
Lupeol-3-carbamate Derivatives: Synthesis and Biological Evaluation as Potential Antitumor Agents

Abstract

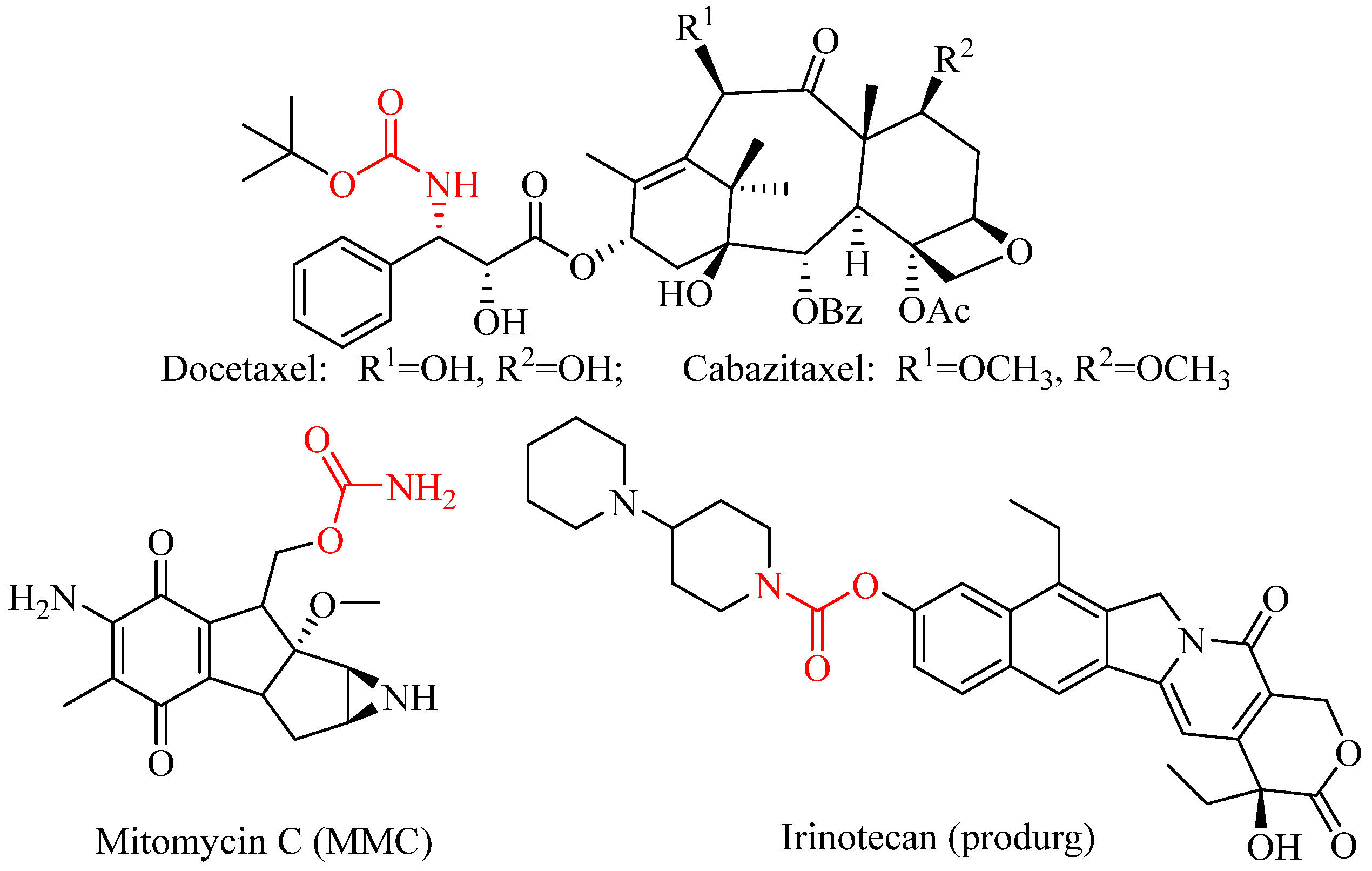
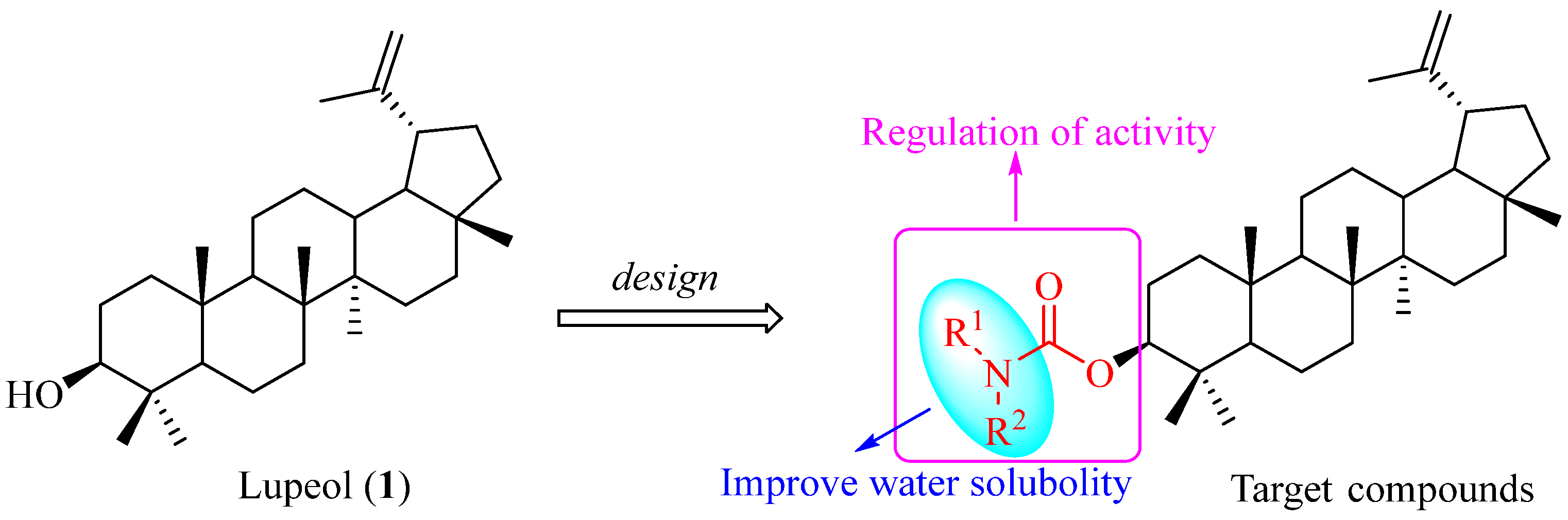
1. Introduction
2. Results and Discussion
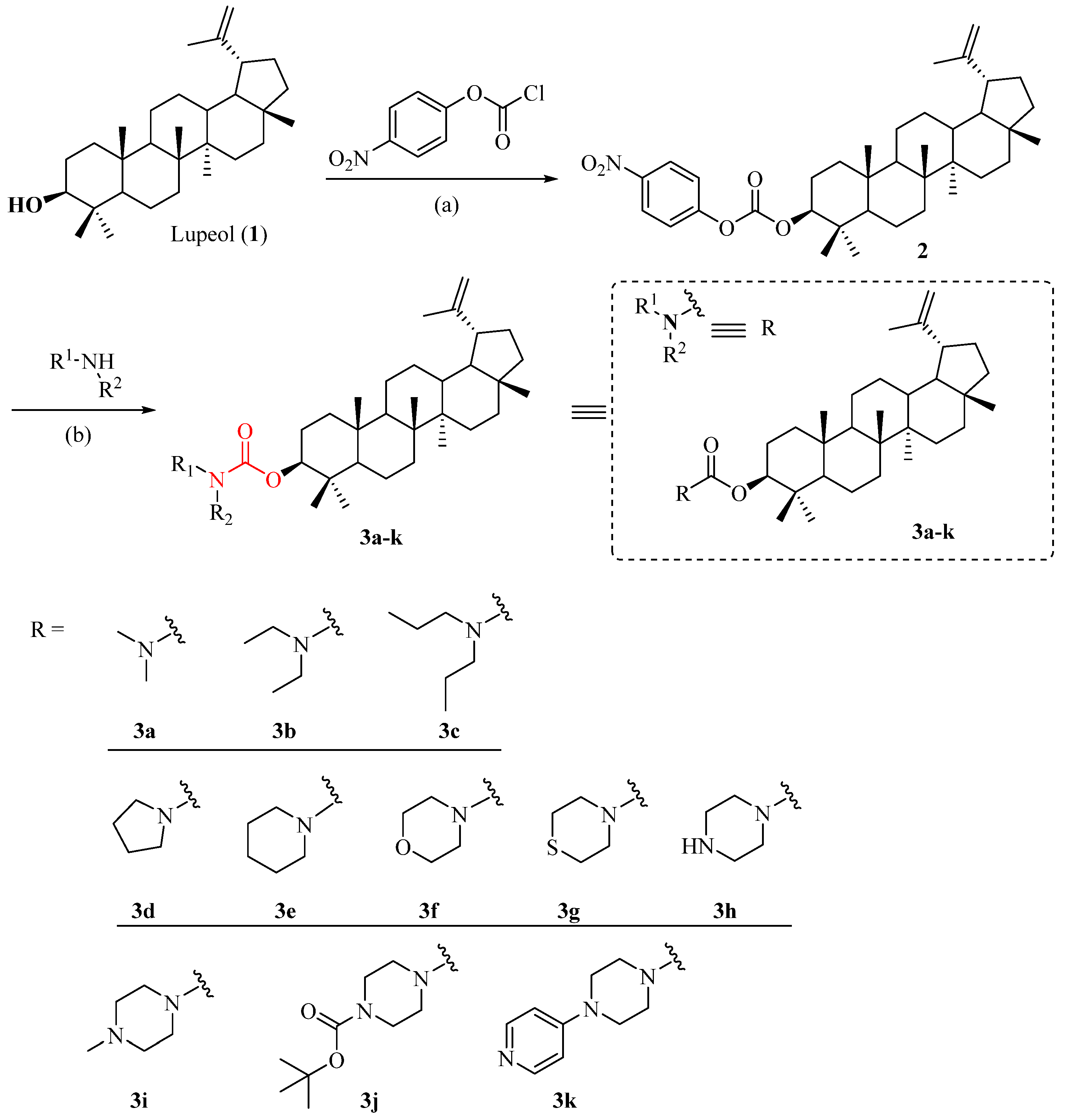
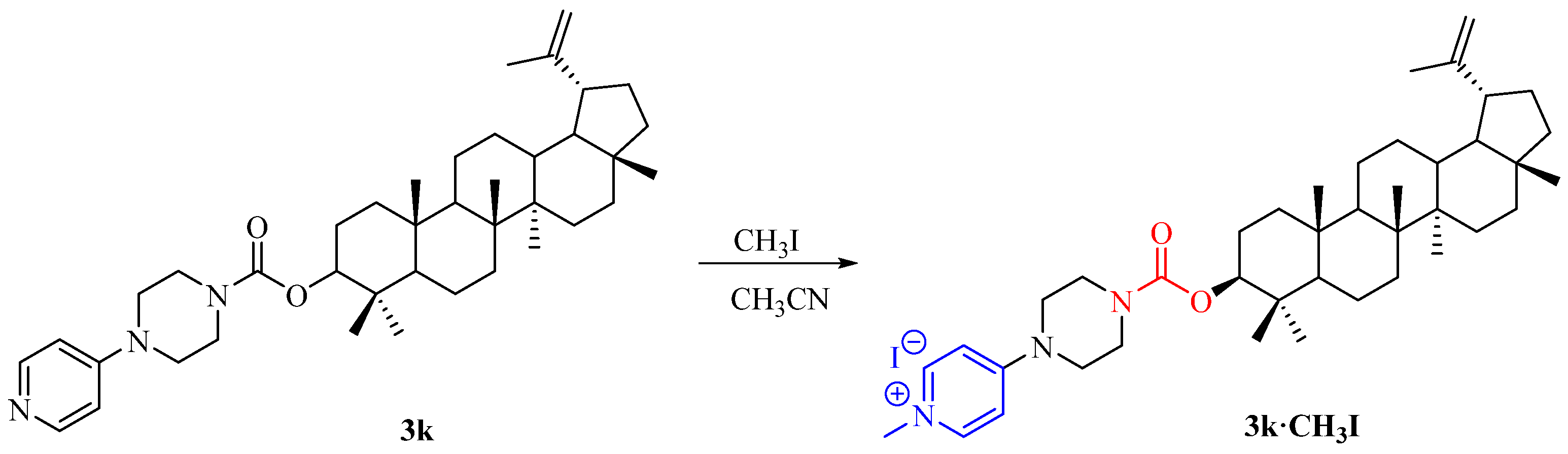
2.1. Synthesis of Lupeol Derivatives
2.2. Biological Evaluation of Lupeol Derivatives
2.2.1. Lupeol Derivatives Inhibited the Proliferation of Various Human Cancer Cell Lines
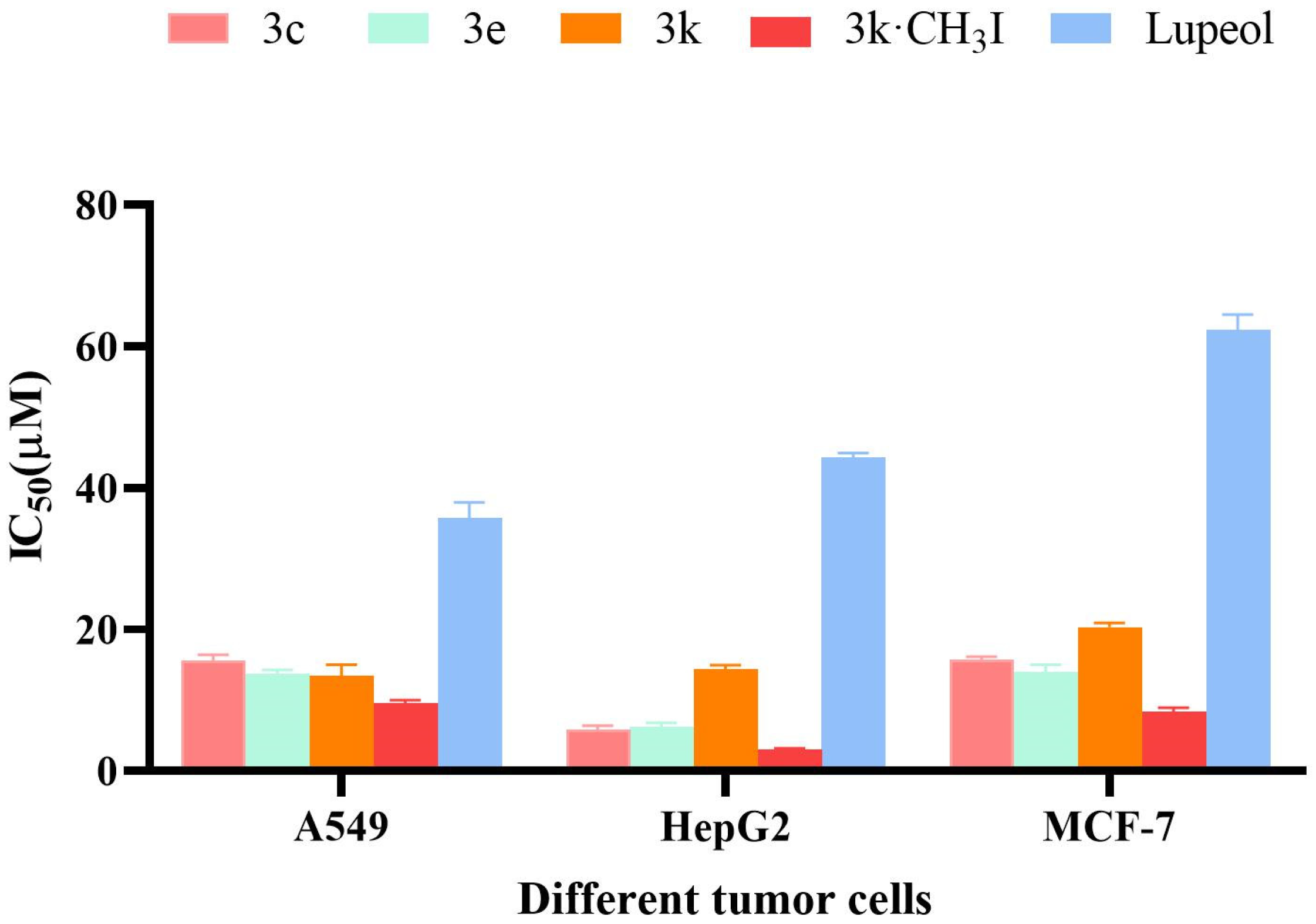
2.2.2. Anti-Proliferative Activity of Selected Compounds in Tumor Cells
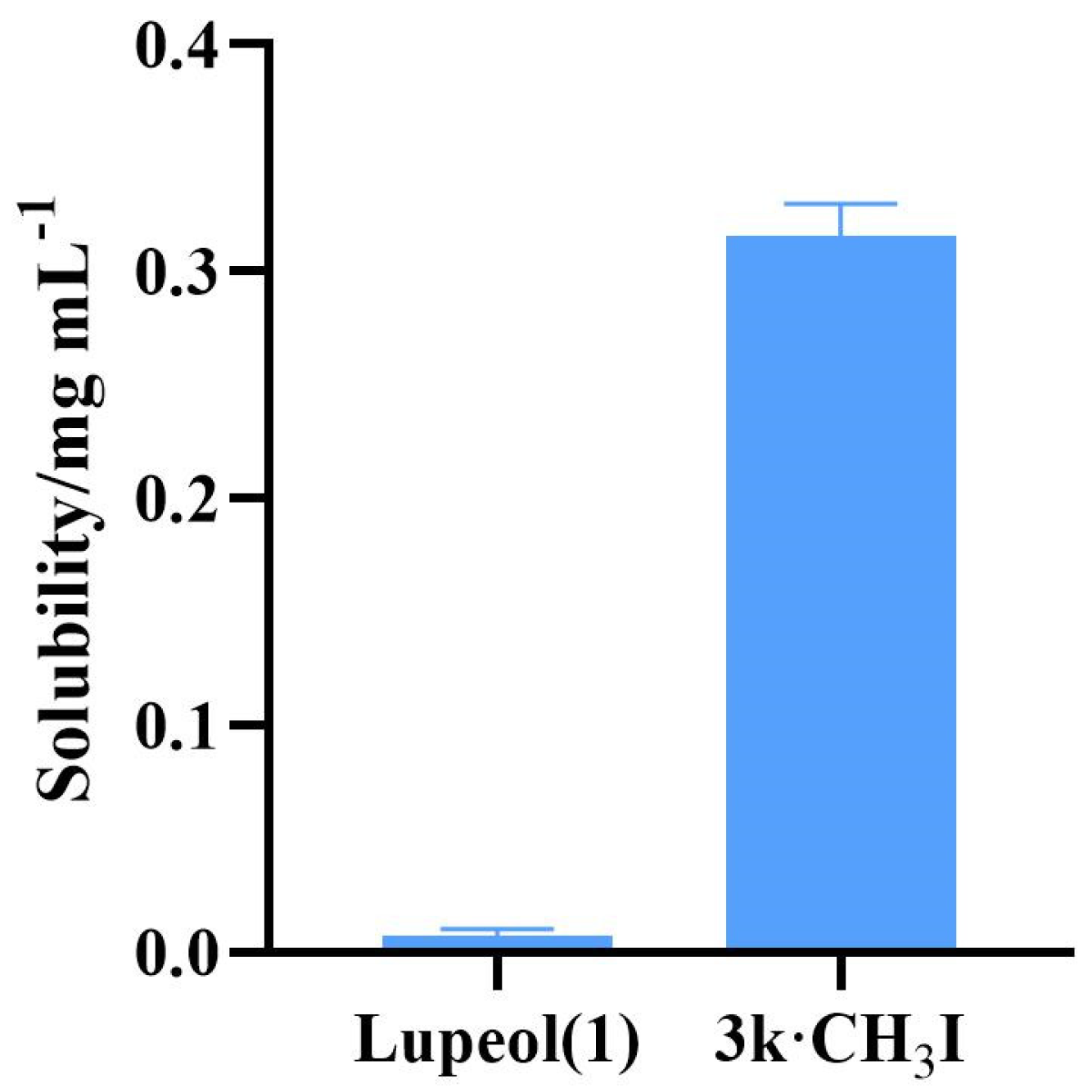
2.3. Water Solubility Testing of Compound 3k·CH3I
2.4. Effect of Compound 3k·CH3I on the Apoptosis Rate of HepG2 Cells
2.5. Effect of Compound 3k·CH3I on Reactive Oxygen Species (ROS) in HepG2 Cells
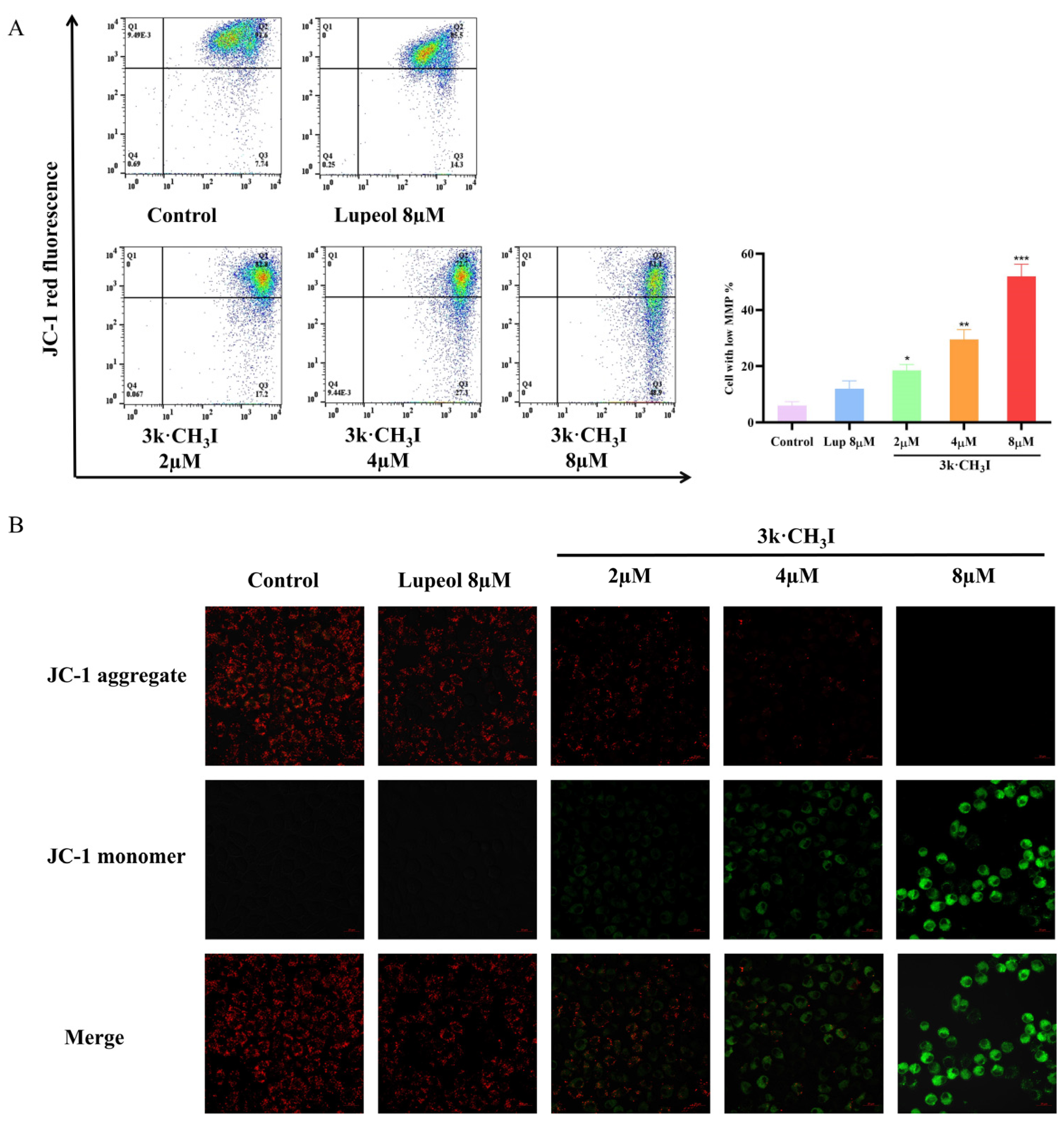
2.6. Effect of Compound 3k·CH3I on the Mitochondrial Membrane Potential of HepG2 Cells
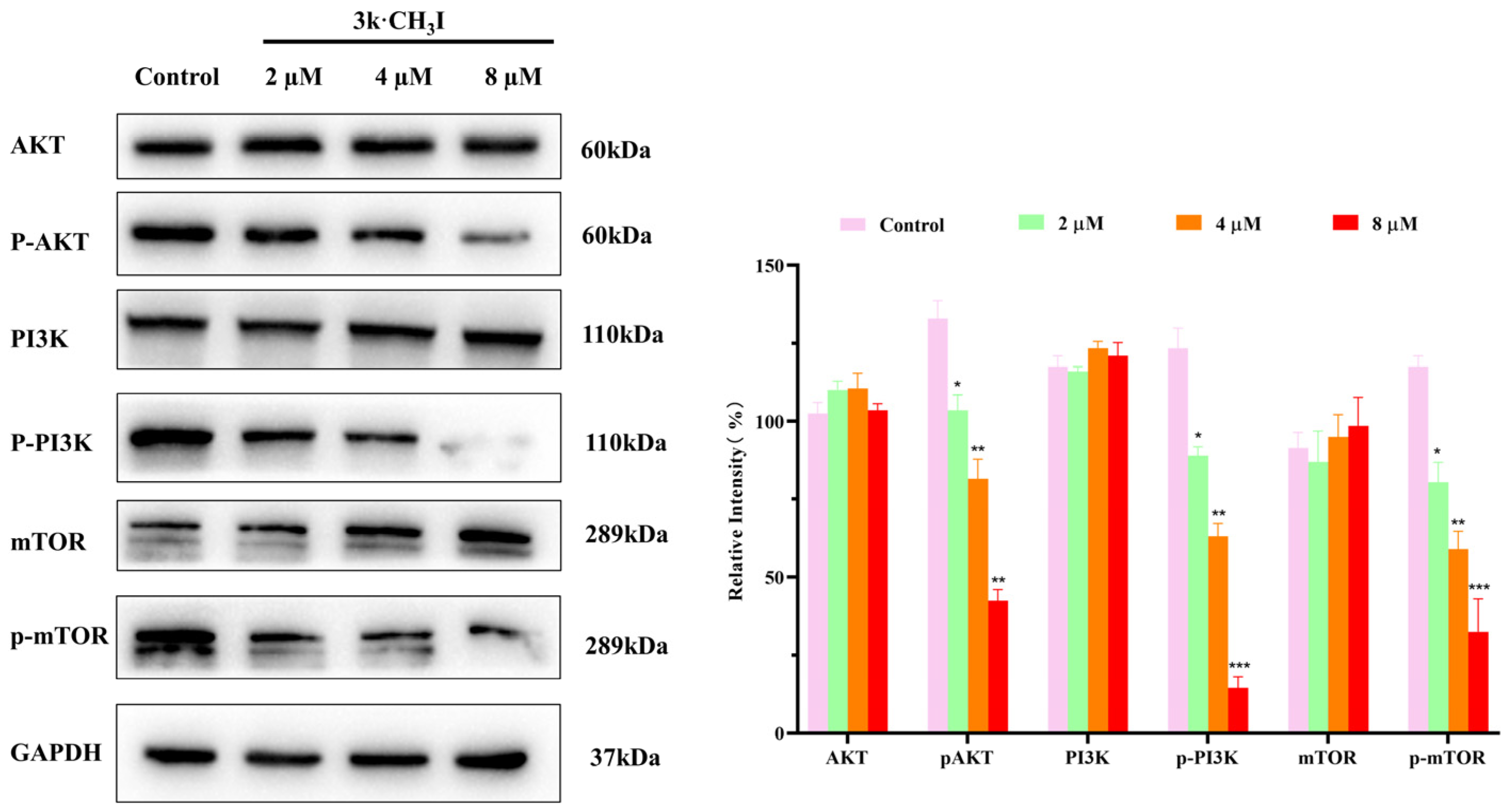
2.7. Effect of Compound 3k·CH3I on the Expression of Related Proteins in HepG2 Cells
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis of Lupeol-3-(4-nitrobenzoate) (2)
3.1.2. General Procedure for Synthesis of Compounds 3a–k
Lupeol-3-(dimethyl) Carbamate (3a)
