
Dual Inhibitors of P-gp and Carbonic Anhydrase XII (hCA XII) against Tumor Multidrug Resistance with Piperazine Scaffold †

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
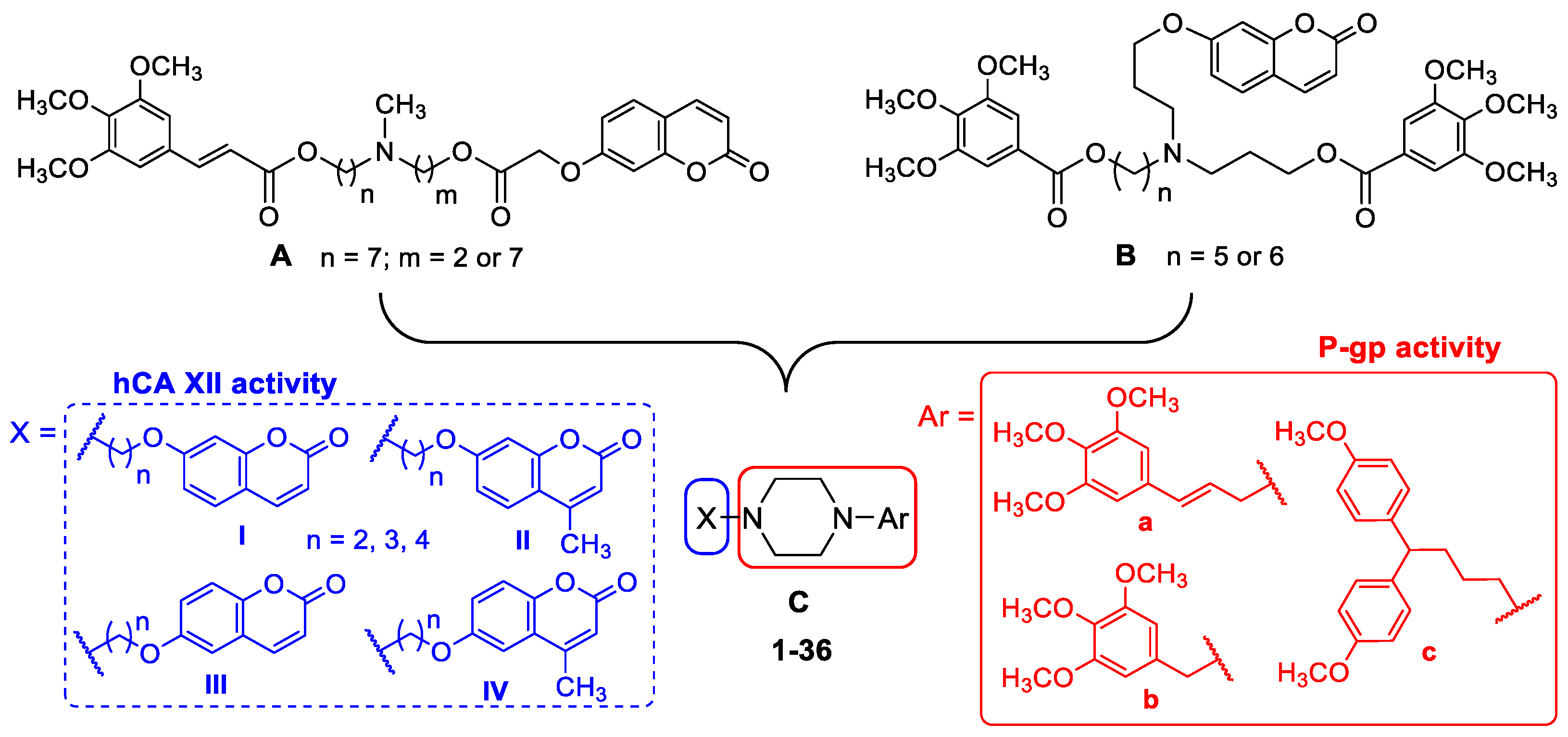
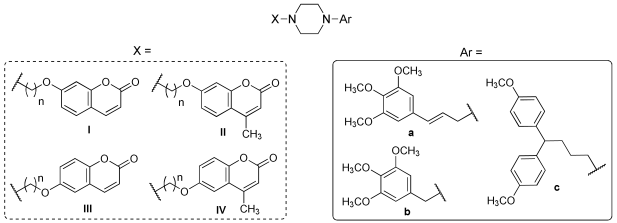
2.1. Chemistry
2.2. CA Inhibitory Activity
2.3. Doxorubicin Cytotoxicity Enhancement Assay in K562/DOX Cells
2.4. Doxorubicin Cytotoxicity Enhancement Assay in HT29/DOX and A549/DOX Cells
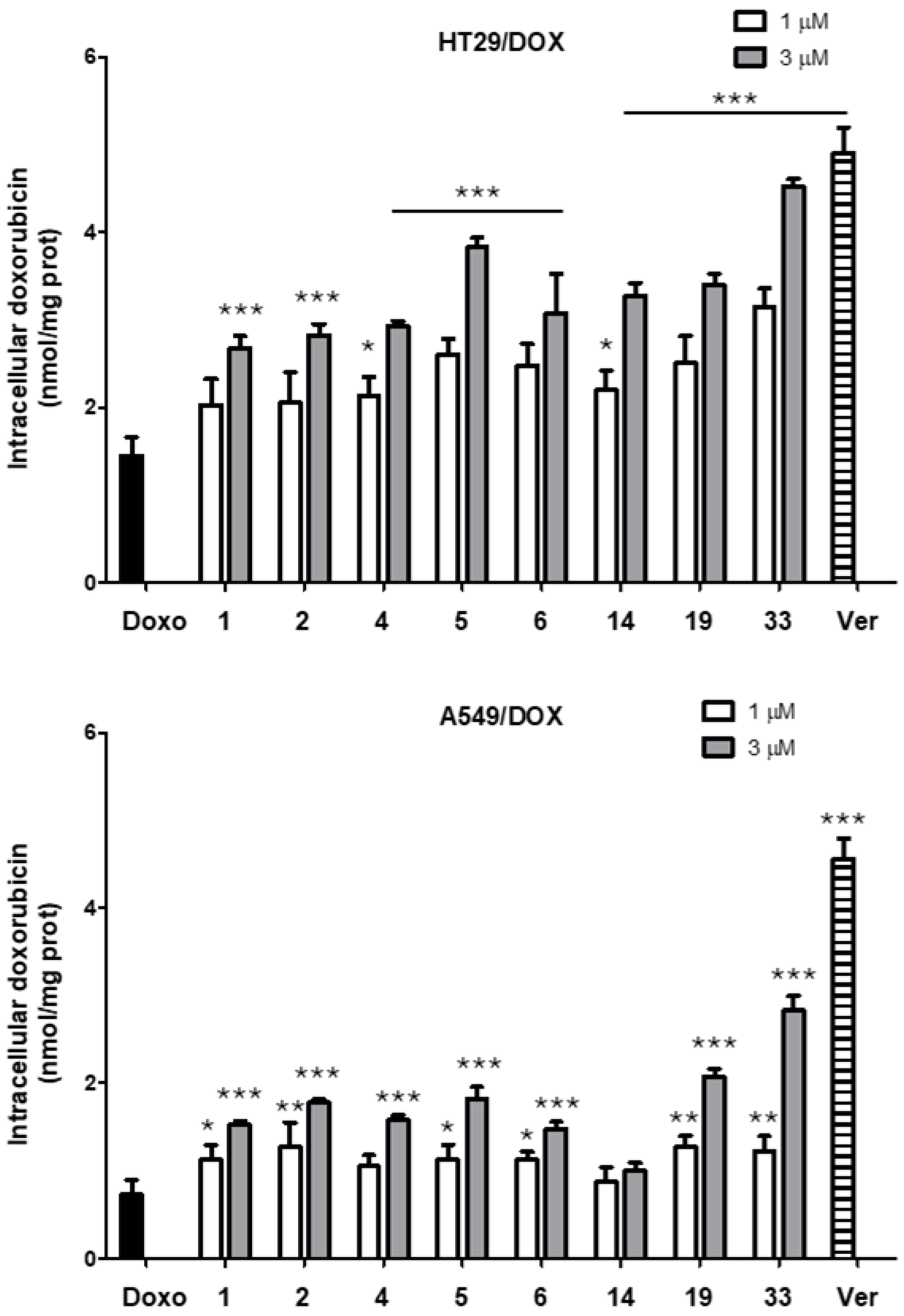
2.5. Doxorubicin Accumulation Assay in HT29/DOX and A549/DOX Cells
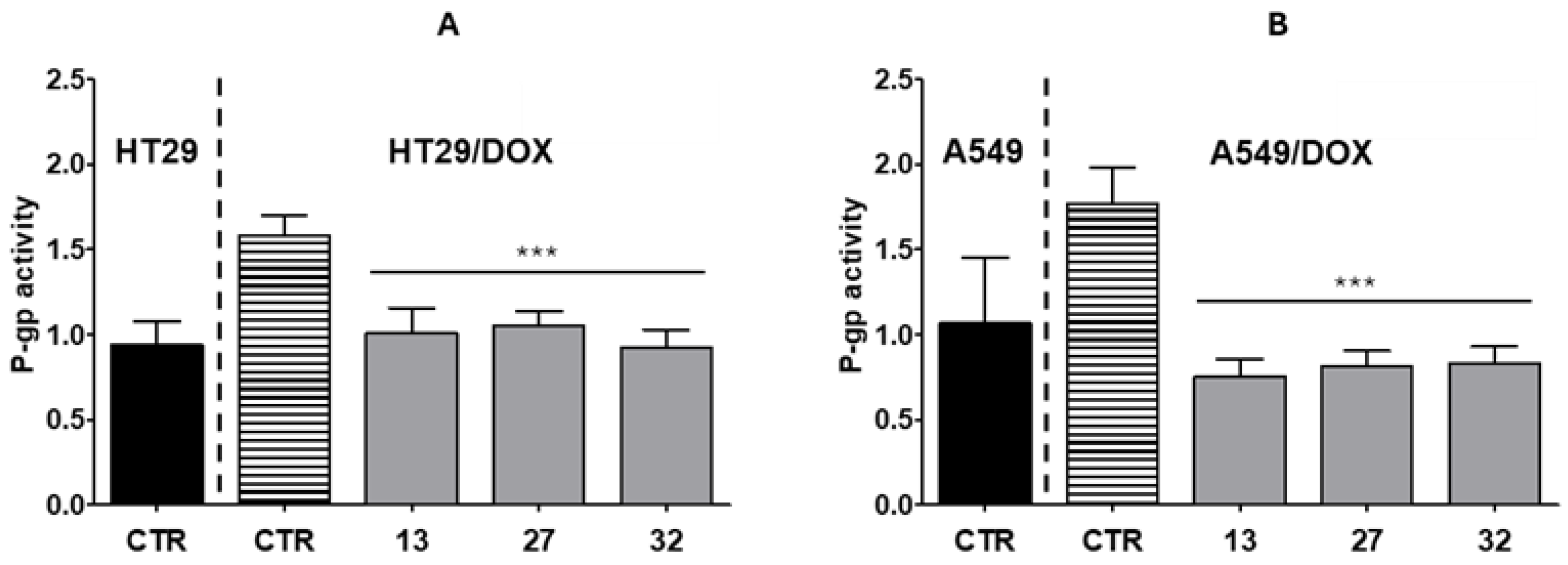
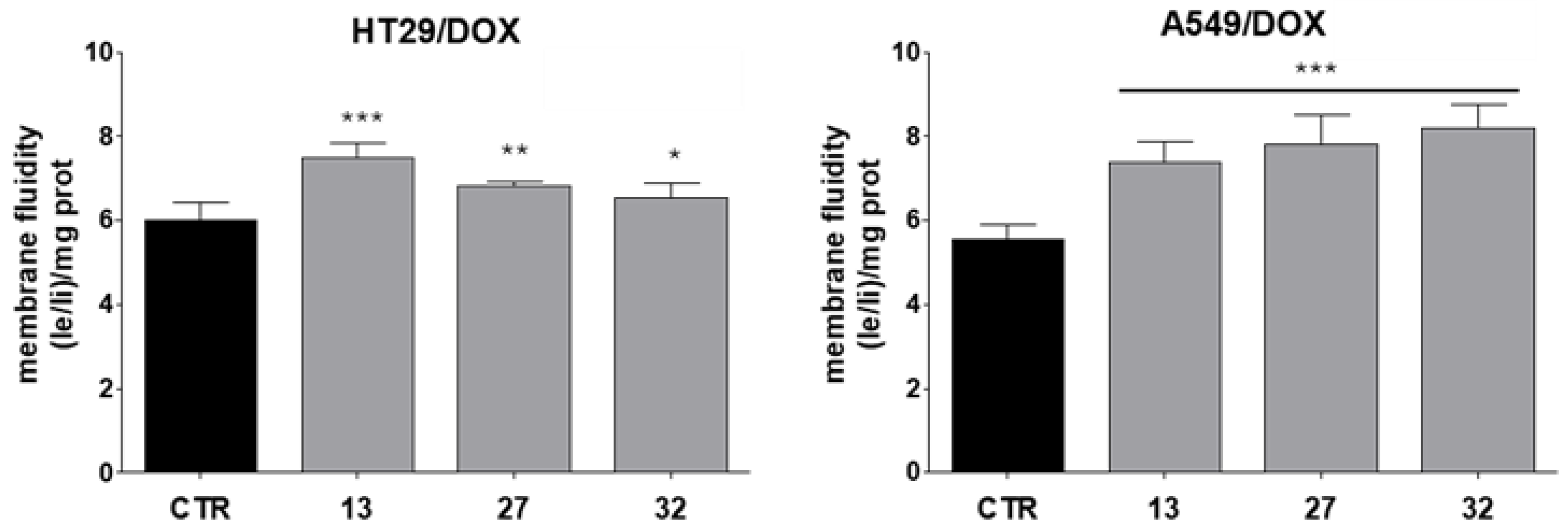
2.6. Collateral Sensitivity Studies
2.7. Transport Inhibition of Fluorescent Probes in MDCK Transfected Cells
3. Discussion and Conclusions
4. Materials and Methods
4.1. Chemistry
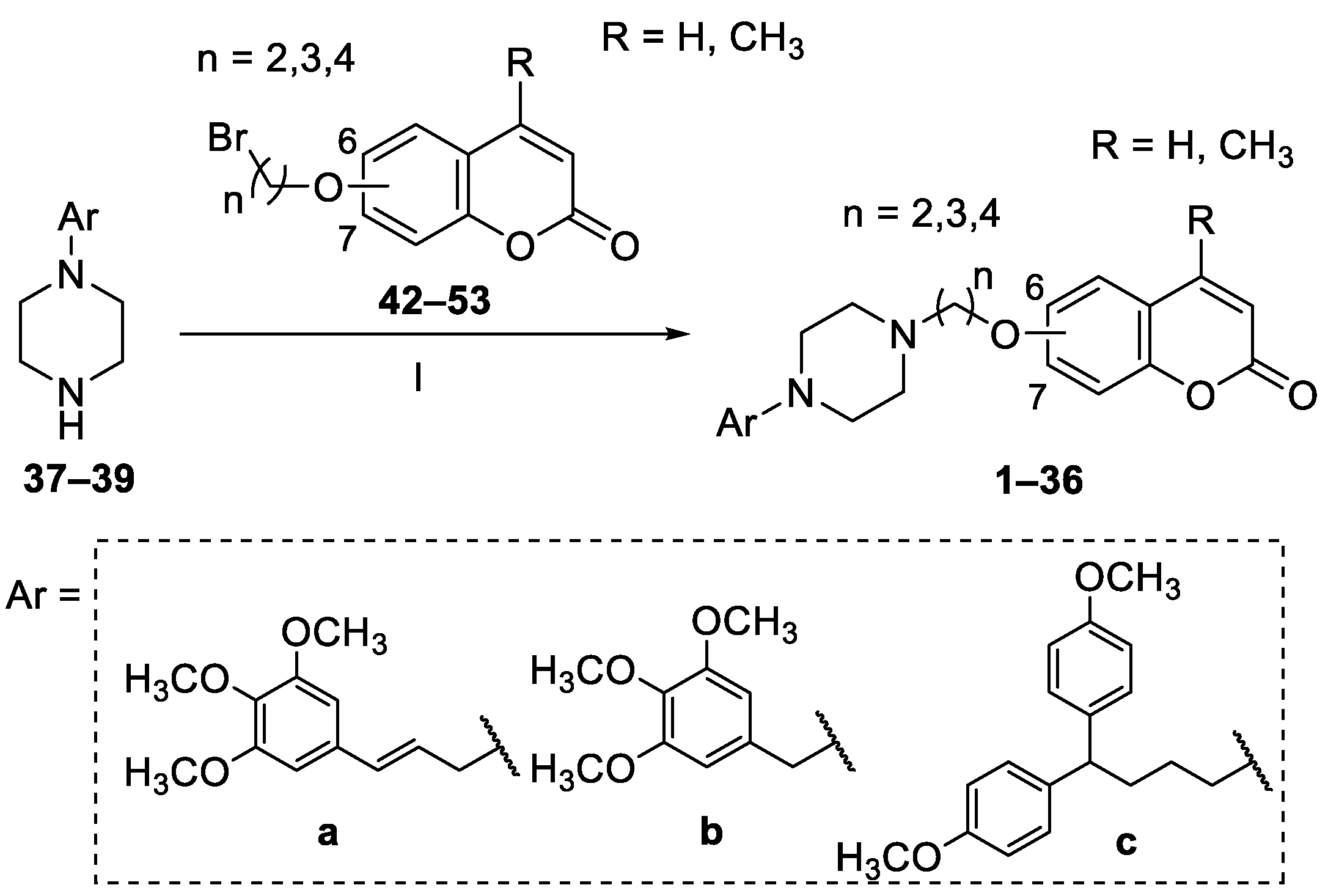
4.1.1. General Procedures for the Synthesis of Piperazine Derivatives 1–36
- (E)-7-(2-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 1. Following the general procedure, compound 1 (0.020 g, yield: 30.6%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.040 g, 0.14 mmol) and 42 (0.044 g, 0.16 mmol) in 2.5 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 9.6 Hz, 1H, CH=CH); 7.35 (d, J = 8.4 Hz, 1H, CH arom.); 6.84–6.80 (m, 2H, CH arom.); 6.60 (s, 2H, CH arom.); 6.44 (d, J = 16.0 Hz, 1H, CH=CH); 6.25–6.15 (m, 2H, CH=CH); 4.15 (t, J = 5.6 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.17 (d, J = 6.4 Hz, 2H, NCH2); 2.86 (t, J = 5.6 Hz, 2H, NCH2); 2.78–2.50 (m, 8H, NCH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.96 (C); 161.15 (C); 155.85 (C); 153.31 (C); 143.36 (CH); 137.81 (C); 133.11 (CH); 132.55 (C); 128.73 (CH); 125.87 (CH); 113.18 (CH); 113.00 (CH) 112.63 (C); 103.37 (CH); 101.50 (CH); 66.56 (CH2); 60.91 (OCH3); 60.87 (CH2); 56.84 (CH2); 56.05 (OCH3); 53.59 (CH2); 53.09 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H33N2O6 = 481.2333, found 481.2339. Dihydrochloride: white solid; mp 140–143 °C.
- (E)-7-(3-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 2. Following the general procedure, compound 2 (0.040 g, yield: 33.9%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.070 g, 0.24 mmol) and 43 [20] (0.081 g, 0.29 mmol) in 4.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.2 Hz, 1H, CH=CH); 7.34 (d, J = 8.4 Hz, 1H, CH arom.); 6.83–6.80 (m, 2H, CH arom.); 6.60 (s, 2H, CH arom.); 6.44 (d, J = 15.6 Hz, 1H, CH=CH); 6.24–6.14 (m, 2H, CH=CH); 4.07 (t, J = 5.6 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.16 (d, J = 6.8 Hz, 2H, NCH2); 2.75–2.38 (m, 10H, NCH2); 2.03–1.95 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.26 (C); 161.26 (C); 155.88 (C); 153.28 (C); 143.46 (CH); 137.67 (C); 132.99 (CH); 132.60 (C); 128.72 (CH); 126.06 (CH); 112.98 (CH); 112.45 (C); 103.27 (CH); 101.34 (CH); 66.84 (CH2); 60.94 (CH2); 60.92 (OCH3); 56.03 (OCH3); 54.87 (CH2); 53.21 (CH2); 26.48 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H35N2O6 = 495.2490, found 495.2481. Dihydrochloride: white solid; mp 240–243 °C.
