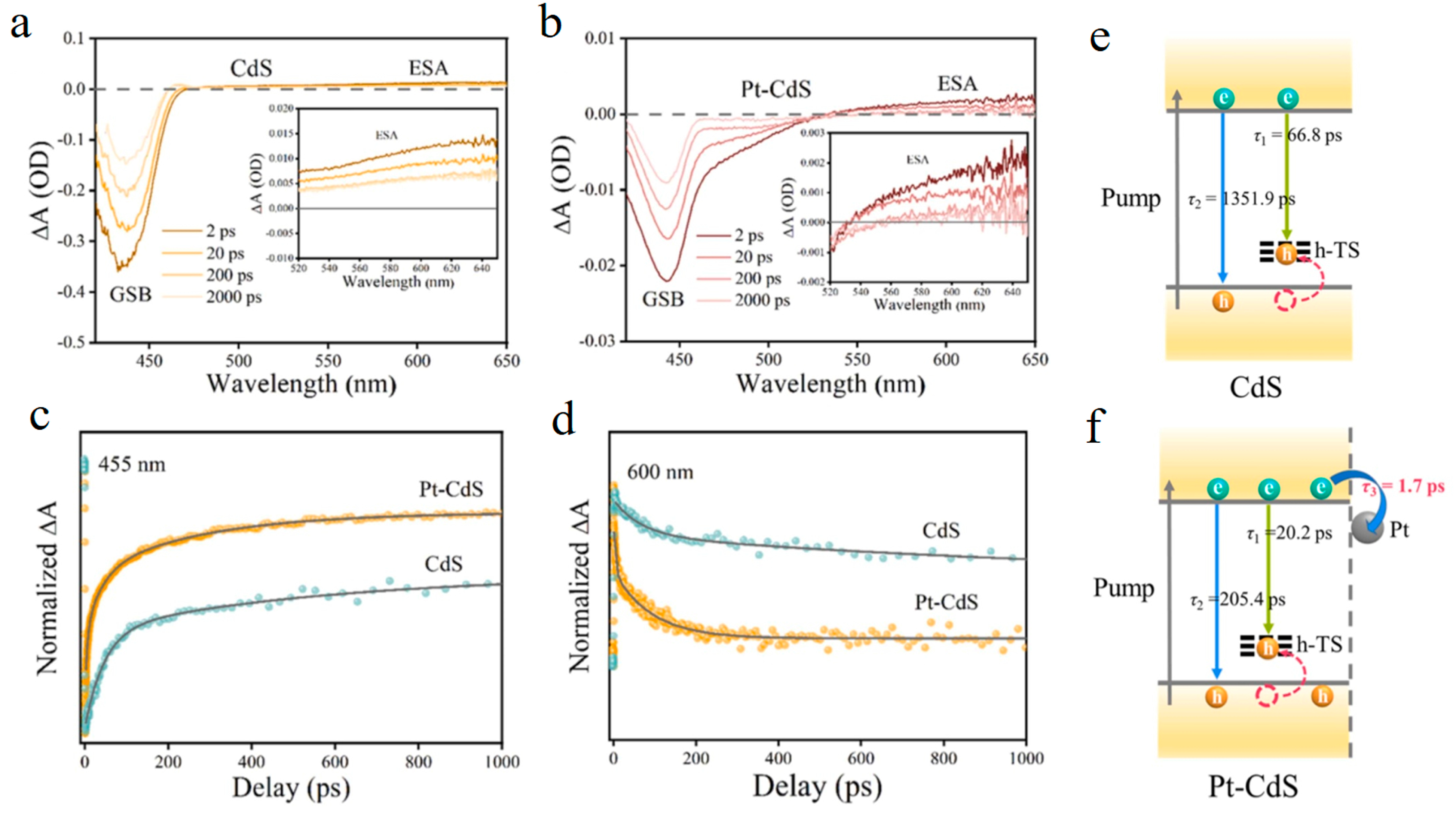
3.1. Ultrafast Kinetics of Common Semiconductors in Photocatalysis
3.2. Ultrafast Kinetics of MOF in Photocatalysis
3.3. Ultrafast Kinetics of Halide Perovskite in Photocatalysis
4. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stolarczyk, J.K.; Bhattacharyya, S.; Polavarapu, L.; Feldmann, J. Challenges and Prospects in Solar Water Splitting and CO2 Reduction with Inorganic and Hybrid Nanostructures. ACS Catal. 2018, 8, 3602–3635. [Google Scholar] [CrossRef]
- Yuan, L.; Xu, Y.-J. Photocatalytic conversion of CO2 into value-added and renewable fuels. Appl. Surf. Sci. 2015, 342, 154–167. [Google Scholar] [CrossRef]
- Lin, H.; Luo, S.; Zhang, H.; Ye, J. Toward solar-driven carbon recycling. Joule 2022, 6, 294–314. [Google Scholar] [CrossRef]
- Valluri, S.; Claremboux, V.; Kawatra, S. Opportunities and challenges in CO2 utilization. J. Environ. Sci. 2022, 113, 322–344. [Google Scholar] [CrossRef] [PubMed]
- Temerov, F.; Baghdadi, Y.; Rattner, E.; Eslava, S. A Review on Halide Perovskite-Based Photocatalysts: Key Factors and Challenges. ACS Appl. Energy Mater. 2022, 5, 14605–14637. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, Z.; Huang, L.; Zaman, S.; Lei, K.; Yue, T.; Li, Z.; You, B.; Xia, B.Y. Engineering 2D Photocatalysts toward Carbon Dioxide Reduction. Adv. Energy Mater. 2021, 11, 2003159. [Google Scholar] [CrossRef]
- Low, J.; Yu, J.; Jaroniec, M.; Wageh, S.; Al-Ghamdi, A.A. Heterojunction Photocatalysts. Adv. Mater. 2017, 29, 1601694. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ren, Z.; Liang, Y.; Zhang, G.; Dittrich, T.; Liu, R.; Liu, Y.; Zhao, Y.; Pang, S.; An, H.; et al. Spatiotemporal imaging of charge transfer in photocatalyst particles. Nature 2022, 610, 296–301. [Google Scholar] [CrossRef]
- Buzzetti, L.; Crisenza, G.E.M.; Melchiorre, P. Mechanistic Studies in Photocatalysis. Angew. Chem. Int. Ed. 2019, 58, 3730–3747. [Google Scholar] [CrossRef]
- Hu, W.; Prasad, P.N.; Huang, W. Manipulating the Dynamics of Dark Excited States in Organic Materials for Phototheranostics. Acc. Chem. Res. 2020, 54, 697–706. [Google Scholar] [CrossRef]
- Cooper, J.K.; Reyes-Lillo, S.E.; Hess, L.H.; Jiang, C.-M.; Neaton, J.B.; Sharp, I.D. Physical Origins of the Transient Absorption Spectra and Dynamics in Thin-Film Semiconductors: The Case of BiVO4. J. Phys. Chem. C 2018, 122, 20642–20652. [Google Scholar] [CrossRef]
- Yu, H.-Z.; Baskin, J.S.; Zewail, A.H. Ultrafast Dynamics of Porphyrins in the Condensed Phase: II. Zinc Tetraphenylporphyrin. J. Phys. Chem. A 2002, 106, 9845–9854. [Google Scholar] [CrossRef]
- Baskin, J.S.; Yu, H.Z.; Zewail, A.H. Ultrafast dynamics of Porphyrins in the condensed phase: I. Free base tetraphenylporphyrin. J. Phys. Chem. A 2002, 106, 9837–9844. [Google Scholar] [CrossRef]
- Yu, H.Z.; Steiger, J.S.B.; Wan, C.Z.; Anson, F.C.; Zewail, A.H. Femtosecond dynamics of metalloporphyrins: Electron transfer and energy redistribution. Chem. Phys. Lett. 1998, 293, 1–8. [Google Scholar] [CrossRef]
- Zhu, J.; Wageh, S.; Al-Ghamdi, A.A. Using the femtosecond technique to study charge transfer dynamics. Chin. J. Catal. 2023, 49, 5–7. [Google Scholar] [CrossRef]
- Zhang, Q.; Luo, Y. Probing the ultrafast dynamics in nanomaterial complex systems by femtosecond transient absorption spectroscopy. High Power Laser Sci. Eng. 2016, 4, e22. [Google Scholar] [CrossRef]
- Miao, T.J.; Tang, J. Characterization of charge carrier behavior in photocatalysis using transient absorption spectroscopy. J. Chem. Phys. 2020, 152, 194201. [Google Scholar] [CrossRef] [PubMed]
- Othonos, A. Probing ultrafast carrier and phonon dynamics in semiconductors. J. Appl. Phys. 1998, 83, 1789–1830. [Google Scholar] [CrossRef]
- Pasanen, H.P.; Khan, R.; Odutola, J.A.; Tkachenko, N.V. Transient Absorption Spectroscopy of Films: Impact of Refractive Index. J. Phys. Chem. C 2024, 128, 6167–6179. [Google Scholar] [CrossRef] [PubMed]
- Berera, R.; van Grondelle, R.; Kennis, J.T.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef]
- Ma, J.; Miao, T.J.; Tang, J. Charge carrier dynamics and reaction intermediates in heterogeneous photocatalysis by time-resolved spectroscopies. Chem. Soc. Rev. 2022, 51, 5777–5794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, B.; Zhang, L.; Yu, J. Femtosecond transient absorption spectroscopy investigation into the electron transfer mechanism in photocatalysis. Chem. Commun. 2023, 59, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Knowles, K.E.; Koch, M.D.; Shelton, J.L. Three applications of ultrafast transient absorption spectroscopy of semiconductor thin films: Spectroelectrochemistry, microscopy, and identification of thermal contributions. J. Mater. Chem. C 2018, 6, 11853–11867. [Google Scholar] [CrossRef]