Lupeol-3-(diethyl) Carbamate (3b)
Lupeol-3-(dipropyl) Carbamate (3c)
Lupeol-3-(pyrrolidine-1-carboxylate) (3d)
Lupeol-3-(piperidine-1-carboxylate) (3e)
Lupeol-3-(morpholine-1-carboxylate) (3f)
Lupeol-3-(thiomorpholine-1-carboxylate) (3g)
Lupeol-3-(piperazine-1-carboxylate) (3h)
Lupeol-3-((4-methylpiperazine)-1-carboxylate) (3i)
Lupeol-3-((4-tert-butyl) piperazine-1)-carbamate (3j)
Lupeol-3-((4-(pyridin-4-yl) piperazine)-1-carboxylate) (3k)
3.1.3. Synthesis of Compound 3k·CH3I
3.2. Biological Evaluation
3.2.1. In Vitro Cytotoxicity
3.2.2. Cell Morphology Detection
3.2.3. Detection of Apoptosis
3.2.4. Detection of Intracellular ROS
3.2.5. Detection of MMP Depolarization
3.2.6. Protein Immunoblot Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goodman, J.E.; Mayfield, D.B.; Becker, R.A.; Hartigan, S.B.; Erraguntla, N.K. Recommendations for further revisions to improve the International Agency for Research on Cancer (IARC) monograph program. Regul. Toxicol. Pharmacol. 2020, 113, 104639. [Google Scholar] [CrossRef] [PubMed]
- Knezevic, C.E.; Clarke, W. Cancer chemotherapy: The case for therapeutic drug monitoring. Ther. Drug Monit. 2020, 42, 6–19. [Google Scholar] [CrossRef]