- (E)-7-(4-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 3. Following the general procedure, compound 3 (0.010 g, yield: 23.0%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.025 g, 0.086 mmol) and 44 (0.030 g, 0.10 mmol) in 2.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 9.6 Hz, 1H, CH=CH); 7.35 (d, J = 8.4 Hz, 1H, CH arom.); 6.83–6.79 (m, 2H, CH arom.); 6.60 (s, 2H, CH arom.); 6.47 (d, J = 16.0 Hz, 1H, CH=CH); 6.25–6.14 (m, 2H, CH=CH); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.86 (s, 6H, OCH3); 3.83 (s, 3H, OCH3); 3.27 (d, J = 6.4 Hz, 2H, NCH2); 2.90–2.52 (m, 10H, NCH2); 1.88–1.75 (m, 4H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.28 (C); 161.24 (C); 155.93 (C); 153.32 (C); 143.41 (CH); 133.17 (CH); 132.56 (C); 128.71 (CH); 125.88 (CH); 112.99 (CH); 112.44 (C); 103.38 (CH); 101.31 (CH); 68.32 (CH2); 60.92 (OCH3); 60.88 (CH2); 58.03 (CH2); 56.06 (OCH3); 53.09 (CH2); 29.68 (CH2); 26.95 (CH2); 23.26 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C29H37N2O6 = 509.2646, found 509.2641. Dihydrochloride: white solid; mp 160–163 °C.
- (E)-4-Methyl-7-(2-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 4. Following the general procedure, compound 4 (0.020 g, yield: 29.5%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.040 g, 0.14 mmol) and 45 (0.046 g, 0.16 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: EtOAc/CH3OH/NH4OH 99:1:0.1. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.48 (d, J = 8.8 Hz, 1H, CH arom.); 6.86 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.81 (d, J = 2.4 Hz, 1H, CH arom.); 6.60 (s, 2H, CH arom.); 6.45 (d, J = 15.6, 1H, CH=CH); 6.18 (dt, J = 15.6, 6.8 Hz, 1H, CH=CH); 6.13 (s, 1H, CH=C); 4.16 (t, J = 5.6 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.83 (s, 3H, OCH3); 3.17 (d, J = 6.8 Hz, 2H, NCH2); 2.86 (t, J = 5.6 Hz, 2H, NCH2); 2.76–2.45 (m, 8H, CH2); 2.39 (s, 3H, CH3) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.77 (C); 161.25 (C); 155.24 (C); 153.32 (C); 152.49 (C); 133.18 (CH); 132.56 (C); 125.51 (CH); 113.70 (C); 112.70 (CH); 112.06 (CH); 103.37 (CH); 101.52 (CH); 66.51 (CH2); 60.93 (OCH3); 60.88 (CH2); 56.87 (CH2); 56.06 (OCH3); 53.58 (CH2); 53.09 (CH2); 18.67 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H35N2O6 = 495.2490, found 495.2490. Dihydrochloride: white solid; mp 139–141 °C.
- (E)-4-Methyl-7-(3-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 5. Following the general procedure, compound 5 (0.030 g, yield: 57.3%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.030 g, 0.10 mmol) and 46 (0.036 g, 0.12 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.47 (d, J = 8.8 Hz, 1H, CH arom.); 6.85–6.81 (m, 2H, CH arom.); 6.61 (s, 2H, CH arom); 6.45 (d, J = 16.0 Hz, 1H, CH=CH); 6.20 (dt, J = 15.6, 6.8 Hz, 1H, CH=CH); 6.12 (s, 1H, CH=C); 4.07 (t, J = 6.4 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.83 (s, 3H, OCH3); 3.16 (d, J = 6.8 Hz, 2H, NCH2); 2.85–2.43 (m, 10H, NCH2); 2.39 (s, 3H, CH3); 2.04–1.98 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.08 (C); 161.37 (C); 155.29 (C); 153.30 (C); 152.59 (C); 133.06 (CH); 132.59 (C); 125.48 (CH); 113.51 (C); 112.70 (CH); 111.90 (CH); 103.29 (CH); 101.35 (CH); 66.80 (CH2); 61.23 (CH2); 60.88 (OCH3); 56.04 (OCH3); 54.90 (CH2); 53.18 (CH2); 26.50 (CH2); 18.70 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C29H37N2O6 = 509.2646, found 509.2646. Dihydrochloride: white solid; mp 70–73 °C.