- Wang, Q.; Yun, L.; Yang, J. Ultrafast molecular movies: Probing chemical dynamics with femtosecond electron and X-ray diffraction. CCS Chem. 2024, 6, 1092–1109. [Google Scholar] [CrossRef]
- Li, N.; Wang, Q.; Zhang, H.L. 2D Materials in Light: Excited-State Dynamics and Applications. Chem. Rec. 2020, 20, 413–428. [Google Scholar] [CrossRef] [PubMed]
- Karaman, C.O.; Bykov, A.Y.; Kiani, F.; Tagliabue, G.; Zayats, A.V. Ultrafast hot-carrier dynamics in ultrathin monocrystalline gold. Nat. Commun 2024, 15, 703. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.A.; Zhang, J.Z. Exciton Dynamics in Semiconductor Nanocrystals. Adv. Mater. 2013, 25, 2878–2896. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ni, C.; Zhu, J.; Fan, F.; Li, C. Surface photovoltage microscopy for mapping charge separation on photocatalyst particles. Nat. Protoc. 2024, 19, 2250–2282. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zhang, J.; Zhu, B.; Liang, G.; Zhang, L.; Yu, J. Verifying the Charge-Transfer Mechanism in S-Scheme Heterojunctions Using Femtosecond Transient Absorption Spectroscopy. Angew. Chem. Int. Ed. 2023, 62, e202218688. [Google Scholar] [CrossRef] [PubMed]
- Magde, D.; Windsor, M.W.; Holten, D.; Gouterman, M. Picosecond flash photolysis: Transient absorption in Sn(IV), Pd(II), and Cu(II) porphyrins. Chem. Phys. Lett. 1974, 29, 183–188. [Google Scholar] [CrossRef]
- Shah, J. Hot electrons and phonons under high intensity photoexcitation of semiconductors. Solid-State Electron. 1978, 21, 43–50. [Google Scholar] [CrossRef]
- He, X.; Ghosh, M.; Yang, D.-S. Impacts of hot electron diffusion, electron–phonon coupling, and surface atoms on metal surface dynamics revealed by reflection ultrafast electron diffraction. J. Chem. Phys 2024, 160, 224701. [Google Scholar] [CrossRef]
- Xiao, J.-D.; Jiang, H.-L. Metal–Organic Frameworks for Photocatalysis and Photothermal Catalysis. Acc. Chem. Res. 2019, 52, 356–366. [Google Scholar] [CrossRef]
- Jing, J.; Yang, J.; Li, W.; Wu, Z.; Zhu, Y. Construction of Interfacial Electric Field via Dual-Porphyrin Heterostructure Boosting Photocatalytic Hydrogen Evolution. Adv. Mater. 2021, e2106807. [Google Scholar] [CrossRef]
- Yuan, J.; Wu, J.; Hardy, W.J.; Loya, P.; Lou, M.; Yang, Y.; Najmaei, S.; Jiang, M.; Qin, F.; Keyshar, K.; et al. Facile Synthesis of Single Crystal Vanadium Disulfide Nanosheets by Chemical Vapor Deposition for Efficient Hydrogen Evolution Reaction. Adv. Mater. 2015, 27, 5605–5609. [Google Scholar] [CrossRef]
- Yuan, H.; Qin, H.; Sun, K.; Sun, X.; Lu, J.; Bian, A.; Hou, J.; Lu, C.; Li, C.; Guo, F. Ultrafast hot electron transfer and trap-state mediated charge separation for boosted photothermal-assisted photocatalytic H2 evolution. Chem. Eng. J. 2024, 494, 153058. [Google Scholar] [CrossRef]
- Schneider, J.; Matsuoka, M.; Takeuchi, M.; Zhang, J.; Horiuchi, Y.; Anpo, M.; Bahnemann, D.W. Understanding TiO2 Photocatalysis: Mechanisms and Materials. Chem. Rev. 2014, 114, 9919–9986. [Google Scholar] [CrossRef]
- Qian, R.; Zong, H.; Schneider, J.; Zhou, G.; Zhao, T.; Li, Y.; Yang, J.; Bahnemann, D.W.; Pan, J.H. Charge carrier trapping, recombination and transfer during TiO2 photocatalysis: An overview. Catal. Today 2019, 335, 78–90. [Google Scholar] [CrossRef]
- Watanabe, M.; Hayashi, T. Time-resolved study of self-trapped exciton luminescence in anatase TiO2 under two-photon excitation. J. Lumin. 2005, 112, 88–91. [Google Scholar] [CrossRef]
- Thompson, T.L.; Yates, J.T. Surface Science Studies of the Photoactivation of TiO2 New Photochemical Processes. Chem. Rev. 2006, 106, 4428–4453. [Google Scholar] [CrossRef]
- Guo, Q.; Zhou, C.; Ma, Z.; Ren, Z.; Fan, H.; Yang, X. Elementary photocatalytic chemistry on TiO2 surfaces. Chem. Soc. Rev. 2016, 45, 3701–3730. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Gao, Y.; Zhang, L.; Zhang, J.; Zhang, Q.; Li, Q.; Bao, H.; Zhou, J.; Miao, S.; Chen, N.; et al. A Promoted Charge Separation/Transfer System from Cu Single Atoms and C3N4 Layers for Efficient Photocatalysis. Adv. Mater. 2020, 32, 2003082. [Google Scholar] [CrossRef]
- Dotan, H.; Sivula, K.; Grätzel, M.; Rothschild, A.; Warren, S.C. Probing the photoelectrochemical properties of hematite (α-Fe2O3) electrodes using hydrogen peroxide as a hole scavenger. Energy Environ. Sci. 2011, 4, 958–964. [Google Scholar] [CrossRef]