- Deng, L.; Feng, Z.; Deng, H.; Jiang, Y.; Song, K.; Shi, Y.; Liu, S.; Zhang, J.; Bai, S.; Qin, Z.; et al. Rational design of nanoparticles to overcome poor tumor penetration and hypoxia-induced chemo-therapy resistance: Combination of optimizing size and self-inducing high level of reactive oxygen species. ACS Appl. Mater. 2019, 11, 31743–31754. [Google Scholar] [CrossRef]
- Castañeda, A.M.; Meléndez, C.M.; Uribe, D.; Pedroza-Díaz, J. Synergistic effects of natural compounds and conventional chemotherapeutic agents: Recent insights for the development of cancer treatment strategies. Heliyon 2022, 8, 09519. [Google Scholar] [CrossRef]
- Greco, G.; Catanzaro, E.; Fimognari, C. Natural Products as Inducers of Non-Canonical Cell Death: A Weapon against Cancer. Cancers 2021, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Fabiani, R. Antitumoral Properties of Natural Products. Molecules 2020, 25, 650. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Yin, Y.; Sheng, L.; Zhang, J.; Zhang, L.; Liu, J.; Wen, X.; Liu, Y.; Si, Y.; Cheng, K. Synthesis and in vitro/in vivo anticancer evaluation of pentacyclic triterpenoid derivatives linked with l-phenylalanine or l-proline. Bioorg. Chem. 2022, 126, 105865. [Google Scholar] [CrossRef]
- Hodon, J.; Borkova, L.; Pokorny, J.; Kazakova, A.; Urban, M. Design and synthesis of pentacyclic triterpene conjugates and their use in medicinal research. Eur. J. Med. Chem. 2019, 182, 111653. [Google Scholar] [CrossRef]