- (E)-4-Methyl-7-(4-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 6. Following the general procedure, compound 6 (0.050 g, yield: 44.3%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.063 g, 0.22 mmol) and 47 (0.080 g, 0.26 mmol) in 3.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 93:7:0.7. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.47 (d, J = 8.8 Hz, 1H, CH arom.); 6.82 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.79 (d, J = 2.4 Hz, 1H, CH arom.); 6.60 (s, 2H, CH arom); 6.45 (d, J = 16.0 Hz, 1H, CH=CH); 6.19 (dt, J = 16.0, 6.8 Hz, 1H, CH=CH); 6.11 (s, 1H, CH=C); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.16 (d, J = 6.8 Hz, 2H, NCH2); 2.78–2.41 (m, 10H, NCH2); 2.38 (s, 3H, CH3); 1.87–1.80 (m, 2H, CH2); 1.73–1.65 (m, 2H, CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C30H39N2O6 = 523.2803, found 523.2811. Dihydrochloride: yellow solid; mp 180–183 °C.
- (E)-6-(2-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 7. Following the general procedure, compound 7 (0.050 g, yield: 55.9%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.054 g, 0.19 mmol) and 48 (0.060 g, 0.22 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.60 (d, J = 9.6 Hz, 1H, CH=CH); 7.20 (d, J = 8.8 Hz, 1H, CH arom); 7.08 (dd, J = 8.8, 2.8 Hz, 1H, CH arom); 6.89 (d, J = 2.8 Hz, 1H, CH arom); 6.57 (s, 2H, CH arom); 6.42 (d, J = 15.6 Hz, 1H, CH=CH); 6.37 (d, J = 9.6 Hz, 1H, CH=CH); 6.17 (dt, J = 15.6, 6.8 Hz, 1H, CH=CH); 4.10 (t, J = 5.6 Hz, 2H, OCH2); 3.82 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.15 (d, J = 6.8 Hz, 2H, NCH2); 2.82 (t, J = 5.6 Hz, 2H, NCH2); 2.77–2.40 (m, 8H, NCH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.91 (C); 155.20 (C); 153.29 (C); 148.49 (C); 143.18 (CH); 137.79 (C); 133.32 (CH); 132.47 (C); 125.53 (CH); 119.98 (CH); 119.15 (C); 117.81 (CH); 117.05 (CH); 110.98 (CH); 103.35 (CH); 66.61 (CH2); 60.89 (OCH3); 60.80 (CH2); 57.03 (CH2); 56.03 (OCH3); 53.45 (CH2); 52.99 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H33N2O6 = 481.2333, found 481.2330. Dihydrochloride: yellow solid; mp 240–243 °C.
- (E)-6-(3-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 8. Following the general procedure, compound 8 (0.050 g, yield: 68.8%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.043 g, 0.15 mmol) and 49 (0.050 g, 0.18 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.6 Hz, 1H, CH=CH); 7.21 (d, J = 8.8 Hz, 1H, CH arom); 7.06 (dd, J = 8.8, 2.8 Hz, 1H, CH arom); 6.88 (d, J = 2.8 Hz, 1H, CH arom); 6.58 (s, 2H, CH arom); 6.42 (d, J = 15.6 Hz, 1H, CH=CH); 6.37 (d, J = 9.6 Hz, 1H, CH=CH); 6.17 (dt, J = 15.6, 6.4 Hz, 1H, CH=CH); 4.00 (t, J = 6.4 Hz, 2H, OCH2); 3.83 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.13 (d, J = 6.4 Hz, 2H, NCH2); 2.70–2.20 (m, 10H, NCH2); 2.00–1.94 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.98 (C); 155.47 (C); 153.28 (C); 148.36 (C); 143.24 (CH); 137.71 (C); 133.04 (CH); 132.56 (C); 125.95 (CH); 119.90 (CH); 119.14 (C); 117.78 (CH); 116.99 (CH); 110.78 (CH); 103.29 (CH); 66.93 (CH2); 60.89 (CH2); 56.03 (OCH3); 55.00 (CH2); 53.16 (CH2); 26.65 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H35N2O6 = 495.2490, found 495.2496. Dihydrochloride: yellow solid; mp 242–244 °C.
- (E)-6-(4-(4-(3-(3,4,5-Trimethoxyphenyl)allyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 9. Following the general procedure, compound 9 (0.060 g, yield: 57.6%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.060 g, 0.21 mmol) and 50 (0.073 g, 0.25 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.63 (d, J = 9.6 Hz, 1H, CH=CH); 7.25 (d, J = 9.2 Hz, 1H, CH arom); 7.09 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.89 (d, J = 2.8 Hz, 1H, CH arom); 6.61 (s, 2H, CH arom); 6.47–6.41 (m, 2H, CH=CH); 6.19 (dt, J = 15.6, 6.4 Hz, 1H, CH=CH); 4.00 (t, J = 6.0 Hz, 2H, OCH2); 3.86 (s, 6H, OCH3); 3.83 (s, 3H, OCH3); 3.16 (d, J = 6.4 Hz, 2H, NCH2); 2.80–2.40 (m, 10H, NCH2); 1.86–1.79 (m, 2H, CH2); 1.74–1.70 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.96 (C); 155.47 (C); 153.26 (C); 148.30 (C); 143.25 (CH); 137.71 (C); 132.99 (CH); 132.57 (C); 125.95 (CH); 119.86 (CH); 119.13 (C); 117.74 (CH); 116.93 (CH); 110.74 (CH); 103.32 (CH); 68.39 (CH2); 60.89 (CH2); 58.11 (CH2); 56.02 (OCH3); 53.11 (CH2); 27.16 (CH2); 23.34 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C29H37N2O6 = 509.2646, found 509.2640. Dihydrochloride: yellow solid; mp 250–252 °C.
- (E)-4-Methyl-6-(2-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 10. Following the general procedure, compound 10 (0.040 g, yield: 33.8%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.070 g, 0.24 mmol) and 51 (0.081 g, 0.29 mmol) in 5.8 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.23 (d, J = 9.2 Hz, 1H, CH arom); 7.08 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 7.01 (d, J = 2.8 Hz, 1H, CH arom); 6.59 (s, 2H, CH arom); 6.44 (d, J = 15.6 Hz, 1H, CH=CH); 6.26 (s, 1H, CH=C); 6.18 (dt, J = 15.6, 6.4 Hz, 1H, CH=CH); 4.13 (t, J = 6.4 Hz, 2H, OCH2); 3.84 (s, 6H, OCH3); 3.81 (s, 3H, OCH3); 3.16 (d, J = 6.4 Hz, 2H, NCH2); 2.85 (t, J = 6.4 Hz, 2H, NCH2); 2.77–2.50 (m, 8H, NCH2); 2.38 (s, 3H, CH3) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.86 (C); 155.11 (C); 153.30 (C); 151.93 (C); 147.95 (C); 137.89 (C); 133.39 (CH); 132.44 (C); 125.41 (CH); 120.43 (C); 119.20 (CH); 117.90 (CH); 115.47 (CH); 108.68 (CH); 103.44 (CH); 66.60 (CH2); 60.88 (OCH3); 60.78 (CH2); 57.07 (CH2); 56.06 (OCH3); 53.44 (CH2); 52.97 (CH2); 18.68 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H35N2O6 = 495.2490, found 495.2482. Dihydrochloride: yellow solid; mp 230–232 (d) °C.
- (E)-4-Methyl-6-(3-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 11. Following the general procedure, compound 11 (0.040 g, yield: 38.4%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.060 g, 0.21 mmol) and 52 (0.073 g, 0.25 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.21 (d, J = 9.2 Hz, 1H, CH arom); 7.07 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.98 (d, J = 2.8 Hz, 1H, CH arom); 6.58 (s, 2H, CH arom); 6.42 (d, J = 15.6 Hz, 1H, CH=CH); 6.25 (s, 1H, CH=C); 6.17 (dt, J = 15.6, 6.4 Hz, 1H, CH=CH); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.83 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.14 (d, J = 6.4 Hz, 2H, NCH2); 2.73–2.43 (m, 10H, NCH2); 2.38 (s, 3H, CH3); 2.02–1.95 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.88 (C); 155.37 (C); 153.32 (C); 151.93 (C); 147.85 (C); 137.89 (C); 133.12 (CH); 132.53 (C); 125.79 (CH); 120.43 (C); 119.09 (CH); 117.86 (CH); 115.43 (CH); 108.56 (CH); 103.46 (CH); 66.98 (CH2); 60.87 (OCH3); 60.84 (CH2); 56.06 (OCH3); 55.00 (CH2); 53.14 (CH2); 53.08 (CH2); 26.68 (CH2); 18.67 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C29H37N2O6 = 509.2646, found 509.2644. Dihydrochloride: yellow solid; mp 234–236 (d) °C.
- (E)-4-Methyl-6-(4-(4-(3-(3,4,5-trimethoxyphenyl)allyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 12. Following the general procedure, compound 12 (0.050 g, yield: 46.7%) was synthesized as a pale-yellow oil, starting from 37 [21] (0.060 g, 0.21 mmol) and 53 (0.077 g, 0.25 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.22 (d, J = 9.2 Hz, 1H, CH arom); 7.06 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.98 (d, J = 2.8 Hz, 1H, CH arom); 6.59 (s, 2H, CH arom); 6.42 (d, J = 15.6 Hz, 1H, CH=CH); 6.26 (s, 1H, CH=C); 6.17 (dt, J = 15.6, 6.4 Hz, 1H, CH=CH); 3.99 (t, J = 6.4 Hz, 2H, OCH2); 3.84 (s, 6H, OCH3); 3.81 (s, 3H, OCH3); 3.14 (d, J = 6.4 Hz, 2H, NCH2); 2.70–2.40 (m, 10H, NCH2); 2.38 (s, 3H, CH3); 1.85–1.78 (m, 2H, CH2); 1.72–1.67 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.95 (C); 155.40 (C); 153.31 (C); 151.96 (C); 147.80 (C); 137.81 (C); 133.03 (CH); 132.57 (C); 125.95 (CH); 120.44 (C); 119.04 (CH); 117.88 (CH); 115.44 (CH); 108.46 (CH); 103.38 (CH); 68.45 (CH2); 60.91 (CH2); 58.15 (CH2); 56.05 (OCH3); 53.14 (CH2); 27.26 (CH2); 23.38 (CH2); 18.70 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C30H39N2O6 = 523.2803, found 523.2797. Dihydrochloride: yellow solid; mp 204–207 °C.
- 7-(2-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 13. Following the general procedure, compound 13 (0.12 g, yield: 78.0%) was synthesized as a yellow oil, starting from 38 [21] (0.090 g, 0.34 mmol) and 42 (0.11 g, 0.40 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.58 (d, J = 9.2 Hz, 1H, CH=CH); 7.31 (d, J = 8.4 Hz, 1H, CH arom.); 6.79 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.75 (d, J = 2.4 Hz, 1H, CH arom.); 6.52 (s, 2H, CH arom.); 6.18 (d, J = 9.2 Hz, 1H, CH=CH); 4.11 (t, J = 5.6 Hz, 2H, OCH2); 3.81 (s, 6H, OCH3); 3.78 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.81 (t, J = 5.6 Hz, 2H, NCH2); 2.70–2.55 (m, 4H, NCH2); 2.54–2.35 (m, 4H, NCH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.94 (C); 161.10 (C); 155.80 (C); 153.07 (C); 143.39 (CH); 136.97 (C); 133.63 (C); 128.76 (CH); 113.08 (CH); 112.94 (CH); 112.60 (C); 105.89 (CH); 101.48 (CH); 66.59 (CH2); 63.11 (CH2); 60.78 (OCH3); 56.81 (CH2); 56.11 (OCH3); 53.57 (CH2); 52.89 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C25H31N2O6 = 455.2177, found 455.2177. Dihydrochloride: white solid; mp 239–241 °C.