- Barroso, M.; Cowan, A.J.; Pendlebury, S.R.; Grätzel, M.; Klug, D.R.; Durrant, J.R. The Role of Cobalt Phosphate in Enhancing the Photocatalytic Activity of α-Fe2O3 toward Water Oxidation. J. Am. Chem. Soc. 2011, 133, 14868–14871. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Chen, S.; Wang, Y.; Zhang, Z.; Li, Z.; Xin, B.; Jing, L. Controlled Construction of Copper Phthalocyanine/α-Fe2O3 Ultrathin S-Scheme Heterojunctions for Efficient Photocatalytic CO2 Reduction under Wide Visible-Light Irradiation. Small Sci. 2021, 1, 2100050. [Google Scholar] [CrossRef]
- Song, G.; Wei, M.; Zhou, J.; Mu, L.; Song, S. Modulation of the phase transformation of Fe2O3 for enhanced water oxidation under a magnetic field. ACS Catal. 2024, 14, 846–856. [Google Scholar] [CrossRef]
- Fitzmorris, B.C.; Patete, J.M.; Smith, J.; Mascorro, X.; Adams, S.; Wong, S.S.; Zhang, J.Z. Ultrafast Transient Absorption Studies of Hematite Nanoparticles: The Effect of Particle Shape on Exciton Dynamics. ChemSusChem 2013, 6, 1907–1914. [Google Scholar] [CrossRef]
- Li, Y.; Jin, R.; Xing, Y.; Li, J.; Song, S.; Liu, X.; Li, M.; Jin, R. Macroscopic Foam-Like Holey Ultrathin g-C3N4 Nanosheets for Drastic Improvement of Visible-Light Photocatalytic Activity. Adv. Energy Mater. 2016, 6, 1601273. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, Z.; Zou, Y.; Chen, J.; Shi, J.-W. The progress of g-C3N4 in photocatalytic H2 evolution: From fabrication to modification. Coord. Chem. Rev. 2024, 500, 215489. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, B.; Wu, D.; Tian, C.; Ran, H.; Chen, W.; Huang, Q.; Zhang, W.; Qi, F.; Zhang, N. Enhanced Photocatalytic Activity of Lead-Free Cs2TeBr6/g-C3N4 Heterojunction Photocatalyst and Its Mechanism. Adv. Funct. Mater. 2024, 34, 2308411. [Google Scholar] [CrossRef]
- Wang, W.; Bai, X.; Ci, Q.; Du, L.; Ren, X.; Phillips, D.L. Near-Field Drives Long-Lived Shallow Trapping of Polymeric C3N4 for Efficient Photocatalytic Hydrogen Evolution. Adv. Funct. Mater. 2021, 31, 2103978. [Google Scholar] [CrossRef]
- Su, H.; Yin, H.; Wang, R.; Wang, Y.; Orbell, W.; Peng, Y.; Li, J. Atomic-level coordination structures meet graphitic carbon nitride (g-C3N4) for photocatalysis: Energy conversion and environmental remediation. Appl. Catal. B Environ. 2024, 348, 123683. [Google Scholar] [CrossRef]
- Ou, M.; Tu, W.; Yin, S.; Xing, W.; Wu, S.; Wang, H.; Wan, S.; Zhong, Q.; Xu, R. Amino-Assisted Anchoring of CsPbBr3 Perovskite Quantum Dots on Porous g-C3N4 for Enhanced Photocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 13570–13574. [Google Scholar] [CrossRef]
- Cao, S.; Yu, J. g-C3N4-Based Photocatalysts for Hydrogen Generation. J. Phys. Chem. Lett. 2014, 5, 2101–2107. [Google Scholar] [CrossRef]
- Zhu, J.; Hu, L.; Zhao, P.; Lee, L.Y.S.; Wong, K.-Y. Recent Advances in Electrocatalytic Hydrogen Evolution Using Nanoparticles. Chem. Rev. 2020, 120, 851–918. [Google Scholar] [CrossRef]
- Zong, S.; Tian, L.; Guan, X.; Zhang, Y.; Cheng, C.; Geng, J.; Jiang, S.; Shi, J. Hierarchical LaTiO2N/Sn3O4 heterojunction with intimate interface contact for enhanced photocatalytic water splitting. Surf. Interfaces 2024, 48, 104285. [Google Scholar] [CrossRef]
- Wolff, C.M.; Frischmann, P.D.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F.; et al. All-in-one visible-light-driven water splitting by combining nanoparticulate and molecular co-catalysts on CdS nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Rafiq, K.; Sabir, M.; Abid, M.Z.; Jalil, M.; Nadeem, M.A.; Iqbal, S.; Rauf, A.; Hussain, E. Tuning of TiO2/CdS Hybrid Semiconductor with Au Cocatalysts: State-of-the-Art Design for Sunlight-Driven H2 Generation from Water Splitting. Energy Fuels 2024, 38, 4625–4636. [Google Scholar] [CrossRef]
- Saleem, F.; Abid, M.Z.; Rafiq, K.; Rauf, A.; Ahmad, K.; Iqbal, S.; Jin, R.; Hussain, E. Synergistic effect of Cu/Ni cocatalysts on CdS for sun-light driven hydrogen generation from water splitting. Int. J. Hydrogen Energy 2024, 52, 305–319. [Google Scholar] [CrossRef]
- Meng, Z.; Zhang, J.; Jiang, C.; Trapalis, C.; Zhang, L.; Yu, J. Dynamics of Electron Transfer in CdS Photocatalysts Decorated with Various Noble Metals. Small 2024, 20, 2308952. [Google Scholar] [CrossRef]
- Wang, F.; Hou, T.; Zhao, X.; Yao, W.; Fang, R.; Shen, K.; Li, Y. Ordered Macroporous Carbonous Frameworks Implanted with CdS Quantum Dots for Efficient Photocatalytic CO2 Reduction. Adv. Mater. 2021, 33, 2102690. [Google Scholar] [CrossRef]
- Liu, Z.-G.; Wei, Y.; Xie, L.; Chen, H.-Q.; Wang, J.; Yang, K.; Zou, L.-X.; Deng, T.; Lu, K.-Q. Decorating CdS with cobaltous hydroxide and graphene dual cocatalyst for photocatalytic hydrogen production coupled selective benzyl alcohol oxidation. Mol. Catal. 2024, 553, 113738. [Google Scholar] [CrossRef]
- Xiang, X.; Zhang, L.; Luo, C.; Zhang, J.; Cheng, B.; Liang, G.; Zhang, Z.; Yu, J. Ultrafast electron transfer from CdS quantum dots to atomically-dispersed Pt for enhanced H2 evolution and value-added chemical synthesis. Appl. Catal. B Environ. 2024, 340, 123196. [Google Scholar] [CrossRef]