- Yu, L.; Xie, X.; Cao, X.; Chen, J.; Chen, G.; Chen, Y.; Li, G.; Qin, J.; Peng, F.; Peng, C. The Anticancer Potential of Maslinic Acid and Its Derivatives: A Review. Drug Des. Dev. Ther. 2021, 15, 3863–3879. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhang, X.; Xie, L.; Deng, M.; Chen, H.; Song, J.; Long, J.; Li, X.; Luo, J. Lupeol and its derivatives as anticancer and anti-inflammatory agents: Molecular mechanisms and therapeutic efficacy. Pharmacol. Res. 2021, 164, 105373. [Google Scholar] [CrossRef] [PubMed]
- Sohag, A.A.M.; Hossain, M.T.; Rahaman, M.A.; Rahman, P.; Hasan, M.S.; Das, R.C.; Khan, M.K.; Sikder, M.H.; Alam, M.; Uddin, M.J.; et al. Molecular pharmacology and therapeutic advances of the pentacyclic triterpene lupeol. Phytomedicine 2022, 99, 154012. [Google Scholar] [CrossRef]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Maurya, S.K.; Shadab, G.G.H.A.; Siddique, H.R. Chemosensitization of Therapy Resistant Tumors: Targeting Multiple Cell Signaling Pathways by Lupeol, A Pentacyclic Triterpene. Curr. Pharm. Des. 2020, 26, 455–465. [Google Scholar] [CrossRef]