- 7-(3-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 14. Following the general procedure, compound 14 (0.11 g, yield: 89.7%) was synthesized as a yellow oil, starting from 38 [21] (0.070 g, 0.26 mmol) and 43 [20] (0.090 g, 0.32 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.60 (d, J = 9.2 Hz, 1H, CH=CH); 7.32 (d, J = 8.4 Hz, 1H, CH arom.); 6.83–6.77 (m, 2H, CH arom.); 6.53 (s, 2H, CH arom.); 6.20 (d, J = 9.2 Hz, 1H, CH=CH); 4.05 (t, J = 5.6 Hz, 2H, OCH2); 3.82 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.66–2.35 (m, 10H, NCH2); 2.00–1.94 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.28 (C); 161.19 (C); 155.88 (C); 153.06 (C); 143.43 (CH); 136.90 (C); 133.98 (C); 128.71 (CH); 112.94 (CH); 112.44 (C); 105.84 (CH); 101.38 (CH); 66.88 (CH2); 63.21 (CH2); 60.81 (OCH3); 56.11 (OCH3); 54.86 (CH2); 53.25 (CH2); 53.07 (CH2); 26.51 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C26H33N2O6 = 469.2333, found 469.2331. Dihydrochloride: white solid; mp 220–223 °C.
- 7-(4-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 15. Following the general procedure, compound 15 (0.12 g, yield: 82.9%) was synthesized as a yellow oil, starting from 38 [21] (0.080 g, 0.30 mmol) and 44 (0.10 g, 0.36 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.2 Hz, 1H, CH=CH); 7.33 (d, J = 8.4 Hz, 1H, CH arom.); 6.81–6.77 (m, 2H, CH arom.); 6.54 (s, 2H, CH arom.); 6.21 (d, J = 9.2 Hz, 1H, CH=CH); 4.01 (t, J = 5.6 Hz, 2H, OCH2); 3.83 (s, 6H, OCH3); 3.81 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.66–2.27 (m, 10H, NCH2); 1.85–1.78 (m, 2H, CH2); 1.70–1.64 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.28 (C); 161.27 (C); 155.90 (C); 153.05 (C); 143.48 (CH); 136.82 (C); 133.98 (C); 128.73 (CH); 112.98 (CH); 112.94 (CH); 112.40 (C); 105.78 (CH); 101.27 (CH); 68.33 (CH2); 63.23 (CH2); 60.84 (OCH3); 58.10 (CH2); 56.11 (OCH3); 53.22 (CH2); 53.06 (CH2); 26.99 (CH2); 23.32 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H35N2O6 = 483.2490, found 483.2484. Dihydrochloride: white solid; mp 215–218 °C.
- 4-Methyl-7-(2-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 16. Following the general procedure, compound 16 (0.090 g, yield: 94.7%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.054 g, 0.20 mmol) and 45 (0.056 g, 0.24 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.44 (d, J = 8.8 Hz, 1H, CH arom.); 6.82 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.77 (d, J = 2.4 Hz, 1H, CH arom.); 6.53 (s, 2H, CH arom.); 6.08 (s, 1H, CH=C); 4.13 (t, J = 5.6 Hz, 2H, OCH2); 3.82 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.82 (t, J = 5.6 Hz, 2H, NCH2); 2.73–2.39 (m, 8H, NCH2); 2.35 (s, 3H, CH3) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.78 (C); 161.21 (C); 155.20 (C); 153.07 (C); 152.51 (C); 136.95 (C); 133.83 (C); 125.51 (CH); 113.64 (C); 112.65 (CH); 111.96 (CH); 105.86 (CH); 101.51 (CH); 66.56 (CH2); 63.16 (CH2); 60.80 (OCH3); 56.87 (CH2); 56.12 (OCH3); 53.66 (CH2); 52.94 (CH2); 18.62 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C26H33N2O6 = 469.2333, found 469.2325. Dihydrochloride: white solid; mp 231–234 °C.
- 4-Methyl-7-(3-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 17. Following the general procedure, compound 17 (0.050 g, yield: 55.4%) was synthesized as a yellow oil, starting from 38 [21] (0.050 g, 0.19 mmol) and 46 (0.066 g, 0.22 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.46 (d, J = 8.8 Hz, 1H, CH arom.); 6.83–6.80 (m, 2H, CH arom.); 6.55 (s, 2H, CH arom.); 6.10 (s, 1H, CH=C); 4.06 (t, J = 6.0 Hz, 2H, OCH2); 3.84 (s, 6H, OCH3); 3.81 (s, 3H, OCH3); 3.43 (s, 2H, NCH2); 2.64–2.40 (m, 10H, NCH2); 2.37 (s, 3H, CH3); 2.00–1.97 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.09 (C); 161.29 (C); 155.28 (C); 153.07 (C); 152.54 (C); 136.94 (C); 133.91 (C); 125.47 (CH); 113.49 (C); 112.64 (CH); 111.87 (CH); 105.86 (CH); 101.40 (CH); 66.84 (CH2); 63.20 (CH2); 60.83 (OCH3); 56.13 (OCH3); 54.88 (CH2); 53.22 (CH2); 53.03 (CH2); 26.50 (CH2); 18.64 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H35N2O6 = 483.2490, found 483.2489. Dihydrochloride: white solid; mp 233–236 °C.
- 4-Methyl-7-(4-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 18. Following the general procedure, compound 18 (0.060 g, yield: 64.6%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.050 g, 0.19 mmol) and 47 (0.069 g, 0.22 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.43 (d, J = 8.8 Hz, 1H, CH arom.); 6.80–6.74 (m, 2H, CH arom.); 6.52 (s, 2H, CH arom.); 6.06 (s, 1H, CH=C); 3.99 (t, J = 6.4 Hz, 2H, OCH2); 3.81 (s, 6H, OCH3); 3.78 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.70–2.37 (m, 10H, NCH2); 2.34 (s, 3H, CH3); 1.83–1.77 (m, 2H, CH2); 1.73–1.64 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.06 (C); 161.26 (C); 155.25 (C); 153.05 (C); 152.57 (C); 136.95 (C); 133.65 (C); 125.49 (CH); 113.43 (C); 112.60 (CH); 111.79 (CH); 105.92 (CH); 101.30 (CH); 68.22 (CH2); 63.07 (CH2); 60.79 (OCH3); 57.92 (CH2); 56.11 (OCH3); 52.97 (CH2); 52.76 (CH2); 26.95 (CH2); 23.09 (CH2); 18.61 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H37N2O6 = 497.2646, found 497.2655. Dihydrochloride: white solid; mp 216–219 °C.
- 6-(2-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 19. Following the general procedure, compound 19 (0.080 g, yield: 67.2%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.070 g, 0.26 mmol) and 48 (0.084 g, 0.32 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 93:7:0.7. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.60 (d, J = 9.6 Hz, 1H, CH=CH); 7.19 (d, J = 8.8 Hz, 1H, CH arom.); 7.06 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.88 (d, J = 2.4 Hz, 1H, CH arom.); 6.52 (s, 2H, CH arom.); 6.36 (d, J = 9.6 Hz, 1H, CH=CH); 4.09 (t, J = 5.6 Hz, 2H, OCH2); 3.81 (s, 6H, OCH3); 3.79 (s, 3H, OCH3); 3.41 (s, 2H, NCH2); 2.80 (t, J = 5.6 Hz, 2H, NCH2); 2.72–2.32 (m, 8H, NCH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.91 (C); 155.24 (C); 153.05 (C); 148.45 (C); 143.23 (CH); 136.85 (C); 133.88 (C); 120.00 (CH); 119.14 (C); 117.78 (CH); 117.01 (CH); 110.95 (CH); 105.77 (CH); 66.71 (CH2); 63.17 (CH2); 60.82 (OCH3); 57.09 (CH2); 56.10 (OCH3); 53.68 (CH2); 52.96 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C25H31N2O6 = 455.2177, found 455.2176. Dihydrochloride: white solid; mp 180–182 °C.
- 6-(3-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 20. Following the general procedure, compound 20 (0.050 g, yield: 71.1%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.040 g, 0.15 mmol) and 49 (0.051 g, 0.18 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.63 (d, J = 9.6 Hz, 1H, CH=CH); 7.24 (d, J = 8.8 Hz, 1H, CH arom.); 7.09 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.90 (d, J = 2.4 Hz, 1H, CH arom.); 6.55 (s, 2H, CH arom.); 6.41 (d, J = 9.6 Hz, 1H, CH=CH); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.44 (s, 2H, NCH2); 2.67–2.40 (m, 10H, NCH2); 2.02–1.94 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.98 (C); 155.51 (C); 153.09 (C); 148.40 (C); 143.19 (CH); 136.94 (C); 133.98 (C); 119.91 (CH); 119.16 (C); 117.82 (CH); 117.05 (CH); 110.81 (CH); 105.84 (CH); 67.03 (CH2); 63.24 (CH2); 60.85 (OCH3); 56.14 (OCH3); 55.05 (CH2); 53.31 (CH2); 53.09 (CH2); 26.74 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C26H33N2O6 = 469.2333, found 469.2332. Dihydrochloride: white solid; mp 243–245 °C.
- 6-(4-(4-(3,4,5-Trimethoxybenzyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 21. Following the general procedure, compound 21 (0.040 g, yield: 55.3%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.040 g, 0.15 mmol) and 50 (0.053 g, 0.18 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.63 (d, J = 9.2 Hz, 1H, CH=CH); 7.25 (d, J = 8.8 Hz, 1H, CH arom.); 7.08 (dd, J = 8.8, 2.8 Hz, 1H, CH arom.); 6.88 (d, J = 2.8 Hz, 1H, CH arom.); 6.55 (s, 2H, CH arom.); 6.41 (d, J = 9.2 Hz, 1H, CH=CH); 3.99 (t, J = 6.0 Hz, 2H, OCH2); 3.85 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.43 (s, 2H, NCH2); 2.67–2.40 (m, 10H, NCH2); 1.83–1.78 (m, 2H, CH2); 1.72–1.66 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.03 (C); 155.51 (C); 153.07 (C); 148.36 (C); 143.23 (CH); 136.85 (C); 133.98 (C); 119.88 (CH); 119.16 (C); 117.84 (CH); 117.05 (CH); 110.71 (CH); 105.78 (CH); 68.43 (CH2); 63.27 (CH2); 60.86 (OCH3); 58.21 (CH2); 56.12 (OCH3); 53.25 (CH2); 53.09 (CH2); 27.23 (CH2); 23.45 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H35N2O6 = 483.2490, found 483.2487. Dihydrochloride: white solid; mp 237–239 °C.
- 4-Methyl-6-(2-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 22. Following the general procedure, compound 22 (0.050 g, yield: 52.0%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.050 g, 0.19 mmol) and 51 (0.064 g, 0.23 mmol) in 4.2 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.23 (d, J = 9.2 Hz, 1H, CH arom); 7.10 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 7.03 (d, J = 2.8 Hz, 1H, CH arom); 6.55 (s, 2H, CH arom); 6.27 (s, 1H, CH=C); 4.13 (t, J = 6.0 Hz, 2H, OCH2); 3.84 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.45 (s, 2H, NCH2); 2.84 (t, J = 6.0 Hz, 2H, NCH2); 2.71–2.58 (m, 4H, NCH2); 2.57–2.47 (m, 4H, NCH2); 2.38 (s, 3H, CH3) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.91 (C); 155.13 (C); 153.10 (C); 151.93 (C); 147.97 (C); 133.66 (C); 120.45 (C); 119.20 (CH); 117.93 (CH); 115.50 (CH); 108.66 (CH); 105.95 (CH); 66.67 (CH2); 63.17 (CH2); 60.83 (OCH3); 57.14 (CH2); 56.15 (OCH3); 53.65 (CH2); 52.90 (CH2); 18.71 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C26H33N2O6 = 469.2333, found 469.2342. Dihydrochloride: yellow solid; mp 240–242 (d) °C.
- 4-Methyl-6-(3-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 23. Following the general procedure, compound 23 (0.050 g, yield: 55.4%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.050 g, 0.19 mmol) and 52 (0.066 g, 0.23 mmol) in 4.2 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.21 (d, J = 9.2 Hz, 1H, CH arom); 7.07 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.98 (d, J = 2.8 Hz, 1H, CH arom); 6.53 (s, 2H, CH arom); 6.25 (s, 1H, CH=C); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.83 (s, 6H, OCH3); 3.80 (s, 3H, OCH3); 3.42 (s, 2H, NCH2); 2.70–2.40 (m, 10H, NCH2); 2.38 (s, 3H, CH3); 2.01–1.94 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.94 (C); 155.38 (C); 153.06 (C); 152.01 (C); 147.80 (C); 136.92 (C); 133.90 (C); 120.41 (C); 119.11 (CH); 117.86 (CH); 115.41 (CH); 108.48 (CH); 105.85 (CH); 67.02 (CH2); 63.20 (CH2); 60.82 (OCH3); 56.12 (OCH3); 55.06 (CH2); 53.27 (CH2); 53.03 (CH2); 26.74 (CH2); 18.71 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C27H35N2O6 = 483.2490, found 483.2497. Dihydrochloride: yellow solid; mp 244–246 (d) °C.