- Jiang, D.; Li, Z.; Li, H.; Cheng, Y.; Du, H.; Zhu, C.; Meng, L.; Fang, Y.; Zhao, C.; Lou, Z.; et al. Achieving Long-Lived Charge Separated State through Ultrafast Interfacial Hole Transfer in Redox Sites-Isolated CdS Nanorods for Enhanced Photocatalysis. Small 2024, 20, 2310414. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tong, X.; Wang, W.; Xu, J.-Y.; Besteiro, L.V.; Channa, A.I.; Lin, F.; Wu, J.; Wang, Q.; Govorov, A.O.; et al. Manipulating the Optoelectronic Properties of Quasi-type II CuInS2/CdS Core/Shell Quantum Dots for Photoelectrochemical Cell Applications. ACS Appl. Mater. Interfaces 2020, 12, 36277–36286. [Google Scholar] [CrossRef] [PubMed]
- Ahmad Beigi, A.; Fatemi, S.; Salehi, Z. Synthesis of nanocomposite CdS/TiO2 and investigation of its photocatalytic activity for CO2 reduction to CO and CH4 under visible light irradiation. J. CO2 Util. 2014, 7, 23–29. [Google Scholar] [CrossRef]
- Chen, C.; Fei, L.; Wang, B.; Xu, J.; Li, B.; Shen, L.; Lin, H. MOF-based photocatalytic membrane for water purification: A review. Small 2024, 20, 2305066. [Google Scholar] [CrossRef]
- Xu, H.-Q.; Yang, S.; Ma, X.; Huang, J.; Jiang, H.-L. Unveiling Charge-Separation Dynamics in CdS/Metal–Organic Framework Composites for Enhanced Photocatalysis. ACS Catal. 2018, 8, 11615–11621. [Google Scholar] [CrossRef]
- Gong, S.; Teng, X.; Niu, Y.; Liu, X.; Xu, M.; Xu, C.; Ji, L.; Chen, Z. Construction of S-scheme 0D/2D heterostructures for enhanced visible-light-driven CO2 reduction. Appl. Catal. B Environ. 2021, 298, 120521. [Google Scholar] [CrossRef]
- Yue, X.; Cheng, L.; Fan, J.; Xiang, Q. 2D/2D BiVO4/CsPbBr3 S-scheme heterojunction for photocatalytic CO2 reduction: Insights into structure regulation and Fermi level modulation. Appl. Catal. B Environ. 2022, 304, 120979. [Google Scholar] [CrossRef]
- Yu, B.; Wu, Y.; Meng, F.; Wang, Q.; Jia, X.; Wasim Khan, M.; Huang, C.; Zhang, S.; Yang, L.; Wu, H. Formation of hierarchical Bi2MoO6/ln2S3 S-scheme heterojunction with rich oxygen vacancies for boosting photocatalytic CO2 reduction. Chem. Eng. J. 2022, 429, 132456. [Google Scholar] [CrossRef]
- Xu, F.; Meng, K.; Cheng, B.; Wang, S.; Xu, J.; Yu, J. Unique S-scheme heterojunctions in self-assembled TiO2/CsPbBr3 hybrids for CO2 photoreduction. Nat. Commun 2020, 11, 4613. [Google Scholar] [CrossRef]
- Ivan Grigioni, K.G.S.; Selli, E.; Kamat, P.V. Dynamics of Photogenerated Charge Carriers in WO3/BiVO4 Heterojunction Photoanodes. J. Phys. Chem. C 2015, 119, 20792–20800. [Google Scholar] [CrossRef]
- Bian, J.; Feng, J.; Zhang, Z.; Li, Z.; Zhang, Y.; Liu, Y.; Ali, S.; Qu, Y.; Bai, L.; Xie, J.; et al. Dimension-Matched Zinc Phthalocyanine/BiVO4 Ultrathin Nanocomposites for CO2 Reduction as Efficient Wide-Visible-Light-Driven Photocatalysts via a Cascade Charge Transfer. Angew. Chem. Int. Ed. 2019, 58, 10873–10878. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, S.; Ouardi, M.E.; Ahsaine, H.A.; Assani, A. Recent progress on the synthesis, morphology and photocatalytic dye degradation of BiVO4 photocatalysts: A review. Catal. Rev. 2024, 66, 214–258. [Google Scholar] [CrossRef]
- Deng, Y.; Zhou, H.; Zhao, Y.; Yang, B.; Shi, M.; Tao, X.; Yang, S.; Li, R.; Li, C. Spatial Separation of Photogenerated Charges on Well-Defined Bismuth Vanadate Square Nanocrystals. Small 2022, 18, 2103245. [Google Scholar] [CrossRef]
- Huang, B.; Fu, X.; Wang, K.; Wang, L.; Zhang, H.; Liu, Z.; Liu, B.; Li, J. Chemically bonded BiVO4/Bi19Cl3S27 heterojunction with fast hole extraction dynamics for continuous CO2 photoreduction. Adv. Powder. Mater 2024, 3, 100140. [Google Scholar] [CrossRef]
- Wan, S.; Ou, M.; Zhong, Q.; Wang, X. Perovskite-type CsPbBr3 quantum dots/UiO-66(NH2) nanojunction as efficient visible-light-driven photocatalyst for CO2 reduction. Chem. Eng. J. 2019, 358, 1287–1295. [Google Scholar] [CrossRef]
- Wang, J.-R.; Song, K.; Luan, T.-X.; Cheng, K.; Wang, Q.; Wang, Y.; Yu, W.W.; Li, P.-Z.; Zhao, Y. Robust links in photoactive covalent organic frameworks enable effective photocatalytic reactions under harsh conditions. Nat. Commun 2024, 15, 1267. [Google Scholar] [CrossRef]
- Cheng, S.; Sun, Z.; Lim, K.H.; Liu, K.; Wibowo, A.A.; Du, T.; Liu, L.; Nguyen, H.T.; Li, G.K.; Yin, Z. Defective heterojunctions in CO2 photoreduction: Enabling ultrafast interfacial charge transfer and selective methanation. Appl. Catal. B Environ. 2024, 343, 123583. [Google Scholar] [CrossRef]
- Lian, Z.; Qu, M.; Xiao, H.; Wang, L.; Wu, H.; Zi, J.; Wang, W.; Li, H. Direct Observation of Z-Scheme Route in Cu31S16/ZnxCd1-xS Heteronanoplates for Highly Efficient Photocatalytic Hydrogen Evolution. Small 2024, 2400611. [Google Scholar] [CrossRef]