- Hsu, M.J.; Peng, S.F.; Chueh, F.S.; Tsai, C.H.; Tsai, F.J.; Huang, C.Y.; Tang, C.H.; Yang, J.S.; Hsu, Y.M.; Huang, W.W.; et al. Lupeol suppresses migration and invasion via p38/MAPK and PI3K/AKT signaling pathways in human osteosarcoma U-2 OS cells. Biosci. Biotechnol. Biochem. 2019, 83, 1729–1739. [Google Scholar] [CrossRef]
- Che, S.; Wu, S.; Yu, P. Lupeol induces autophagy and apoptosis with reduced cancer stem-like properties in retinoblastoma via phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin inhibition. J. Pharm. Pharmacol. 2022, 74, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jin, X.; Zhang, J.; Wang, K.; Jin, X.; Xu, D.; Tian, X.; Liu, L. Antitumor Activity of a Novel Double-Targeted System for Folate Receptor-Mediated Delivery of Mitomycin C. ACS Omega 2020, 5, 26864–26870. [Google Scholar] [CrossRef]
- Matošević, A.; Bosak, A. Carbamate group as structural motif in drugs: A review of carbamate derivatives used as therapeutic agents. Arch. Ind. Hyg. Toxicol. 2020, 71, 285–299. [Google Scholar] [CrossRef]
- Pacheco, D.F.; González Ceballos, L.; Castro, A.Z.; Conde González, M.R.; González, D.L.; Torre, L.; Rostgaard, L.P.; Espinoza, L.; Díaz, K.; Olea, A.F.; et al. Synthesis of New Steroidal Carbamates with Plant-Growth-Promoting Activity: Theoretical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 2330. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; De, V.D.; Bortolami, M.; Coluccia, A.; Di, S.R.; Costi, R.; Andrisano, V.; Alabiso, F.; Bergamini, C.; Fato, R.; et al. New pyridine derivatives as inhibitors of acetylcholinesterase and amyloid aggregation. Eur. J. Med. Chem. 2017, 141, 197–210. [Google Scholar] [CrossRef]
- Bajda, M.; Łątka, K.; Hebda, M.; Jończyk, J.; Malawska, B. Novel carbamate derivatives as selective butyrylcholinesterase inhibitors. Bioorg. Chem. 2018, 78, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.P.; Kumar, N.; Chauhan, B.S.; Garg, P. Carbamate as a potential anti-Alzheimer’s pharmacophore: A review. Drug Dev. Res. 2023, 84, 1624–1651. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, J.; Long, H.; Zhang, Z.; Lei, M.; Wu, W. Design, synthesis and anti-tumor activities of carbamate derivatives of cinobufagin. Steroids 2020, 164, 108749. [Google Scholar] [CrossRef]
- Bu, M.; Cao, T.; Li, H.; Guo, M.; Yang, B.B.; Zeng, C.; Hu, L. Synthesis of 5α,8α-Ergosterol Peroxide 3-Carbamate Derivatives and a Fluorescent Mitochondria-Targeting Conjugate for Enhanced Anticancer Activities. ChemMedChem 2017, 12, 466–474. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide erived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, S.; Radhakrishnan, J. Anticancer efficacy of lupeol incorporated electrospun Polycaprolactone/gelatin nanocomposite nanofibrous mats. Nanotechnology 2022, 33, 295104. [Google Scholar] [CrossRef]