- 4-Methyl-6-(4-(4-(3,4,5-trimethoxybenzyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 24. Following the general procedure, compound 24 (0.060 g, yield: 64.4%) was synthesized as a pale-yellow oil, starting from 38 [21] (0.050 g, 0.19 mmol) and 53 (0.070 g, 0.23 mmol) in 4.2 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.24 (d, J = 9.2 Hz, 1H, CH arom); 7.07 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.99 (d, J = 2.8 Hz, 1H, CH arom); 6.55 (s, 2H, CH arom); 6.28 (s, 1H, CH=C); 4.00 (t, J = 6.4 Hz, 2H, OCH2); 3.84 (s, 6H, OCH3); 3.82 (s, 3H, OCH3); 3.43 (s, 2H, NCH2); 2.60–2.39 (m, 10H, NCH2); 2.38 (s, 3H, CH3); 1.86–1.79 (m, 2H, CH2); 1.73–1.65 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.93 (C); 155.39 (C); 153.04 (C); 152.01 (C); 147.75 (C); 136.90 (C); 133.91 (C); 120.40 (C); 119.06 (CH); 117.83 (CH); 115.37 (CH); 108.41 (CH); 105.85 (CH); 68.44 (CH2); 63.20 (CH2); 60.80 (OCH3); 58.16 (CH2); 56.11 (OCH3); 53.19 (CH2); 53.02 (CH2); 27.26 (CH2); 23.39 (CH2); 18.69 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C28H37N2O6 = 497.2646, found 497.2653. Dihydrochloride: yellow solid; mp 236–238 (d) °C.
- 7-(2-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 25. Following the general procedure, compound 25 (0.060 g, yield: 78.5%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.050 g, 0.14 mmol) and 42 (0.045 g, 0.17 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 96:4:0.6. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.2 Hz, 1H, CH=CH); 7.34 (d, J = 8.4 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.84–6.78 (m, 6H, CH arom.); 6.23 (d, J = 9.2 Hz, 1H, CH=CH); 4.13 (t, J = 6.0 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.82 (t, J = 6.0 Hz, 2H, NCH2); 2.71–2.54 (m, 4H, NCH2); 2.53–2.42 (m, 4H, NCH2); 2.41–2.34 (m, 2H, NCH2); 2.00–1.94 (m, 2H, CH2); 1.50–1.42 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.97 (C); 161.20 (C); 157.81 (C); 155.85 (C); 143.40 (CH); 137.54 (C); 128.75 (CH); 128.60 (CH); 113.79 (CH); 113.16 (CH); 113.01 (CH); 112.62 (C); 101.50 (CH); 66.55 (CH2); 58.47 (CH2); 56.83 (CH2); 55.22 (OCH3); 53.49 (CH2); 53.01 (CH2); 49.55 (CH); 33.89 (CH2); 25.25 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C33H39N2O5 = 543.2853, found 543.2859. Dihydrochloride: white solid; mp 206–209 °C.
- 7-(3-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 26. Following the general procedure, compound 26 (0.040 g, yield: 57.4%) was synthesized as a yellow oil, starting from 39 [21] (0.050 g, 0.14 mmol) and 43 [20] (0.045 g, 0.17 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 96:4:0.6. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.2 Hz, 1H, CH=CH); 7.34 (d, J = 8.4 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.83–6.70 (m, 6H, CH arom.); 6.23 (d, J = 9.2 Hz, 1H, CH=CH); 4.06 (t, J = 6.0 Hz, 2H, OCH2); 3.79 (t, J = 8.0 Hz, 1H, CH); 3.76 (s, 6H, OCH3); 2.63–2.26 (m, 12H, NCH2); 2.05–1.88 (m, 4H, CH2); 1.50–1.36 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.27 (C); 161.19 (C); 157.83 (C); 155.91 (C); 143.40 (CH); 137.53 (C); 128.71 (CH); 128.60 (CH); 113.80 (CH); 113.01 (CH); 112.94 (CH); 112.47 (C); 101.40 (CH); 66.83 (CH2); 58.45 (CH2); 55.22 (OCH3); 54.82 (CH2); 53.02 (CH2); 49.54 (CH); 33.89 (CH2); 26.43 (CH2); 25.18 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C34H41N2O5 = 557.3010, found 557.3006. Dihydrochloride: white solid; mp 196–199 °C.
- 7-(4-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 27. Following the general procedure, compound 27 (0.040 g, yield: 62.6%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.040 g, 0.11 mmol) and 44 (0.039 g, 0.13 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.2 Hz, 1H, CH=CH); 7.34 (d, J = 8.4 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.81–6.78 (m, 6H, CH arom.); 6.23 (d, J = 9.2 Hz, 1H, CH=CH); 4.01 (t, J = 6.0 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.66–2.33 (m, 12H, NCH2); 2.00–1.94 (m, 2H, CH2); 1.85–1.78 (m, 2H, CH2); 1.70–1.63 (m, 2H, CH2); 1.48–1.42 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.29 (C); 161.28 (C); 157.80 (C); 155.91 (C); 143.46 (CH); 137.56 (C); 128.73 (CH); 128.60 (CH); 113.78 (CH); 112.97 (CH); 112.42 (C); 101.31 (CH); 68.33 (CH2); 58.53 (CH2); 58.07 (CH2); 55.23 (OCH3); 53.06 (CH2); 49.55 (CH); 33.93 (CH2); 26.98 (CH2); 25.30 (CH2); 23.28 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C35H43N2O5 = 571.3166, found 571.3157. Dihydrochloride: white solid; mp 188–191 °C.
- 7-(2-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)ethoxy)-4-methyl-2H-chromen-2-one 28. Following the general procedure, compound 28 (0.070 g, yield: 74.0%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.060 g, 0.17 mmol) and 45 (0.057 g, 0.20 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 97:3:0.3. 1H-NMR (400 MHz, CDCl3) δ: 7.47 (d, J = 8.8 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.85 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.82–6.76 (m, 5H, CH arom.); 6.12 (s, 1H, CH=C); 4.13 (t, J = 5.6 Hz, 2H, OCH2); 3.79 (t, J = 7.6 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.82 (t, J = 5.6 Hz, 2H, NCH2); 2.73–2.51 (m, 4H, NCH2); 2.50–2.31 (m, 9H, CH2 and CH3); 2.00–1.95 (m, 2H, CH2); 1.50–1.38 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.83 (C); 161.21 (C); 157.83 (C); 155.26 (C); 152.44 (C); 137.62 (C); 128.60 (CH); 125.48 (CH); 114.55 (C); 113.79 (CH); 113.66 (C); 112.67 (CH); 112.02 (CH); 101.56 (CH); 66.56 (CH2); 58.54 (CH2); 56.90 (CH2); 55.22 (OCH3); 53.68 (CH2); 53.11 (CH2); 49.59 (CH); 33.95 (CH2); 25.41 (CH2); 18.61 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C34H41N2O5= 557.3010, found 557.3006. Dihydrochloride: white solid; mp 147–150 °C.
- 7-(3-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)propoxy)-4-methyl-2H-chromen-2-one 29. Following the general procedure, compound 29 (0.050 g, yield: 51.8%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.060 g, 0.17 mmol) and 46 (0.059 g, 0.20 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.46 (d, J = 8.8 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.84 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.80–6.76 (m, 5H, CH arom.); 6.11 (s, 1H, CH=C); 4.05 (t, J = 5.6 Hz, 2H, OCH2); 3.79 (t, J = 7.6 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.65–2.30 (m, 15H, CH2 and CH3); 2.00–1.95 (m, 4H, CH2); 1.47–1.40 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.12 (C); 161.27 (C); 157.82 (C); 155.29 (C); 152.51 (C); 137.62 (C); 128.60 (CH); 125.46 (CH); 114.55 (C); 113.79 (CH); 113.49 (C); 112.63 (CH); 111.87 (CH); 101.43 (CH); 66.87 (CH2); 58.56 (CH2); 55.21 (OCH3); 54.90 (CH2); 53.20 (CH2); 49.57 (CH); 33.96 (CH2); 26.52 (CH2); 25.38 (CH2); 18.61 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C35H43N2O5= 571.3166, found 571.3174. Dihydrochloride: white solid; mp 176–178 °C.
- 7-(4-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)butoxy)-4-methyl-2H-chromen-2-one 30. Following the general procedure, compound 30 (0.050 g, yield: 75.7%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.040 g, 0.11 mmol) and 47 (0.042 g, 0.14 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.46 (d, J = 8.8 Hz, 1H, CH arom.); 7.11 (d, J = 8.8 Hz, 4H, CH arom.); 6.87–6.75 (m, 6H, CH arom.); 6.11 (s, 1H, CH=C); 4.02 (t, J = 5.6 Hz, 2H, OCH2); 3.78 (t, J = 7.6 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.50–2.40 (m, 13H, CH2 and CH3); 2.03–1.90 (m, 4H, CH2); 1.86–1.76 (m, 2H, CH2); 1.70–1.60 (m, 2H, CH2); 1.50–1.39 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 162.12 (C); 161.35 (C); 157.80 (C); 155.30 (C); 152.57 (C); 137.61 (C); 128.60 (CH); 125.47 (CH); 113.77 (CH); 113.45 (C); 112.66 (CH); 111.85 (CH); 101.33 (CH); 68.32 (CH2); 58.59 (CH2); 58.14 (CH2); 55.22 (OCH3); 53.19 (CH2); 49.57 (CH); 33.97 (CH2); 27.03 (CH2); 25.40 (CH2); 23.35 (CH2); 18.65 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C36H45N2O5 = 585.3323, found 585.3322. Dihydrochloride: white solid; mp 58–61 °C.
- 6-(2-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)ethoxy)-2H-chromen-2-one 31. Following the general procedure, compound 31 (0.040 g, yield: 65.2%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.040 g, 0.11 mmol) and 48 (0.036 g, 0.14 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 9.6 Hz, 1H, CH=CH); 7.24 (d, J = 8.8 Hz, 1H, CH arom); 7.12–7.08 (m, 5H, CH arom); 6.90 (d, J = 2.8 Hz, 1H, CH arom); 6.79 (d, J = 8.4 Hz, 4H, CH arom); 6.41 (d, J = 9.6 Hz, 1H, CH=CH); 4.10 (t, J = 6.0 Hz, 2H, OCH2); 3.79 (t, J = 8.0 Hz, 1H, CH); 3.75 (s, 6H, OCH3); 2.82 (t, J = 6.0 Hz, 2H, NCH2); 2.74–2.34 (m, 10H, CH2); 2.01–1.95 (m, 2H, CH2); 1.53–1.41 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.94 (C); 157.83 (C); 155.22 (C); 148.54 (C); 143.18 (CH); 137.47 (C); 128.59 (CH); 120.00 (CH); 119.17 (C); 117.87 (CH); 117.11 (CH); 114.52 (C); 113.80 (CH); 111.00 (CH); 66.65 (CH2); 58.41 (CH2); 57.02 (CH2); 55.23 (OCH3); 53.30 (CH2); 52.95 (CH2); 49.52 (CH); 33.84 (CH2); 25.10 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C33H39N2O5 = 543.2853, found 543.2845. Dihydrochloride: yellow solid; mp 238–241 °C.