- Dissegna, S.; Epp, K.; Heinz, W.R.; Kieslich, G.; Fischer, R.A. Defective Metal-Organic Frameworks. Adv. Mater. 2018, 30, e1704501. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Wang, Y.; Jiang, H.L.; Xu, Q. Metal-Organic Frameworks as Platforms for Catalytic Applications. Adv. Mater. 2018, 30, e1703663. [Google Scholar] [CrossRef]
- Liang, Z.; Qu, C.; Guo, W.; Zou, R.; Xu, Q. Pristine Metal-Organic Frameworks and their Composites for Energy Storage and Conversion. Adv. Mater. 2018, 30, e1702891. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, Y.; Kaskel, S. Porphyrin-Based Metal–Organic Frameworks for Biomedical Applications. Angew. Chem. Int. Ed. 2021, 60, 5010–5035. [Google Scholar] [CrossRef]
- Chen, X.; Xiao, S.; Wang, H.; Wang, W.; Cai, Y.; Li, G.; Qiao, M.; Zhu, J.; Li, H.; Zhang, D.; et al. MOFs Conferred with Transient Metal Centers for Enhanced Photocatalytic Activity. Angew. Chem. Int. Ed. 2020, 59, 17182–17186. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, W.; Zhou, K. Metal–Organic-Framework-Based Catalysts for Photoreduction of CO2. Adv. Mater. 2018, 30, 1705512. [Google Scholar] [CrossRef]
- Han, B.; Ou, X.; Deng, Z.; Song, Y.; Tian, C.; Deng, H.; Xu, Y.-J.; Lin, Z. Nickel Metal–Organic Framework Monolayers for Photoreduction of Diluted CO2: Metal-Node-Dependent Activity and Selectivity. Angew. Chem. Int. Ed. 2018, 57, 16811–16815. [Google Scholar] [CrossRef]
- Kolobov, N.; Goesten, M.G.; Gascon, J. Metal–Organic Frameworks: Molecules or Semiconductors in Photocatalysis? Angew. Chem. Int. Ed. 2021, 60, 26038–26052. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, J.; Liu, J.-J.; Dong, L.-Z.; Xin, Z.-F.; Teng, Y.-L.; Lan, Y.-Q. Adenine Components in Biomimetic Metal–Organic Frameworks for Efficient CO2 Photoconversion. Angew. Chem. Int. Ed. 2019, 58, 5226–5231. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, Q.-L.; Zou, R.; Xu, Q. Metal-Organic Frameworks for Energy Applications. Chem 2017, 2, 52–80. [Google Scholar] [CrossRef]
- Wei, Y.S.; Zhang, M.; Zou, R.; Xu, Q. Metal-Organic Framework-Based Catalysts with Single Metal Sites. Chem. Rev. 2020, 120, 12089–12174. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Yue, X.; Li, F.; Xiang, Q. Functionalized MOF-Based Photocatalysts for CO2 Reduction. Chem. Eur. J. 2023, 29, e202203706. [Google Scholar] [CrossRef]
- Fang, X.; Shang, Q.; Wang, Y.; Jiao, L.; Yao, T.; Li, Y.; Zhang, Q.; Luo, Y.; Jiang, H.-L. Single Pt Atoms Confined into a Metal–Organic Framework for Efficient Photocatalysis. Adv. Mater. 2018, 30, 1705112. [Google Scholar] [CrossRef]
- Xu, H.-Q.; Hu, J.; Wang, D.; Li, Z.; Zhang, Q.; Luo, Y.; Yu, S.-H.; Jiang, H.-L. Visible-Light Photoreduction of CO2 in a Metal–Organic Framework: Boosting Electron–Hole Separation via Electron Trap States. J. Am. Chem. Soc. 2015, 137, 13440–13443. [Google Scholar] [CrossRef]
- Zhong, H.; Sa, R.; Lv, H.; Yang, S.; Yuan, D.; Wang, X.; Wang, R. Covalent Organic Framework Hosting Metalloporphyrin-Based Carbon Dots for Visible-Light-Driven Selective CO2 Reduction. Adv. Funct. Mater. 2020, 30, 2002654. [Google Scholar] [CrossRef]
- Chen, E.X.; Qiu, M.; Zhang, Y.F.; He, L.; Sun, Y.Y.; Zheng, H.L.; Wu, X.; Zhang, J.; Lin, Q. Energy Band Alignment and Redox-active Sites in Metalloporphyrin-Spaced Metal-Catechol Frameworks for Enhanced CO2 Photoreduction. Angew. Chem. Int. Ed. 2021, 61, e202111622. [Google Scholar] [CrossRef]
- Feng, D.; Gu, Z.-Y.; Li, J.-R.; Jiang, H.-L.; Wei, Z.; Zhou, H.-C. Zirconium-Metalloporphyrin PCN-222: Mesoporous Metal–Organic Frameworks with Ultrahigh Stability as Biomimetic Catalysts. Angew. Chem. Int. Ed. 2012, 51, 10307–10310. [Google Scholar] [CrossRef] [PubMed]
- Min Park, J.; Lee, J.H.; Jang, W.-D. Applications of porphyrins in emerging energy conversion technologies. Coord. Chem. Rev. 2020, 407, 213157. [Google Scholar] [CrossRef]
- Lian, S.; Kodaimati, M.S.; Weiss, E.A. Photocatalytically Active Superstructures of Quantum Dots and Iron Porphyrins for Reduction of CO2 to CO in Water. ACS Nano 2018, 12, 568–575. [Google Scholar] [CrossRef]
- Gong, X.; Shu, Y.; Jiang, Z.; Lu, L.; Xu, X.; Wang, C.; Deng, H. Metal-Organic Frameworks for the Exploitation of Distance between Active Sites in Efficient Photocatalysis. Angew. Chem. Int. Ed. 2020, 59, 5326–5331. [Google Scholar] [CrossRef]
- Ghosh, M.; Mora, A.K.; Nath, S.; Chandra, A.K.; Hajra, A.; Sinha, S. Photophysics of Soret-excited free base tetraphenylporphyrin and its zinc analog in solution. Spectrochim. Acta Part A 2013, 116, 466–472. [Google Scholar] [CrossRef]