- Park, J.S.; Rehman, I.U.; Choe, K.; Ahmad, R.; Lee, H.J.; Kim, M.O. A Triterpenoid Lupeol as an Antioxidant and Anti-Neuroinflammatory Agent: Impacts on Oxidative Stress in Alzheimer’s Disease. Nutrients 2023, 15, 3059. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Urea derivatives in modern drug discovery and medicinal chemistry. J. Med. Chem. 2020, 63, 2751–2788. [Google Scholar] [CrossRef]
- Sharma, K.; Kumar, H.; Priyanka. Formation of nitrogen-containing six-membered heterocycles on steroidal ring system: A review. Steroids 2023, 191, 109171. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) a | ||
|---|---|---|---|
| A549 | HepG2 | MCF-7 | |
| 3a | 14.04 ± 1.03 | 11.11 ± 1.45 | 10.18 ± 1.24 |
| 3b | 14.04 ± 1.25 | 13.83 ± 1.23 | 12.68 ± 1.43 |
| 3c | 15.51 ± 1.20 | 5.39 ± 0.77 | 15.29 ± 1.11 |
| 3d | 11.34 ± 1.14 | 17.68 ± 1.35 | 12.39 ± 1.55 |
| 3e | 13.18 ± 1.56 | 6.91 ± 1.60 | 13.99 ± 1.58 |
| 3f | 6.76 ± 0.96 | 10.82 ± 1.52 | 11.26 ± 1.21 |
| 3g | 8.19 ± 1.02 | 10.12 ± 1.78 | 7.77 ± 1.09 |
| 3h | 12.03 ± 1.47 | 11.19 ± 1.45 | 9.78 ± 1.20 |