- 6-(3-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)propoxy)-2H-chromen-2-one 32. Following the general procedure, compound 32 (0.050 g, yield: 79.5%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.040 g, 0.11 mmol) and 49 (0.038 g, 0.14 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.61 (d, J = 9.6 Hz, 1H, CH=CH); 7.23 (d, J = 9.2 Hz, 1H, CH arom); 7.11 (d, J = 8.4 Hz, 4H, CH arom); 7.08 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.89 (d, J = 2.8 Hz, 1H, CH arom); 6.79 (d, J = 8.4 Hz, 4H, CH arom); 6.39 (d, J = 9.6 Hz, 1H, CH=CH); 4.01 (t, J = 6.4 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.74 (s, 6H, OCH3); 2.68–2.32 (m, 12H, CH2); 2.00–1.94 (m, 4H, CH2); 1.48–1.40 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.01 (C); 157.80 (C); 155.48 (C); 148.38 (C); 143.25 (CH); 137.55 (C); 128.60 (CH); 119.93 (CH); 119.16 (C); 117.80 (CH); 117.01 (CH); 114.55 (C); 113.78 (CH); 110.79 (CH); 66.96 (CH2); 58.54 (CH2); 55.23 (OCH3); 55.02 (CH2); 53.12 (CH2); 49.56 (CH); 33.92 (CH2); 26.65 (CH2); 25.33 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C34H41N2O5 = 557.3010, found 557.3019. Dihydrochloride: yellow solid; mp 225–227 °C.
- 6-(4-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)butoxy)-2H-chromen-2-one 33. Following the general procedure, compound 33 (0.050 g, yield: 51.8%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.060 g, 0.17 mmol) and 50 (0.060 g, 0.20 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 9.6 Hz, 1H, CH=CH); 7.23 (d, J = 8.8 Hz, 1H, CH arom); 7.11 (d, J = 8.8 Hz, 4H, CH arom); 7.07 (dd, J = 8.8, 2.8 Hz, 1H, CH arom); 6.88 (d, J = 2.8 Hz, 1H, CH arom); 6.79 (d, J = 8.8 Hz, 4H, CH arom); 6.40 (d, J = 9.6 Hz, 1H, CH=CH); 3.97 (t, J = 6.0 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.74 (s, 6H, OCH3); 2.68–2.12 (m, 12H, CH2); 2.00–1.94 (m, 2H, CH2); 1.82–1.77 (m, 2H, CH2); 1.70–1.62 (m, 2H, CH2); 1.47–1.39 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 161.02 (C); 157.80 (C); 155.50 (C); 148.36 (C); 143.25 (CH); 137.57 (C); 128.60 (CH); 119.89 (CH); 119. 16 (C); 117.83 (CH); 117.03 (CH); 114.52 (C); 113.77 (CH); 110.73 (CH); 68.43 (CH2); 58.57 (CH2); 58.17 (CH2); 55.23 (OCH3); 53.13 (CH2); 49.57 (CH); 33.94 (CH2); 27.20 (CH2); 25.37 (CH2); 23.39 (CH2) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C35H43N2O5 = 571.3166, found 571.3156. Dihydrochloride: yellow solid; mp 234–236 °C.
- 6-(2-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)ethoxy)-4-methyl-2H-chromen-2-one 34. Following the general procedure, compound 34 (0.070 g, yield: 63.8%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.070 g, 0.20 mmol) and 51 (0.067 g, 0.24 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.23 (d, J = 8.8 Hz, 1H, CH arom); 7.12–7.07 (m, 5H, CH arom); 7.03 (d, J = 2.8 Hz, 1H, CH arom); 6.79 (d, J = 8.8 Hz, 4H, CH arom); 6.27 (s, 1H, CH=C); 4.11 (t, J = 6.0 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.74 (s, 6H, OCH3); 2.81 (t, J = 6.0 Hz, 2H, NCH2); 2.70–2.52 (m, 4H, NCH2); 2.51–2.32 (m, 9H, CH2 and CH3); 2.00–1.94 (m, 2H, CH2); 1.47–1.42 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.94 (C); 157.81 (C); 155.16 (C); 151.97 (C); 147.95 (C); 137.58 (C); 128.59 (CH); 120.44 (C); 119.24 (CH); 117.92 (CH); 115.48 (CH); 114.54 (C); 113.79 (CH); 108.67 (CH); 66.62 (CH2); 58.53 (CH2); 57.15 (CH2); 55.22 (OCH3); 53.64 (CH2); 53.08 (CH2); 49.56 (CH); 33.92 (CH2); 25.36 (CH2); 18.72 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C34H41N2O5 = 557.3010, found 557.3017. Dihydrochloride: yellow solid; mp 218–221 °C.
- 6-(3-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)propoxy)-4-methyl-2H-chromen-2-one 35. Following the general procedure, compound 35 (0.070 g, yield: 72.6%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.060 g, 0.17 mmol) and 52 (0.060 g, 0.20 mmol) in 5.0 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.22 (d, J = 9.2 Hz, 1H, CH arom); 7.12–7.06 (m, 5H, CH arom); 6.99 (d, J = 2.8 Hz, 1H, CH arom); 6.79 (d, J = 8.8 Hz, 4H, CH arom); 6.26 (s, 1H, CH=C); 4.03 (t, J = 6.4 Hz, 2H, OCH2); 3.78 (t, J = 8.0 Hz, 1H, CH); 3.74 (s, 6H, OCH3); 2.55–2.31 (m, 15H, CH2 and CH3); 2.00–1.94 (m, 4H, CH2); 1.47–1.39 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 160.95 (C); 157.81 (C); 155.40 (C); 152.01 (C); 147.82 (C); 137.60 (C); 128.60 (CH); 120.43 (C); 119.13 (CH); 117.87 (CH); 115.42 (CH); 114.56 (C); 113.78 (CH); 108.53 (CH); 67.05 (CH2); 58.57 (CH2); 55.21 (OCH3); 55.07 (CH2); 53.26 (CH2); 53.18 (CH2); 49.57 (CH); 33.96 (CH2); 26.74 (CH2); 25.39 (CH2); 18.71 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C35H43N2O5 = 571.3166, found 571.3163. Dihydrochloride: yellow solid; mp 236–239 °C.
- 6-(4-(4-(4,4-Bis(4-methoxyphenyl)butyl)piperazin-1-yl)butoxy)-4-methyl-2H-chromen-2-one 36. Following the general procedure, compound 36 (0.030 g, yield: 45.4%) was synthesized as a pale-yellow oil, starting from 39 [21] (0.040 g, 0.11 mmol) and 53 (0.042 g, 0.14 mmol) in 3.4 mL of dry acetonitrile. Free base: chromatographic eluent: CH2Cl2/2-propanol/NH4OH 95:5:0.5. TLC: CH2Cl2/CH3OH/NH4OH 95:5:0.5. 1H-NMR (400 MHz, CDCl3) δ: 7.25 (d, J = 9.2 Hz, 1H, CH arom); 7.12–7.08 (m, 5H, CH arom); 6.99 (d, J = 2.8 Hz, 1H, CH arom); 6.80 (d, J = 8.8 Hz, 4H, CH arom); 6.29 (s, 1H, CH=C); 4.00 (t, J = 6.4 Hz, 2H, OCH2); 3.78 (t, J = 6.4 Hz, 1H, CH); 3.76 (s, 6H, OCH3); 2.66–2.41 (m, 15H, CH2 and CH3); 2.00–1.95 (m, 2H, CH2); 1.85–1.78 (m, 2H, CH2); 1.75–1.69 (m, 2H, CH2); 1.50–1.42 (m, 2H, CH2) ppm. 13C-NMR (100 MHz, CDCl3) δ: 157.84 (C); 155.36 (C); 151.97 (C); 147.86 (C); 137.43 (C); 128.57 (CH); 120.46 (C); 119.06 (CH); 117.92 (CH); 115.47 (CH); 113.81 (CH); 108.46 (CH); 68.36 (CH2); 58.20 (CH2); 57.87 (CH2); 55.22 (OCH3); 52.51 (CH2); 49.47 (CH); 33.80 (CH2); 27.18 (CH2); 24.91 (CH2); 23.05 (CH2); 18.72 (CH3) ppm. ESI-HRMS (m/z) calculated for [M+H]+ ion species C36H45N2O5 = 585.3323, found 585.3313. Dihydrochloride: yellow solid; mp 240–242 °C.
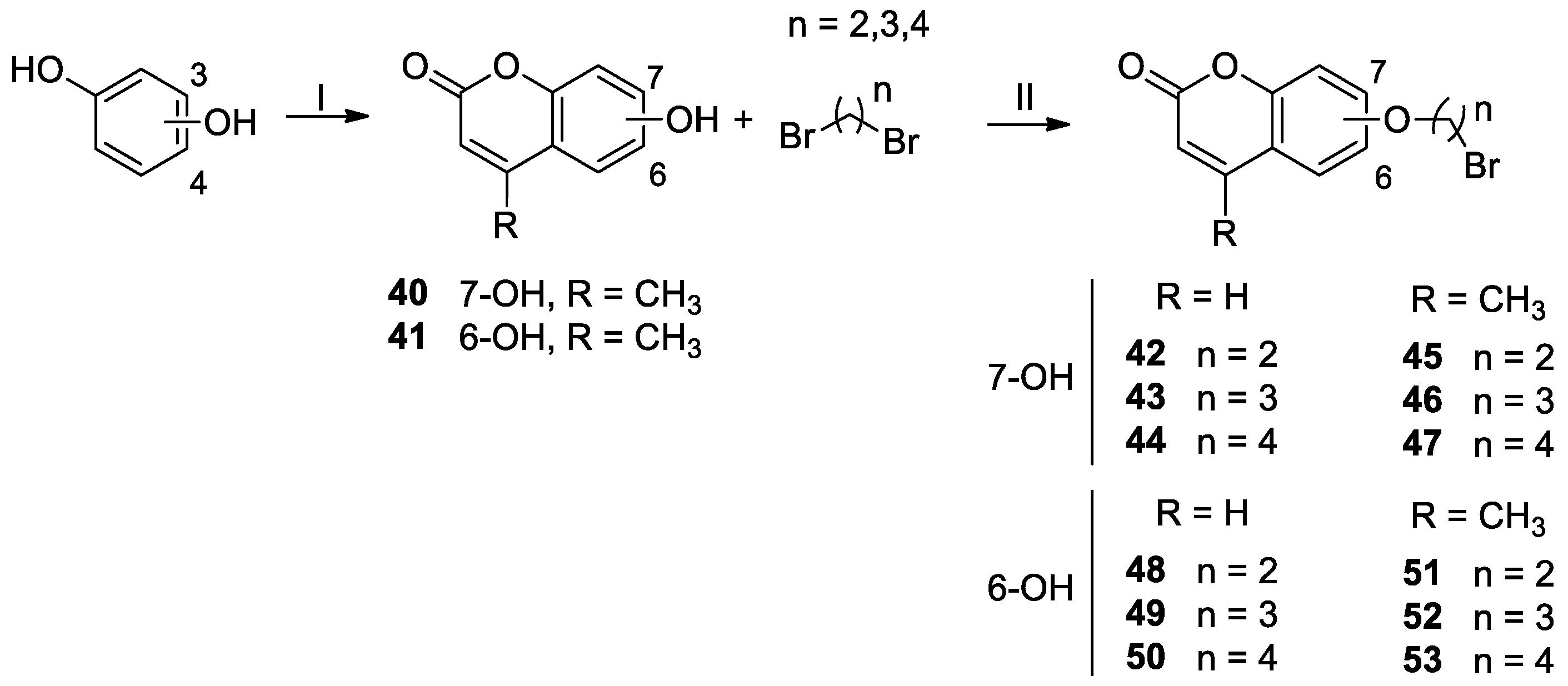
4.1.2. 7-Hydroxy-4-methyl-2H-chromen-2-one 40 [30]
4.1.3. 6-Hydroxy-4-methyl-2H-chromen-2-one 41 [31]
4.1.4. General Procedure for the Synthesis of the Bromoalkoxy-2H-chromen-2-ones 42 and 44–53
- 7-(2-Bromoethoxy)-2H-chromen-2-one 42 [25]. Following the general procedure, compound 42 (0.30 g, yield: 90.7%) was synthesized as a white solid, from 7-hydroxy-2H-chromen-2-one (0.20 g, 1.23 mmol) and 1,2-dibromoethane (0.61 mL, 6.50 mmol) in 15.0 mL of acetone. TLC: CH2Cl2/CH3OH 90:10. Mp: 134–136 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.63 (d, J = 9.2 Hz, 1H, CH=CH); 7.38 (d, J = 8.4 Hz, 1H, CH arom.); 6.86 (dd, J = 8.4, 2.4 Hz, 1H, CH arom.); 6.80 (d, J = 2.4 Hz, 1H, CH arom.); 6.26 (d, J = 9.2 Hz, 1H, CH=CH); 4.35 (t, J = 6.4 Hz, 2H, OCH2); 3.67 (t, J = 6.4 Hz, 2H, CH2Br) ppm.