- Kumble, R.; Palese, S.; Lin, V.S.Y.; Therien, M.J.; Hochstrasser, R.M. Ultrafast Dynamics of Highly Conjugated Porphyrin Arrays. J. Am. Chem. Soc. 1998, 120, 11489–11498. [Google Scholar] [CrossRef]
- Li, X.; Gong, C.; Gurzadyan, G.G.; Gelin, M.F.; Liu, J.; Sun, L. Ultrafast Relaxation Dynamics in Zinc Tetraphenylporphyrin Surface-Mounted Metal Organic Framework. J. Phys. Chem. C 2018, 122, 50–61. [Google Scholar] [CrossRef]
- Kano, H.; Kobayashi, T. Time-resolved fluorescence and absorption spectroscopies of porphyrin J-aggregates. J. Chem. Phys 2002, 116, 184. [Google Scholar] [CrossRef]
- Akimoto, S.; Yamazaki, T.; Yamazaki, I.; Osuka, A. Excitation relaxation of zinc and free-base porphyrin probed by femtosecond fluorescence spectroscopy. Chem. Phys. Lett. 1999, 309, 177–182. [Google Scholar] [CrossRef]
- Rajasree, S.S.; Li, X.; Deria, P. Physical properties of porphyrin-based crystalline metal-organic frameworks. Commun. Chem. 2021, 4, 47. [Google Scholar] [CrossRef]
- Mataga, N.; Shibata, Y.; Chosrowjan, H.; Yoshida, N.; Osuka, A. Internal Conversion and Vibronic Relaxation from Higher Excited Electronic State of Porphyrins: Femtosecond Fluorescence Dynamics Studies. J. Phys. Chem. B 2000, 104, 4001–4004. [Google Scholar] [CrossRef]
- Sun, K.; Huang, Y.; Wang, Q.; Zhao, W.; Zheng, X.; Jiang, J.; Jiang, H.-L. Manipulating the Spin State of Co Sites in Metal–Organic Frameworks for Boosting CO2 Photoreduction. J. Am. Chem. Soc. 2024, 146, 3241–3249. [Google Scholar] [CrossRef]
- Fang, Z.-B.; Liu, T.-T.; Liu, J.; Jin, S.; Wu, X.-P.; Gong, X.-Q.; Wang, K.; Yin, Q.; Liu, T.-F.; Cao, R.; et al. Boosting Interfacial Charge-Transfer Kinetics for Efficient Overall CO2 Photoreduction via Rational Design of Coordination Spheres on Metal–Organic Frameworks. J. Am. Chem. Soc. 2020, 142, 12515–12523. [Google Scholar] [CrossRef]
- Yuan, Y.-J.; Chen, D.; Zhong, J.; Yang, L.-X.; Wang, J.-J.; Yu, Z.-T.; Zou, Z.-G. Construction of a Noble-Metal-Free Photocatalytic H2 Evolution System Using MoS2/Reduced Graphene Oxide Catalyst and Zinc Porphyrin Photosensitizer. J. Phys. Chem. C 2017, 121, 24452–24462. [Google Scholar] [CrossRef]
- Shu, Y.; Liu, X.; Zhang, M.; Liu, B.; Wang, Z. Deactivation of porphyrin metal-organic framework in advanced oxidation process: Photobleaching and underlying mechanism. Appl. Catal. B Environ. Energy 2024, 346, 123746. [Google Scholar] [CrossRef]
- Hu, H.; Wang, Z.; Cao, L.; Zeng, L.; Zhang, C.; Lin, W.; Wang, C. Metal-organic frameworks embedded in a liposome facilitate overall photocatalytic water splitting. Nat. Chem. 2021, 13, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-M.; Gu, Y.; Zhang, X.-Y.; Dao, X.-Y.; Wang, S.-Q.; Ma, J.; Zhao, J.; Sun, W.-Y. Crystallographic facet heterojunction of MIL-125-NH2(Ti) for carbon dioxide photoreduction. Appl. Catal. B Environ. 2021, 298, 120524. [Google Scholar] [CrossRef]
- Santaclara, J.G.; Nasalevich, M.A.; Castellanos, S.; Evers, W.H.; Spoor, F.C.M.; Rock, K.; Siebbeles, L.D.A.; Kapteijn, F.; Grozema, F.; Houtepen, A.; et al. Organic Linker Defines the Excited-State Decay of Photocatalytic MIL-125(Ti)-Type Materials. ChemSusChem 2016, 9, 388–395. [Google Scholar] [CrossRef]
- Zheng, C.; Qiu, X.; Han, J.; Wu, Y.; Liu, S. Zero-Dimensional-g-CNQD-Coordinated Two-Dimensional Porphyrin MOF Hybrids for Boosting Photocatalytic CO2 Reduction. ACS Appl. Mater. Interfaces 2019, 11, 42243–42249. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiri, A.M.; Garcia, H. 2D Metal-Organic Frameworks as Multifunctional Materials in Heterogeneous Catalysis and Electro/Photocatalysis. Adv. Mater. 2019, 31, e1900617. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, M.; Han, S.; Lai, Z.; Yang, J.; Tan, C.; Ma, Q.; Lu, Q.; Chen, J.; Zhang, X.; et al. Growth of Au Nanoparticles on 2D Metalloporphyrinic Metal-Organic Framework Nanosheets Used as Biomimetic Catalysts for Cascade Reactions. Adv. Mater. 2017, 29, 1700102. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Liu, J.; Leng, J.; Yin, Z.; Yin, Y.; Zhang, F.; Sun, C.; Jin, S. Long-Lived Internal Charge-Separated State in Two-Dimensional Metal–Organic Frameworks Improving Photocatalytic Performance. ACS Energy Lett. 2022, 7, 2323–2330. [Google Scholar] [CrossRef]
- Bai, Y.; Dou, Y.; Xie, L.-H.; Rutledge, W.; Li, J.-R.; Zhou, H.-C. Zr-based metal–organic frameworks: Design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. [Google Scholar] [CrossRef]
- Li, D.D.; Kassymova, M.; Cai, X.C.; Zang, S.Q.; Jiang, H.L. Photocatalytic CO2 reduction over metal-organic framework-based materials. Coord. Chem. Rev. 2020, 412, 213262. [Google Scholar] [CrossRef]