| 3i | 5.74 ± 0.83 | 9.43 ± 1.21 | 8.20 ± 1.46 |
| 3j | 9.84 ± 1.21 | 14.46 ± 1.57 | 18.36 ± 1.78 |
| 3k | 12.12 ± 1.35 | 13.98 ± 1.65 | 19.88 ± 1.82 |
| Lupeol | 35.86 ± 1.58 | 43.62 ± 1.37 | 62.03 ± 1.79 |
| Cisplatin | 4.65 ± 0.52 | 4.05 ± 0.45 | 5.27 ± 0.68 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, S.; Zhao, Y.; Deng, S.; Hou, L.; Song, J.; Wang, M.; Bu, M. Lupeol-3-carbamate Derivatives: Synthesis and Biological Evaluation as Potential Antitumor Agents. Molecules 2024, 29, 3990. https://doi.org/10.3390/molecules29173990
Tian S, Zhao Y, Deng S, Hou L, Song J, Wang M, Bu M. Lupeol-3-carbamate Derivatives: Synthesis and Biological Evaluation as Potential Antitumor Agents. Molecules. 2024; 29(17):3990. https://doi.org/10.3390/molecules29173990
Chicago/Turabian StyleTian, Shuang, Yinxu Zhao, Siqi Deng, Liman Hou, Juan Song, Ming Wang, and Ming Bu. 2024. "Lupeol-3-carbamate Derivatives: Synthesis and Biological Evaluation as Potential Antitumor Agents" Molecules 29, no. 17: 3990. https://doi.org/10.3390/molecules29173990
APA StyleTian, S., Zhao, Y., Deng, S., Hou, L., Song, J., Wang, M., & Bu, M. (2024). Lupeol-3-carbamate Derivatives: Synthesis and Biological Evaluation as Potential Antitumor Agents. Molecules, 29(17), 3990. https://doi.org/10.3390/molecules29173990