- 7-(4-Bromobutoxy)-2H-chromen-2-one 44 [25]. Following the general procedure, compound 44 (0.33 g, yield: 86.0%) was synthesized as a white solid, from 7-hydroxy-2H-chromen-2-one (0.21 g, 1.30 mmol) and 1,4-dibromobutane (0.77 mL, 6.50 mmol) in 15.0 mL of acetone. TLC: CH2Cl2/CH3OH 90:10. Mp: 105–107 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.62 (d, J = 9.2 Hz, 1H, CH=CH); 7.36 (d, J = 8.4 Hz, 1H, CH arom.); 6.82 (dd, J = 8.4, 2.4 Hz, 1H, CH arom.); 6.78 (d, J = 2.4 Hz, 1H, CH arom.); 6.24 (d, J = 9.2 Hz, 1H, CH=CH); 4.04 (t, J = 6.0 Hz, 2H, OCH2); 3.49 (t, J = 6.0 Hz, 2H, CH2Br); 2.10–2.03 (m, 2H, CH2); 2.02–1.96 (m, 2H, CH2) ppm.
- 7-(2-Bromoethoxy)-4-methyl-2H-chromen-2-one 45 [26]. Following the general procedure, compound 45 (0.13 g, yield: 62.4%) was synthesized as a white solid, from 40 (0.13 g, 0.74 mmol) and 1,2-dibromoethane (0.51 mL, 5.95 mmol) in 10.0 mL of acetone. TLC: CH2Cl2/CH3OH 90:10. Mp: 139–141 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.51 (d, J = 8.8 Hz, 1H, CH arom.); 6.91 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.81 (d, J = 2.4 Hz, 1H, CH arom.); 6.15 (s, 1H, CH=C); 4.35 (t, J = 6.0 Hz, 2H, OCH2); 3.61 (t, J = 6.0 Hz, 2H, CH2Br); 2.39 (s, 3H, CH3) ppm.
- 7-(3-Bromopropoxy)-4-methyl-2H-chromen-2-one 46 [26]. Following the general procedure, compound 46 (0.19 g, yield: 74.8%) was synthesized as a white solid, from 40 (0.15 g, 0.85 mmol) and 1,3-dibromopropane (0.43 mL, 4.26 mmol) in 11.0 mL of acetone. TLC: CH2Cl2/CH3OH 90:10. Mp: 75–78 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.50 (d, J = 8.8 Hz, 1H, CH arom.); 6.86 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.82 (d, J = 2.4 Hz, 1H, CH arom.); 6.13 (s, 1H, CH=C); 4.23 (t, J = 6.0 Hz, 2H, OCH2); 3.61 (t, J = 6.0 Hz, 2H, CH2Br); 2.39 (s, 3H, CH3); 2.36–2.32 (m, 2H, CH2) ppm.
- 7-(4-Bromobutoxy)-4-methyl-2H-chromen-2-one 47 [26]. Following the general procedure, compound 47 (0.16 g, yield: 76.3%) was synthesized as a white solid, from 40 (0.12 g, 0.68 mmol) and 1,4-dibromobutane (0.40 mL, 3.40 mmol) in 9.0 mL of acetone. TLC: CH2Cl2/CH3OH 90:10. Mp: 63–65 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.49 (d, J = 8.4 Hz, 1H, CH arom.); 6.85 (dd, J = 8.8, 2.4 Hz, 1H, CH arom.); 6.79 (d, J = 2.4 Hz, 1H, CH arom.); 6.13 (s, 1H, CH=C); 4.05 (t, J = 6.0 Hz, 2H, OCH2); 3.49 (t, J = 6.0 Hz, 2H, CH2Br); 2.39 (s, 3H, CH3); 2.09–2.03 (m, 2H, CH2); 2.02–1.96 (m, 2H, CH2) ppm.
- 6-(2-Bromoethoxy)-2H-chromen-2-one 48 [27]. Following the general procedure, compound 48 (0.26 g, yield: 64.7%) was synthesized as a white solid, from 6-hydroxy-2H-chromen-2-one (0.25 g, 1.51 mmol) and 1,2-dibromoethane (0.66 mL, 7.72 mmol) in 10.0 mL of acetonitrile. TLC: CH2Cl2/CH3OH 95:5. Mp: 96–98 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.64 (d, J = 9.6 Hz, 1H, CH=CH); 7.27 (d, J = 9.2 Hz, 1H, CH arom.); 7.13 (dd, J = 9.2, 2.8 Hz, 1H, CH arom.); 6.95 (d, J = 2.8 Hz, 1H, CH arom.); 6.44 (d, J = 9.6 Hz, 1H, CH=CH); 4.32 (t, J = 5.6 Hz, 2H, OCH2); 3.66 (t, J = 5.6 Hz, 2H, CH2Br) ppm.
- 6-(3-Bromopropoxy)-2H-chromen-2-one 49 [28]. Following the general procedure, compound 49 (0.15 g, yield: 74.6%) was synthesized as a white solid, from 6-hydroxy-2H-chromen-2-one (0.12 g, 0.71 mmol) and 1,3-dibromopropane (0.36 mL, 3.57 mmol) in 10.0 mL of acetonitrile. TLC: CH2Cl2/CH3OH 95:5. Mp: 97–99 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.65 (d, J = 9.6 Hz, 1H, CH=CH); 7.27 (d, J = 9.2 Hz, 1H, CH arom.); 7.11 (dd, J = 9.2, 2.8 Hz, 1H, CH arom.); 6.93 (d, J = 2.8 Hz, 1H, CH arom.); 6.43 (d, J = 9.6 Hz, 1H, CH=CH); 4.14 (t, J = 5.6 Hz, 2H, OCH2); 3.62 (t, J = 5.6 Hz, 2H, CH2Br); 2.37–2.31 (m, 2H, CH2) ppm.
- 6-(4-Bromobutoxy)-2H-chromen-2-one 50 [28]. Following the general procedure, compound 50 (0.20 g, yield: 87.6%) was synthesized as a white solid, from 6-hydroxy-2H-chromen-2-one (0.13 g, 0.77 mmol) and 1,4-dibromobutane (0.46 mL, 3.85 mmol) in 10.0 mL of acetonitrile. TLC: CH2Cl2/CH3OH 95:5. Mp: 114–116 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.64 (d, J = 9.6 Hz, 1H, CH=CH); 7.26 (d, J = 9.2 Hz, 1H, CH arom); 7.09 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 6.90 (d, J = 2.8 Hz, 1H, CH arom); 6.43 (d, J = 9.6 Hz, 1H, CH=CH); 4.02 (t, J = 6.4 Hz, 2H, CH2O); 3.50 (t, J = 6.4 Hz, 2H, CH2Br); 2.10–2.06 (m, 2H, CH2); 2.00–1.95 (m, 2H, CH2) ppm.
- 6-(2-Bromoethoxy)-4-methyl-2H-chromen-2-one 51 [29]. Following the general procedure, compound 51 (0.34 g, yield: 64.2%) was synthesized as a yellow solid, from 41 (0.33 g, 1.87 mmol) and 1,2-dibromoethane (0.80 mL, 9.37 mmol) in 50.0 mL of acetone. TLC: CH2Cl2/CH3OH 95:5. Mp: 133–135 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.26 (d, J = 9.2 Hz, 1H, CH arom); 7.14 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 7.07 (d, J = 2.8 Hz, 1H, CH arom); 6.31 (s, 1H, CH=C); 4.34 (t, J = 6.0 Hz, 2H, CH2O); 3.66 (t, J = 6.0 Hz, 2H, CH2Br); 2.42 (s, 3H, CH3) ppm.
- 6-(3-Bromopropoxy)-4-methyl-2H-chromen-2-one 52 [29]. Following the general procedure, compound 52 (0.53 g, yield: 96.0%) was synthesized as a white solid, from 41 (0.33 g, 1.87 mmol) and 1,3-dibromopropane (0.95 mL, 9.37 mmol) in 50.0 mL of acetone. TLC: CH2Cl2/CH3OH 95:5. Mp: 90–92 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.27 (d, J = 8.8 Hz, 1H, CH arom); 7.12 (dd, J = 8.8, 2.8 Hz, 1H, CH arom); 7.04 (d, J = 2.8 Hz, 1H, CH arom); 6.30 (s, 1H, CH=C); 4.16 (t, J = 6.0 Hz, 2H, CH2O); 3.63 (t, J = 6.0 Hz, 2H, CH2Br); 2.40 (s, 3H, CH3); 2.36–2.32 (m, 2H, CH2) ppm.
- 6-(4-Bromobutoxy)-4-methyl-2H-chromen-2-one 53 [29]. Following the general procedure, compound 53 (0.53 g, yield: 91.1%) was synthesized as a white solid, from 41 (0.33 g, 1.87 mmol) and 1,4-dibromobutane (1.12 mL, 9.37 mmol) in 50.0 mL of acetone. TLC: CH2Cl2/CH3OH 95:5. Mp: 100–102 °C. 1H-NMR (400 MHz, CDCl3) δ: 7.26 (d, J = 9.2 Hz, 1H, CH arom); 7.09 (dd, J = 9.2, 2.8 Hz, 1H, CH arom); 7.01 (d, J = 2.8 Hz, 1H, CH arom); 6.30 (s, 1H, CH=C); 4.04 (t, J = 6.4 Hz, 2H, CH2O); 3.50 (t, J = 6.4 Hz, 2H, CH2Br); 2.42 (s, 3H, CH3); 2.11–2.07 (m, 2H, CH2); 2.01–1.96 (m, 2H, CH2) ppm.