- Shi, X.; Lian, X.; Yang, D.; Hu, X.; Zhang, J.; Bu, X.-H. Facet-engineering of NH2-UiO-66 with enhanced photocatalytic hydrogen production performance. Dalton Trans. 2021, 50, 17953–17959. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.D.; Li, R.; Jiang, H.L. Metal-Organic Framework-Based Photocatalysis for Solar Fuel Production. Small Methods 2023, 7, e2201258. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Kong, L.H.; Wang, P.; Yao, S.; Lu, T.B.; Zhang, Z.M. Switching Excited State Distribution of Metal-Organic Framework for Dramatically Boosting Photocatalysis. Angew. Chem. Int. Ed. 2022, 134, e202206193. [Google Scholar] [CrossRef]
- Chen, K.-K.; Qin, C.-C.; Ding, M.-J.; Guo, S.; Lu, T.-B.; Zhang, Z.-M. Broadband and strong visible-light-absorbing cuprous sensitizers for boosting photosynthesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2213479119. [Google Scholar] [CrossRef]
- Wang, S.; Guan, B.Y.; Lu, Y.; Lou, X.W.D. Formation of Hierarchical In2S3–CdIn2S4 Heterostructured Nanotubes for Efficient and Stable Visible Light CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 17305–17308. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; He, C.-T.; Huang, R.; Mao, J.; Wang, D.; Li, Y. Photoinduction of Cu Single Atoms Decorated on UiO-66-NH2 for Enhanced Photocatalytic Reduction of CO2 to Liquid Fuels. J. Am. Chem. Soc. 2020, 142, 19339–19345. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chong, K.C.; Qi, G.; Xiao, Y.; Chen, G.; Li, B.; Tang, Y.; Zhang, X.; Yao, Y.; Lin, Z.; et al. Unraveling the Transformation from Type-II to Z-Scheme in Perovskite-Based Heterostructures for Enhanced Photocatalytic CO2 Reduction. J. Am. Chem. Soc. 2024, 146, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, Y.; Fujisawa, Y.; Satake, A. Photocatalytic CO2 Reduction Mediated by Electron Transfer via the Excited Triplet State of Zn(II) Porphyrin. J. Am. Chem. Soc. 2020, 142, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Dong, L.-Z.; Liu, J.; Li, S.-L.; Lan, Y.-Q. From molecular metal complex to metal-organic framework: The CO2 reduction photocatalysts with clear and tunable structure. Coord. Chem. Rev. 2019, 390, 86–126. [Google Scholar] [CrossRef]
- Bhosale, S.S.; Kharade, A.K.; Jokar, E.; Fathi, A.; Chang, S.M.; Diau, E.W. Mechanism of Photocatalytic CO2 Reduction by Bismuth-Based Perovskite Nanocrystals at the Gas-Solid Interface. J. Am. Chem. Soc. 2019, 141, 20434–20442. [Google Scholar] [CrossRef]
- Liu, M.; Liu, M.; Wang, X.; Kozlov, S.M.; Cao, Z.; De Luna, P.; Li, H.; Qiu, X.; Liu, K.; Hu, J.; et al. Quantum-Dot-Derived Catalysts for CO2 Reduction Reaction. Joule 2019, 3, 1703–1718. [Google Scholar] [CrossRef]
- Wang, S.; Guan, B.Y.; Lou, X.W.D. Construction of ZnIn2S4–In2O3 Hierarchical Tubular Heterostructures for Efficient CO2 Photoreduction. J. Am. Chem. Soc. 2018, 140, 5037–5040. [Google Scholar] [CrossRef]
- Wu, D.; Zhao, X.; Huang, Y.; Lai, J.; Li, H.; Yang, J.; Tian, C.; He, P.; Huang, Q.; Tang, X. Lead-Free Perovskite Cs2AgBiX6 Nanocrystals with a Band Gap Funnel Structure for Photocatalytic CO2 Reduction under Visible Light. Chem. Mater. 2021, 33, 4971–4976. [Google Scholar] [CrossRef]
- Sheng, J.; He, Y.; Huang, M.; Yuan, C.; Wang, S.; Dong, F. Frustrated Lewis Pair Sites Boosting CO2 Photoreduction on Cs2CuBr4 Perovskite Quantum Dots. ACS Catal. 2022, 12, 2915–2926. [Google Scholar] [CrossRef]
- Song, W.T.; Qi, G.B.; Liu, B. Halide perovskite quantum dots for photocatalytic CO2 reduction. J. Mater. Chem. A 2023, 11, 12482–12498. [Google Scholar] [CrossRef]
- Lee, S.; Jang, G.Y.; Kim, J.K.; Park, J.H. Solar-harvesting lead halide perovskite for artificial photosynthesis. J. Energy Chem. 2021, 62, 11–26. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, Y.; Shu, M.; Li, L.; Dong, Z.; Xu, J. Artificial Photosynthesis over Metal Halide Perovskites: Achievements, Challenges, and Prospects. J. Phys. Chem. Lett. 2021, 12, 5864–5870. [Google Scholar] [CrossRef]
- Hong, J.; Zhang, W.; Ren, J.; Xu, R. Photocatalytic reduction of CO2: A brief review on product analysis and systematic methods. Anal. Methods 2013, 5, 1086–1097. [Google Scholar] [CrossRef]
- Mitzi, D.B. Introduction: Perovskites. Chem. Rev. 2019, 119, 3033–3035. [Google Scholar] [CrossRef]
- DuBose, J.T.; Kamat, P.V. Efficacy of Perovskite Photocatalysis: Challenges to Overcome. ACS Energy Lett. 2022, 7, 1994–2011. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.; Du, Z.; Li, Z. Recent advances in metal halide perovskite photocatalysts: Properties, synthesis and applications. J. Energy Chem. 2021, 54, 770–785. [Google Scholar] [CrossRef]
- Huang, H.; Pradhan, B.; Hofkens, J.; Roeffaers, M.B.J.; Steele, J.A. Solar-Driven Metal Halide Perovskite Photocatalysis: Design, Stability, and Performance. ACS Energy Lett. 2020, 5, 1107–1123. [Google Scholar] [CrossRef]
- Schanze, K.S.; Kamat, P.V.; Yang, P.; Bisquert, J. Progress in Perovskite Photocatalysis. ACS Energy Lett. 2020, 5, 2602–2604. [Google Scholar] [CrossRef]
- Chen, S.; Yin, H.; Liu, P.; Wang, Y.; Zhao, H. Stabilization and Performance Enhancement Strategies for Halide Perovskite Photocatalysts. Adv. Mater. 2023, 35, 2203836. [Google Scholar] [CrossRef]
- Mai, H.; Chen, D.; Tachibana, Y.; Suzuki, H.; Abe, R.; Caruso, R.A. Developing sustainable, high-performance perovskites in photocatalysis: Design strategies and applications. Chem. Soc. Rev. 2021, 50, 13692–13729. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, H.-Y.; Li, J.-Y.; Liao, J.-F.; Zhang, H.-H.; Wang, X.-D.; Kuang, D.-B. Z-Scheme 2D/2D Heterojunction of CsPbBr3/Bi2WO6 for Improved Photocatalytic CO2 Reduction. Adv. Funct. Mater. 2020, 30, 2004293. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, J.; Du, Y.; Zhou, C.; Zhang, M.; Wang, Z.; Weng, Y.; Long, J.; Hofkens, J.; Steele, J.A.; et al. Direct Z-Scheme Heterojunction of Semicoherent FAPbBr3/Bi2WO6 Interface for Photoredox Reaction with Large Driving Force. ACS Nano 2020, 14, 16689–16697. [Google Scholar] [CrossRef]
- Wu, L.-Y.; Mu, Y.-F.; Guo, X.-X.; Zhang, W.; Zhang, Z.-M.; Zhang, M.; Lu, T.-B. Encapsulating Perovskite Quantum Dots in Iron-Based Metal–Organic Frameworks (MOFs) for Efficient Photocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 9491–9495. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Li, Y.; Wang, J.; Zhang, G. Efficient photocatalytic CO2 conversion over 2D/2D Ni-doped CsPbBr3/Bi3O4Br Z-scheme heterojunction: Critical role of Ni doping, boosted charge separation and mechanism study. Appl. Catal. B Environ. 2022, 319, 121895. [Google Scholar] [CrossRef]
- Jiang, Y.; Liao, J.-F.; Chen, H.-Y.; Zhang, H.-H.; Li, J.-Y.; Wang, X.-D.; Kuang, D.-B. All-Solid-State Z-Scheme α-Fe2O3/Amine-RGO/CsPbBr3 Hybrids for Visible-Light-Driven Photocatalytic CO2 Reduction. Chem 2020, 6, 766–780. [Google Scholar] [CrossRef]
- Yuan, C.; He, Y.; Chen, R.; Sun, Y.; Li, J.; Cui, W.; Chen, P.; Sheng, J.; Dong, F. Perovskite Nanocrystals-Based Heterostructures: Synthesis Strategies, Interfacial Effects, and Photocatalytic Applications. Sol. RRL 2021, 5, 2000419. [Google Scholar] [CrossRef]
- Wang, X.; Li, K.; He, J.; Yang, J.; Dong, F.; Mai, W.; Zhu, M. Defect in reduced graphene oxide tailored selectivity of photocatalytic CO2 reduction on Cs4PbBr6 pervoskite hole-in-microdisk structure. Nano Energy 2020, 78, 105388. [Google Scholar] [CrossRef]
- Li, N.; Zhai, X.-P.; Yan, W.-K.; Zhang, Y.-J.; Zhang, Z.-T.; Xiao, M.-J.; Zhang, X.-D.; Wang, Q.; Zhang, H.-L. Boosting Cascade Electron Transfer for Highly Efficient CO2 Photoreduction. Sol. RRL 2021, 5, 2100558. [Google Scholar] [CrossRef]
- Wang, X.-D.; Huang, Y.-H.; Liao, J.-F.; Jiang, Y.; Zhou, L.; Zhang, X.-Y.; Chen, H.-Y.; Kuang, D.-B. In Situ Construction of a Cs2SnI6 Perovskite Nanocrystal/SnS2 Nanosheet Heterojunction with Boosted Interfacial Charge Transfer. J. Am. Chem. Soc. 2019, 141, 13434–13441. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, M.; Chi, Z.; Li, W.; Yu, H.; Yang, N.; Yu, H. Internal electric field engineering step-scheme–based heterojunction using lead-free Cs3Bi2Br9 perovskite–modified In4SnS8 for selective photocatalytic CO2 reduction to CO. Appl. Catal. B Environ. 2022, 313, 121426. [Google Scholar] [CrossRef]
- Li, N.; Zhai, X.P.; Ma, B.; Zhang, H.J.; Xiao, M.J.; Wang, Q.; Zhang, H.L. Highly selective photocatalytic CO2 reduction via a lead-free perovskite/MOF catalyst. J. Mater. Chem. A 2023, 11, 4020–4029. [Google Scholar] [CrossRef]

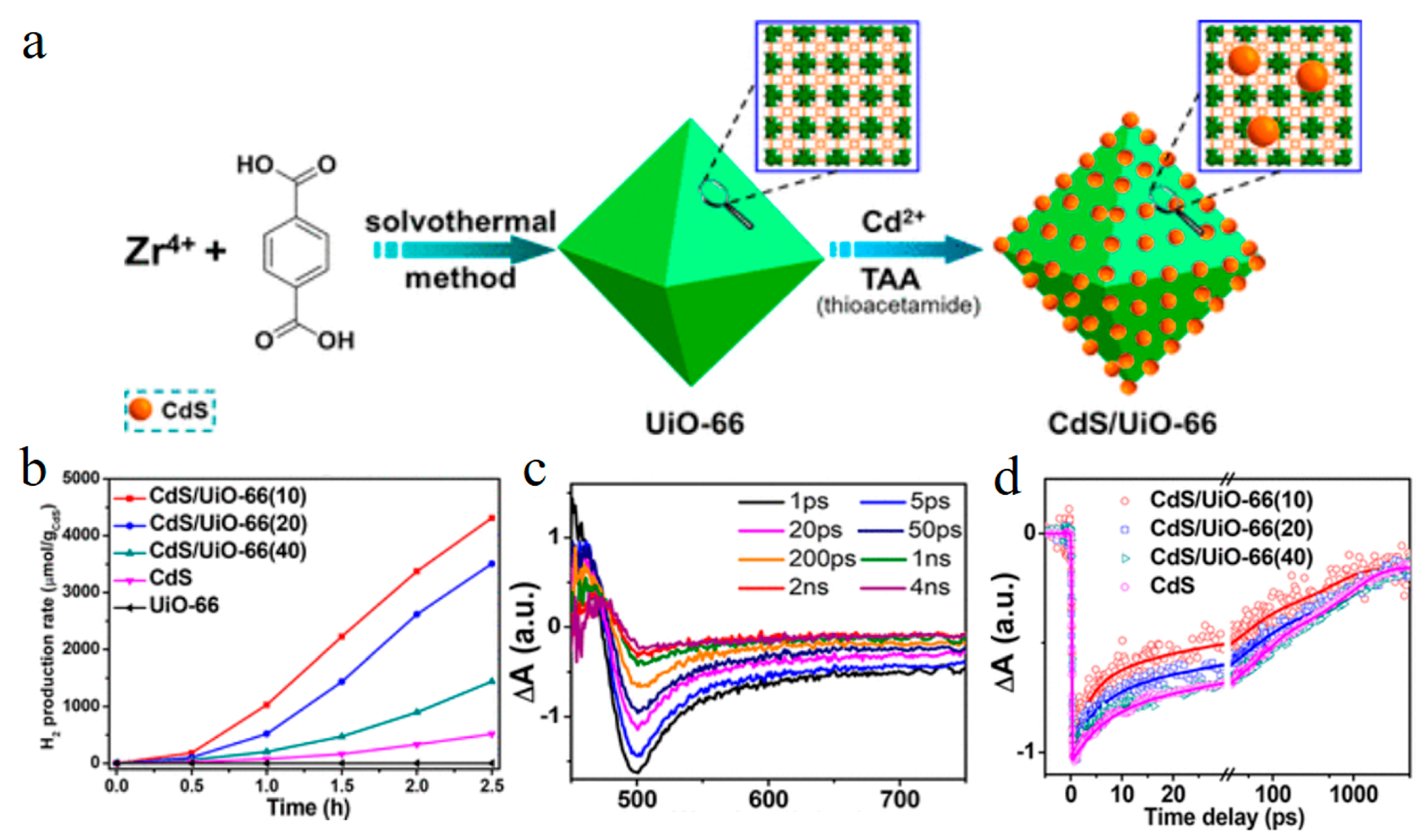

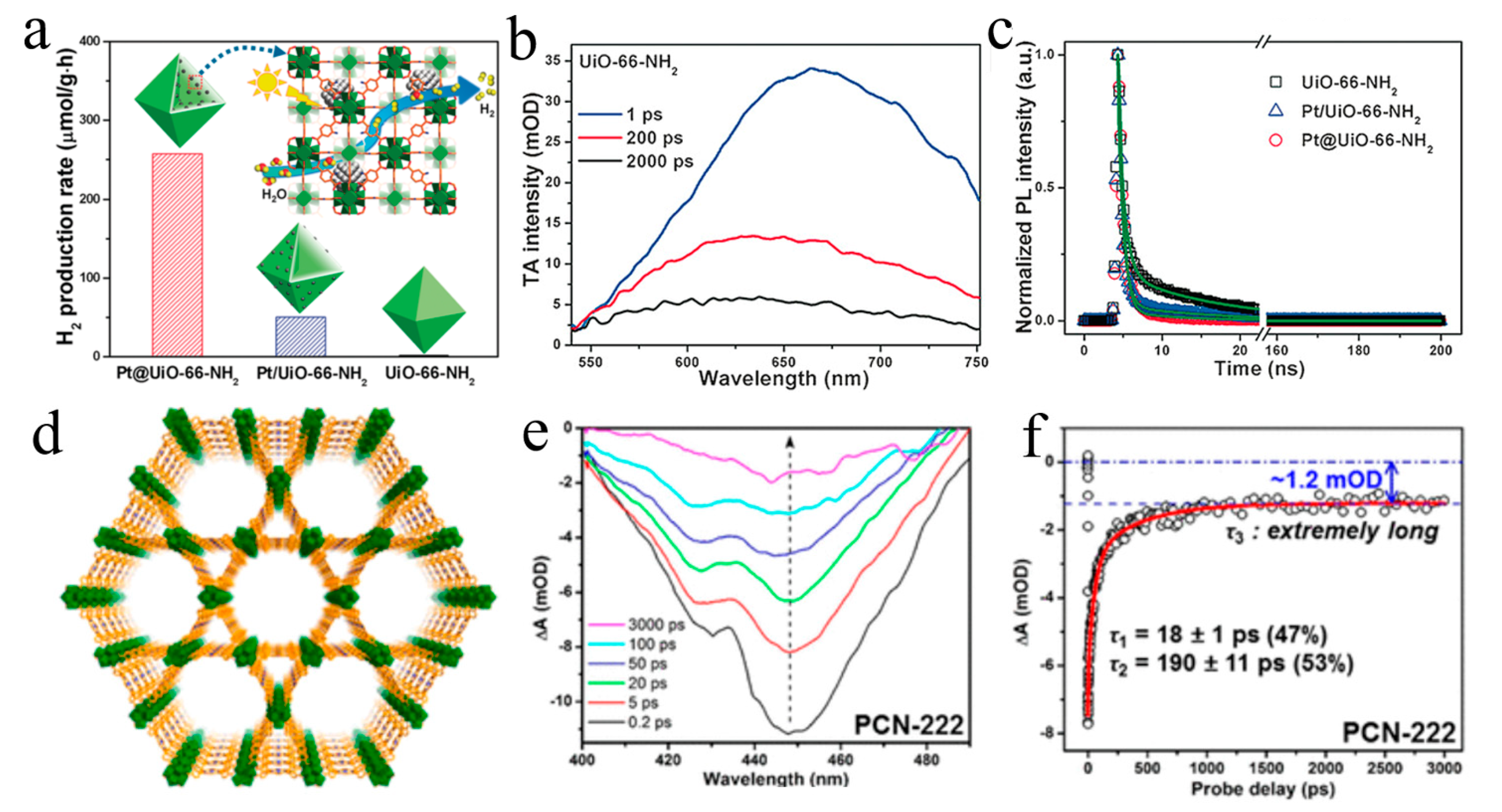
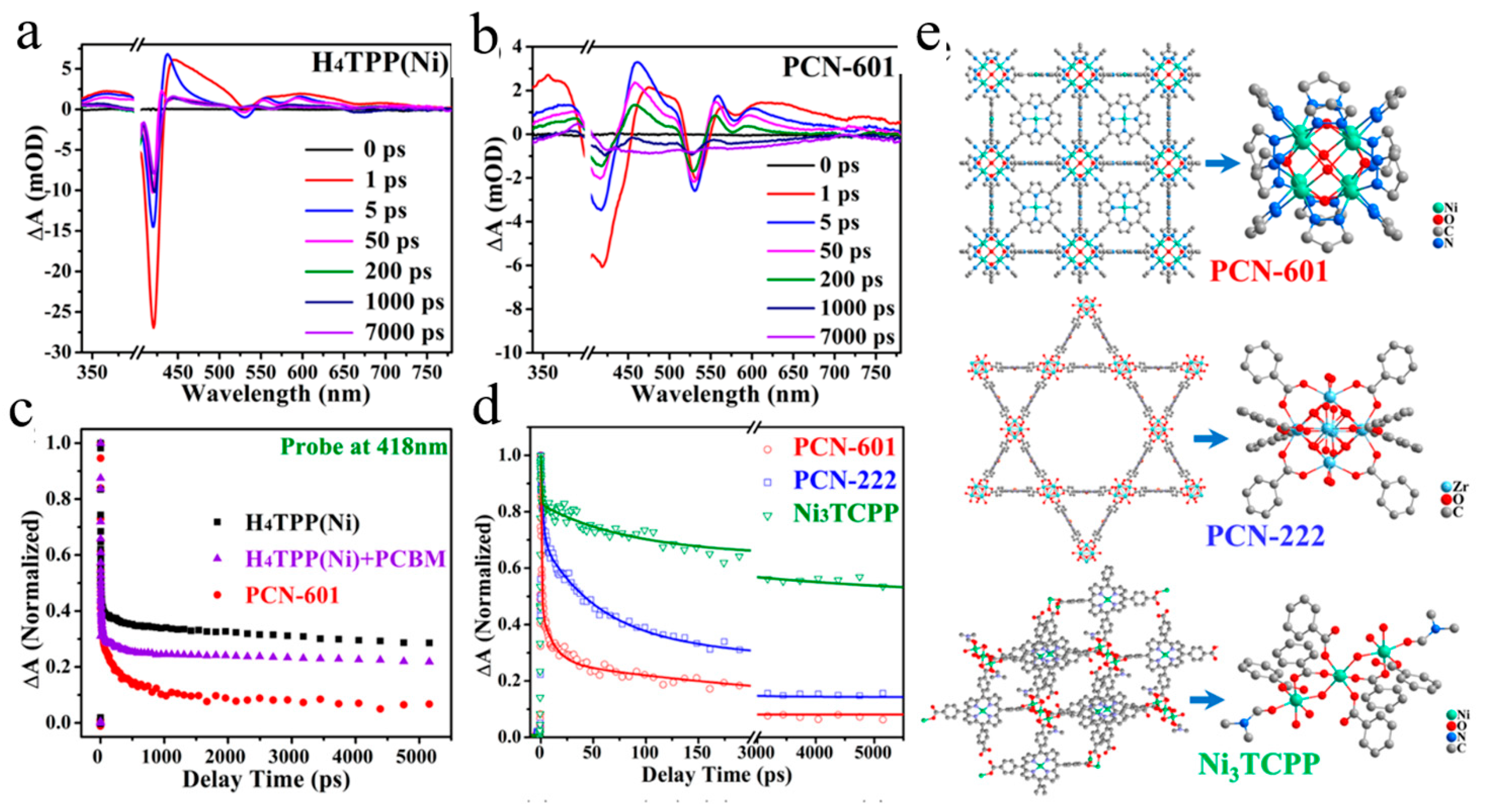
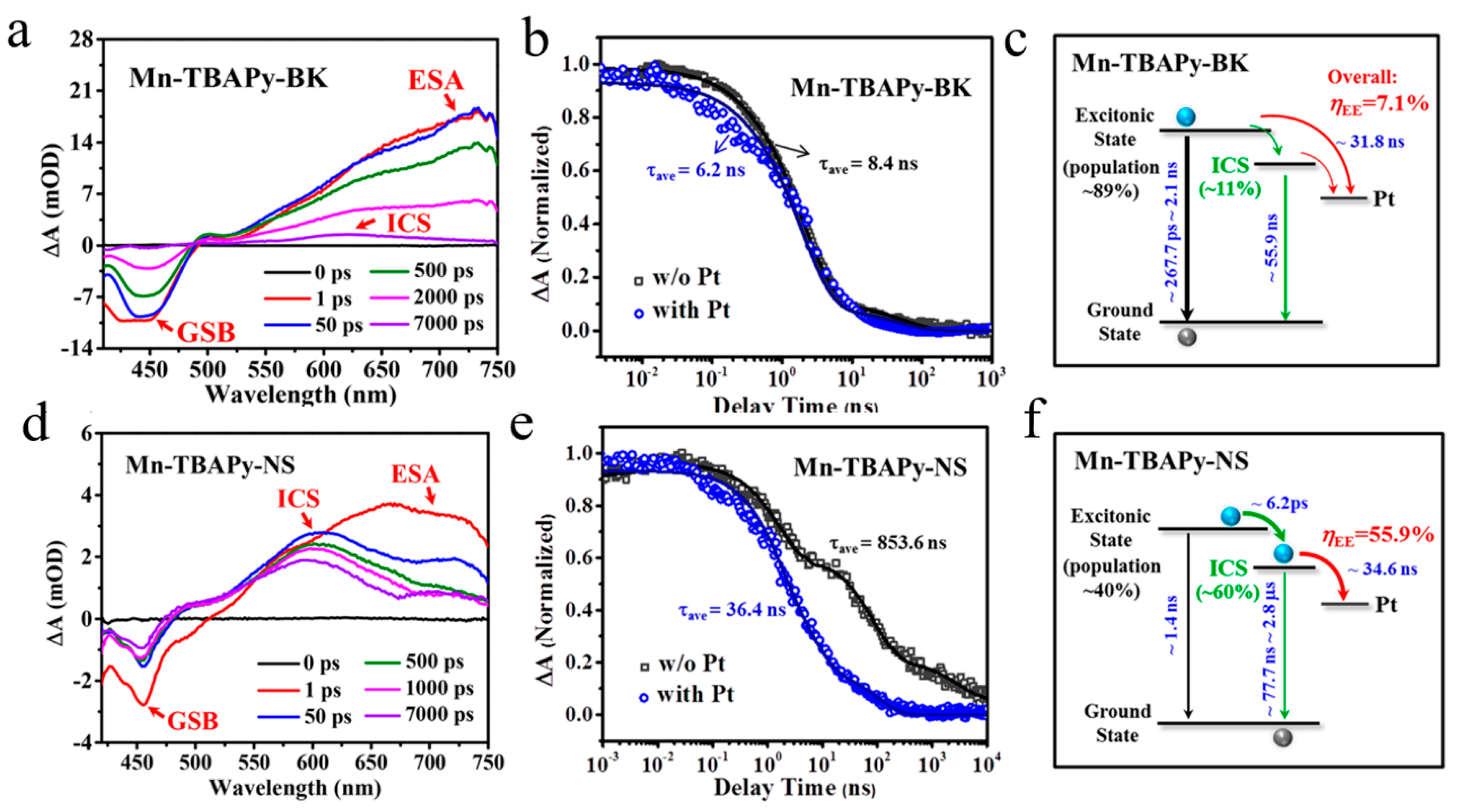


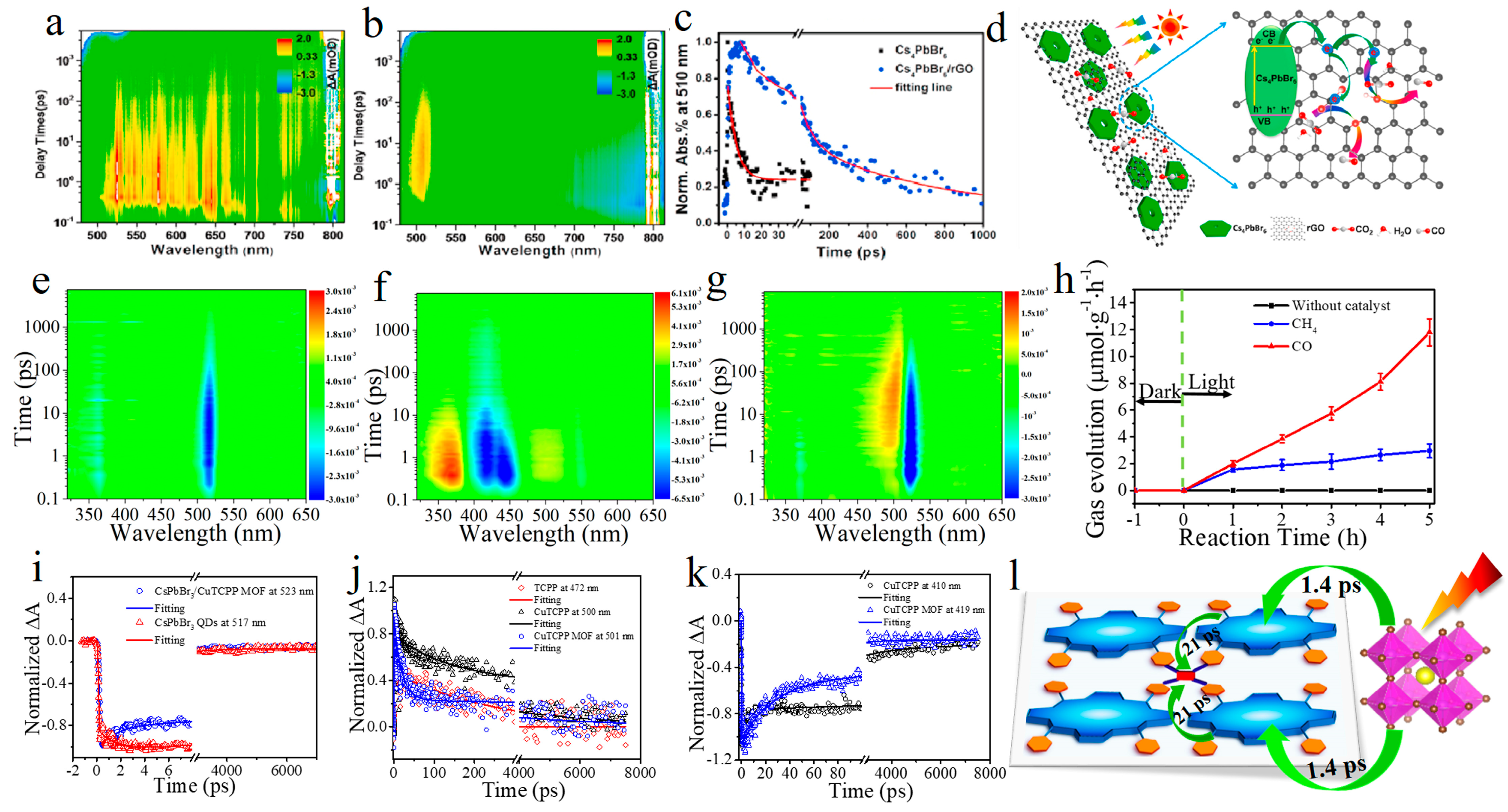
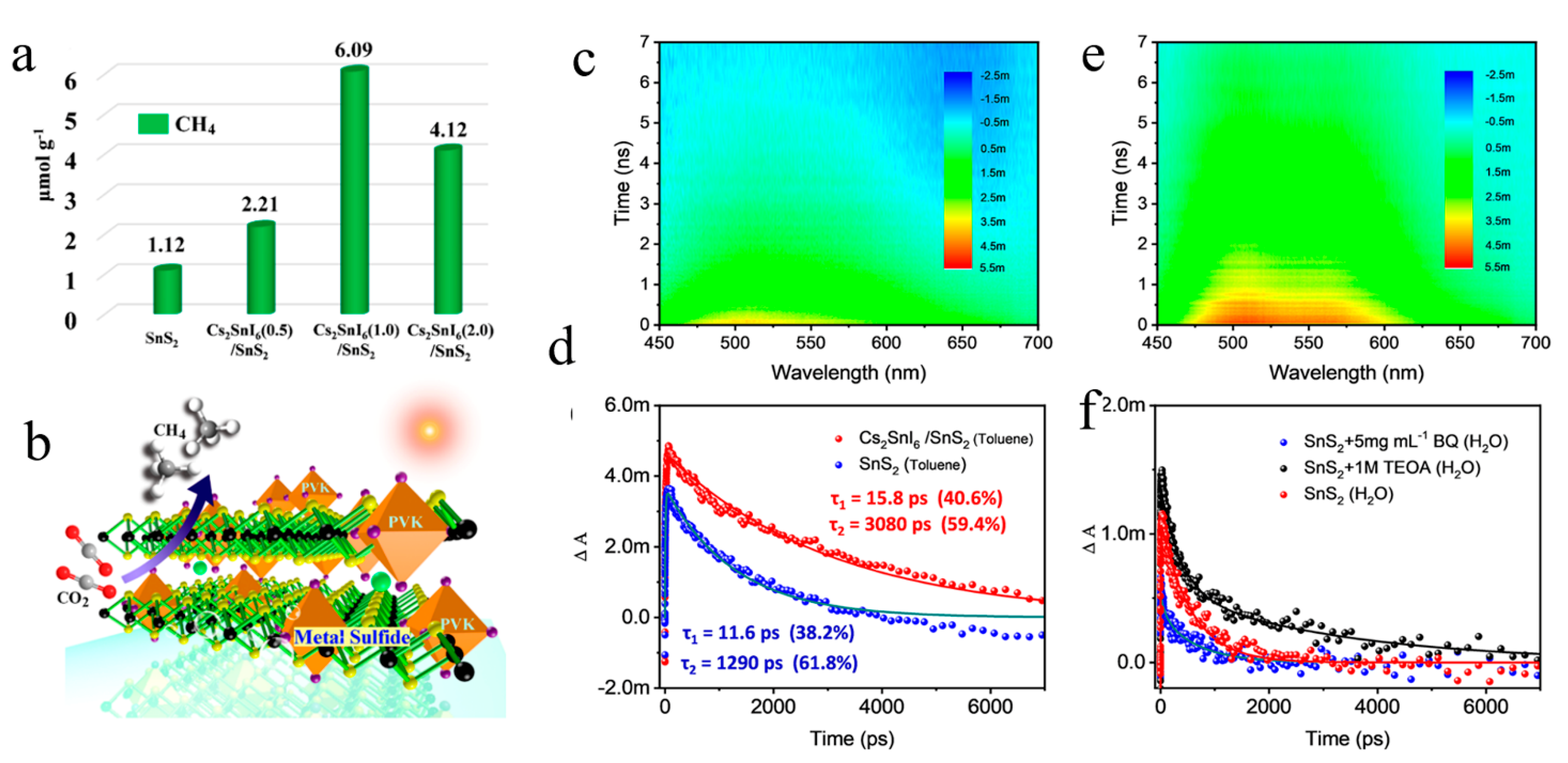
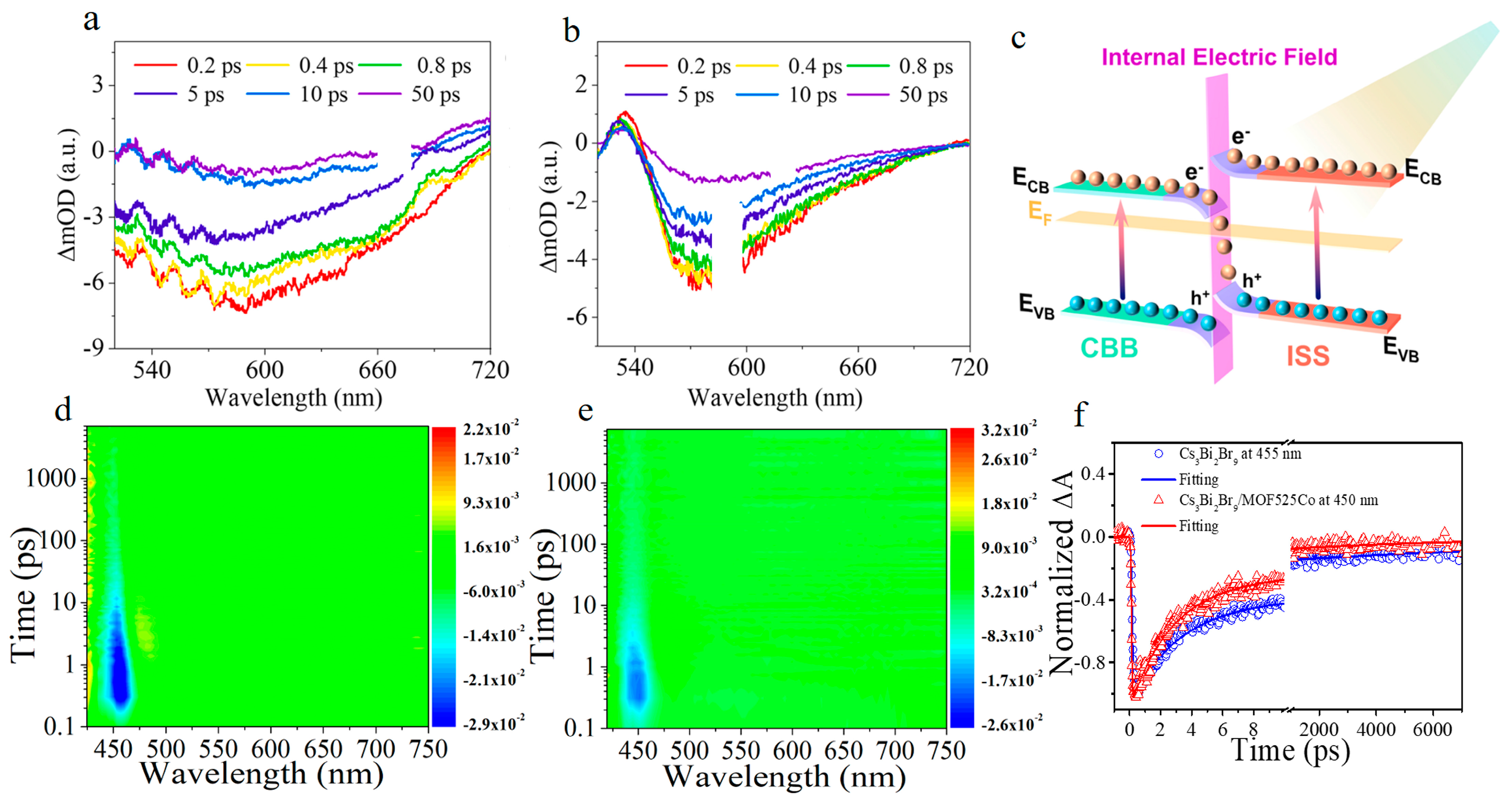
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Ma, Y.; Sun, W. Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy. Molecules 2024, 29, 3995. https://doi.org/10.3390/molecules29173995
Li N, Ma Y, Sun W. Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy. Molecules. 2024; 29(17):3995. https://doi.org/10.3390/molecules29173995
Chicago/Turabian StyleLi, Na, Yanlong Ma, and Wanjun Sun. 2024. "Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy" Molecules 29, no. 17: 3995. https://doi.org/10.3390/molecules29173995
APA StyleLi, N., Ma, Y., & Sun, W. (2024). Exploring the Dynamics of Charge Transfer in Photocatalysis: Applications of Femtosecond Transient Absorption Spectroscopy. Molecules, 29(17), 3995. https://doi.org/10.3390/molecules29173995