4.2. Biology and Enzymology
4.2.1. CA Inhibition Assay
4.2.2. Cell Lines and Cultures
4.2.3. Drugs and Chemicals
4.2.4. Intrinsic Cytotoxicity
4.2.5. Co-Administration Assays in K562/DOX, HT29, A549, HT29/DOX and A549/DOX Cells
4.2.6. Intracellular Doxorubicin Accumulation in HT29, A549, HT29/DOX and A549/DOX Cells
4.2.7. Calcein-AM Experiment
4.2.8. Hoechst 33342 Experiment
4.2.9. Oxidative Stress Measurement
4.2.10. P-gp ATPase Activity
4.2.11. Membrane Fluidity
4.2.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gottesman, M.M. Mechanisms of Cancer Drug Resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug Resistance in Cancer: Role of ATP-Dependent Transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Annereau, J.P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting Drug Sensitivity and Resistance: Profiling ABC Transporter Genes in Cancer Cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Gupta, P.; Ashby, C.R.; Chen, Z.S. The Modulation of ABC Transporter-Mediated Multidrug Resistance in Cancer: A Review of the Past Decade. Drug Resist. Updat. 2015, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three Decades of P-gp Inhibitors: Skimming Through Several Generations and Scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC Transporter-Mediated Multidrug Resistance: Molecular Mechanisms and Novel Therapeutic Drug Strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, H.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.S.; Liu, H.M. Chemical Molecular-Based Approach to Overcome Multidrug Resistance in Cancer by Targeting P-Glycoprotein (P-gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef]
- Coley, H.M. Overcoming Multidrug Resistance in Cancer: Clinical Studies of P-Glycoprotein Inhibitors. Methods Mol. Biol. 2010, 596, 341–358. [Google Scholar]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A Pharmacodynamic Study of Docetaxel in Combination with the P-Glycoprotein Antagonist Tariquidar (XR9576) in Patients with Lung, Ovarian, and Cervical Cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef]
- Ueda, K. ABC Proteins Protect the Human Body and Maintain Optimal Health. Biosci. Biotechnol. Biochem. 2011, 75, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Darby, R.A.J.; Callaghan, R.; McMahon, R.M. P-Glycoprotein Inhibition: The Past, the Present and the Future. Curr. Drug Metab. 2011, 12, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T. Carbonic Anhydrase Inhibitors as Antitumor/Antimetastatic Agents: A Patent Review (2008–2018). Expert Opin. Ther. Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Hynninen, P.; Vaskivuo, L.; Saarnio, J.; Haapasalo, H.; Kivelä, J.; Pastoreková, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Puistola, U.; et al. Expression of Transmembrane Carbonic Anhydrases IX and XII in Ovarian Tumours. Histopathology 2006, 49, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Rafalko, A.; Iliopoulos, O.; Fusaro, V.A.; Hancock, W.; Hincapie, M. Immunoaffinity Enrichment and Liquid Chromatography-Selected Reaction Monitoring Mass Spectrometry for Quantitation of Carbonic Anhydrase 12 in Cultured Renal Carcinoma Cells. Anal. Chem. 2010, 82, 8998–9005. [Google Scholar] [CrossRef]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Anticancer Carbonic Anhydrase Inhibitors: A Patent Review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibitors as Emerging Agents for the Treatment and Imaging of Hypoxic Tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef]
- Chiche, J.; Ilc, K.; Laferrière, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef]
- Teodori, E.; Braconi, L.; Bua, S.; Lapucci, A.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Dei, S.; Supuran, C.T.; Coronnello, M. Dual P-Glycoprotein and CA XII Inhibitors: A New Strategy to Reverse the P-gp Mediated Multidrug Resistance (MDR) in Cancer Cells. Molecules 2020, 25, 1748. [Google Scholar] [CrossRef]
- Braconi, L.; Teodori, E.; Riganti, C.; Coronnello, M.; Nocentini, A.; Bartolucci, G.; Pallecchi, M.; Contino, M.; Manetti, D.; Romanelli, M.N.; et al. New Dual P-Glycoprotein (P-gp) and Human Carbonic Anhydrase XII (hCA XII) Inhibitors as Multidrug Resistance (MDR) Reversers in Cancer Cells. J. Med. Chem. 2022, 65, 14655–14672. [Google Scholar] [CrossRef]
- Dei, S.; Coronnello, M.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Udomtanakunchai, C.; Salerno, M.; Teodori, E. Design and Synthesis of New Potent N,N-Bis(Arylalkyl)Piperazine Derivatives as Multidrug Resistance (MDR) Reversing Agents. Eur. J. Med. Chem. 2018, 147, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Buran, K.; Bua, S.; Poli, G.; Bayram, F.E.Ö.; Tuccinardi, T.; Supuran, C.T. Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII. Int. J. Mol. Sci. 2019, 20, 1208. [Google Scholar] [CrossRef]
- De Luca, L.; Mancuso, F.; Ferro, S.; Buemi, M.R.; Angeli, A.; Del Prete, S.; Capasso, C.; Supuran, C.T.; Gitto, R. Inhibitory Effects and Structural Insights for a Novel Series of Coumarin-Based Compounds That Selectively Target Human CA IX and CA XII Carbonic Anhydrases. Eur. J. Med. Chem. 2018, 143, 276–282. [Google Scholar] [CrossRef]
- Yalçintepe, L.; Halis, E.; Ulku, S. Effect of CD38 on the Multidrug Resistance of Human Chronic Myelogenous Leukemia K562 Cells to Doxorubicin. Oncol. Lett. 2016, 11, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.L.; Cai, P.; Liu, Q.H.; Yang, X.L.; Li, F.; Wang, J.; Wu, J.J.; Wang, X.B.; Kong, L.Y. Design, Synthesis and Evaluation of Coumarin-Pargyline Hybrids as Novel Dual Inhibitors of Monoamine Oxidases and Amyloid-β Aggregation for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2017, 138, 715–728. [Google Scholar] [CrossRef]
- Jiang, N.; Huang, Q.; Liu, J.; Liang, N.; Li, Q.; Li, Q.; Xie, S.S. Design, Synthesis and Biological Evaluation of New Coumarin-Dithiocarbamate Hybrids as Multifunctional Agents for the Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2018, 146, 287–298. [Google Scholar] [CrossRef]
- Pisani, L.; Catto, M.; Giangreco, I.; Leonetti, F.; Nicolotti, O.; Stefanachi, A.; Cellamare, S.; Carotti, A. Design, Synthesis, and Biological Evaluation of Coumarin Derivatives Tethered to an Edrophonium-like Fragment as Highly Potent and Selective Dual Binding Site Acetylcholinesterase Inhibitors. ChemMedChem 2010, 5, 1616–1630. [Google Scholar] [CrossRef]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl Derivatives of Bi- and Tricyclic Systems as Potent Inhibitors of Acetyl- and Butyrylcholinesterase with Potential in Alzheimer’s Disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Thakar, K.A.; Pathak, R.V.; Dumir, A.B. Synthesis of Unsymmetric Bis-Coumarinoxy-Alkanes. J. Indian Chem. Soc. 1980, 57, 89–91. [Google Scholar]
- Narella, S.G.; Shaik, M.G.; Mohammed, A.; Alvala, M.; Angeli, A.; Supuran, C.T. Synthesis and Biological Evaluation of Coumarin-1,3,4-Oxadiazole Hybrids as Selective Carbonic Anhydrase IX and XII Inhibitors. Bioorg. Chem. 2019, 87, 765–772. [Google Scholar] [CrossRef]
- Romanelli, G.P.; Bennardi, D.; Ruiz, D.M.; Baronetti, G.; Thomas, H.J.; Autino, J.C. A Solvent-Free Synthesis of Coumarins Using a Wells-Dawson Heteropolyacid as Catalyst. Tetrahedron Lett. 2004, 45, 8935–8939. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Supuran, C.T. How Many Carbonic Anhydrase Inhibition Mechanisms Exist? J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Lozzio, C.; Lozzio, B. Human Chronic Myelogenous Leukemia Cell Line with Positive Philadelphia Chromosome. Blood 1975, 45, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.; Boyd, M.R. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Riganti, C.; Kopecka, J.; Panada, E.; Barak, S.; Rubinstein, M. The Role of C/EBP-β LIP in Multidrug Resistance. J. Natl. Cancer Inst. 2015, 107, djv046. [Google Scholar] [CrossRef]
- Qian, J.; Cui, J.; Li, S.; Chen, J.; Jia, J. Anticancer Natural Products with Collateral Sensitivity: A Review. Mini-Rev. Med. Chem. 2021, 21, 1465–1486. [Google Scholar] [CrossRef] [PubMed]
- Furedi, A.; Toth, S.; Szebenyi, K.; Pape, V.F.S.; Türk, D.; Kucsma, N.; Cervenak, L.; Tovari, J.; Szakacs, G. Identification and Validation of Compounds Selectively Killing Resistant Cancer: Delineating Cell Line-Specific Effects from P-Glycoprotein-Induced Toxicity. Mol. Cancer Ther. 2017, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Türk, D.; Hall, M.D.; Chu, B.F.; Ludwig, J.A.; Fales, H.M.; Gottesman, M.M.; Szakács, G. Identification of Compounds Selectively Killing Multidrug-Resistant Cancer Cells. Cancer Res. 2009, 69, 8293–8301. [Google Scholar] [CrossRef]
- Podolski-Renić, A.; Čipak Gašparović, A.; Valente, A.; López, Ó.; Bormio Nunes, J.H.; Kowol, C.R.; Heffeter, P.; Filipović, N.R. Schiff Bases and Their Metal Complexes to Target and Overcome (Multidrug) Resistance in Cancer. Eur. J. Med. Chem. 2024, 270, 116363. [Google Scholar] [CrossRef]
- Szakács, G.; Hall, M.D.; Gottesman, M.M.; Boumendjel, A.; Kachadourian, R.; Day, B.J.; Baubichon-Cortay, H.; Di Pietro, A. Targeting the Achilles Heel of Multidrug-Resistant Cancer by Exploiting the Fitness Cost of Resistance. Chem. Rev. 2014, 114, 5753–5774. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, K.M.; Hall, M.D.; Goldsborough, A.S.; Callaghan, R.; Gottesman, M.M. Collateral Sensitivity as a Strategy against Cancer Multidrug Resistance. Drug Resist. Updat. 2012, 15, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Marshall, T.S.; Kwit, A.D.T.; Miller Jenkins, L.M.; Dulcey, A.E.; Madigan, J.P.; Pluchino, K.M.; Goldsborough, A.S.; Brimacombe, K.R.; Griffiths, G.L.; et al. Inhibition of Glutathione Peroxidase Mediates the Collateral Sensitivity of Multidrug-Resistant Cells to Tiopronin. J. Biol. Chem. 2014, 289, 21473–21489. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Legato, M.; Umbach, A.; Riganti, C.; Jones, R.; Martini, B.; Boido, M.; Medana, C.; Facchinetti, I.; Barni, D.; et al. Treatment with ROS Detoxifying Gold Quantum Clusters Alleviates the Functional Decline in a Mouse Model of Friedreich Ataxia. Sci. Transl. Med. 2021, 13, eabe1633. [Google Scholar] [CrossRef] [PubMed]
- Nikitjuka, A.; Ozola, M.; Jackevica, L.; Bobrovs, R.; Žalubovskis, R. Exploration of 3,4-Unsubstituted Coumarins as Thioredoxin Reductase 1 Inhibitors for Cancer Therapy. Org. Biomol. Chem. 2023, 21, 9630–9639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Dong, J.; Gao, J. Design, Synthesis and Bioactivity Evaluation of Coumarin-Chalcone Hybrids as Potential Anticancer Agents. Bioorg. Chem. 2020, 95, 103530. [Google Scholar] [CrossRef]
- Lai, L.; Tan, M.; Hu, M.; Yue, X.; Tao, L.; Zhai, Y.; Li, Y. Important Molecular Mechanisms in Ferroptosis. Mol. Cell. Biochem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist. Updat. 2020, 49, 100670. [Google Scholar] [CrossRef] [PubMed]
- Gelsomino, G.; Corsetto, P.A.; Campia, I.; Montorfano, G.; Kopecka, J.; Castella, B.; Gazzano, E.; Ghigo, D.; Rizzo, A.M.; Riganti, C. Omega 3 Fatty Acids Chemosensitize Multidrug Resistant Colon Cancer Cells by Down-Regulating Cholesterol Synthesis and Altering Detergent Resistant Membranes Composition. Mol. Cancer 2013, 12, 137. [Google Scholar] [CrossRef]
- Kopecka, J.; Campia, I.; Olivero, P.; Pescarmona, G.; Ghigo, D.; Bosia, A.; Riganti, C. A LDL-Masked Liposomal-Doxorubicin Reverses Drug Resistance in Human Cancer Cells. J. Control. Release 2011, 149, 196–205. [Google Scholar] [CrossRef]
- Braconi, L.; Dei, S.; Contino, M.; Riganti, C.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Perrone, M.G.; Colabufo, N.A.; Guglielmo, S.; et al. Tetrazole and Oxadiazole Derivatives as Bioisosteres of Tariquidar and Elacridar: New Potent P-gp Modulators Acting as MDR Reversers. Eur. J. Med. Chem. 2023, 259, 115716. [Google Scholar] [CrossRef]
- Marshall, A.G.; Hendrickson, C.L. High-Resolution Mass Spectrometers. Annu. Rev. Anal. Chem. 2008, 1, 579–599. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Tars, K.; Vullo, D.; Kazaks, A.; Leitans, J.; Lends, A.; Grandane, A.; Zalubovskis, R.; Scozzafava, A.; Supuran, C.T. Sulfocoumarins (1,2-Benzoxathiine-2,2-Dioxides): A Class of Potent and Isoform-Selective Inhibitors of Tumor-Associated Carbonic Anhydrases. J. Med. Chem. 2013, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Gazzano, E.; Gulino, G.R.; Volante, M.; Ghigo, D.; Kopecka, J. Two Repeated Low Doses of Doxorubicin Are More Effective than a Single High Dose against Tumors Overexpressing P-Glycoprotein. Cancer Lett. 2015, 360, 219–226. [Google Scholar] [CrossRef]
- Kopecka, J.; Salzano, G.; Campia, I.; Lusa, S.; Ghigo, D.; De Rosa, G.; Riganti, C. Insights in the Chemical Components of Liposomes Responsible for P-Glycoprotein Inhibition. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 77–87. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| KI (nM) 1,2 | RF 3 | ||||||||
| Cmpd | n | X | Ar | CA I | CA II | CA IX | CA XII | 1 µM | 3 µM |
| 1 | 2 | I | a | >10,000 | >10,000 | 51.3 | 13.0 | 1.6 | 2.3 |
| 2 | 3 | I | a | >10,000 | >10,000 | 62.6 | 29.4 | 3.8 | 4.5 |
| 3 | 4 | I | a | >10,000 | >10,000 | 82.0 | 34.6 | 3.1 | 3.7 |
| 4 | 2 | II | a | >10,000 | >10,000 | 25.2 | 32.8 | 5.7 | 15.8 |
| 5 | 3 | II | a | >10,000 | >10,000 | 34.7 | 27.2 | 5.5 | 27.6 |
| 6 | 4 | II | a | >10,000 | >10,000 | 58.9 | 44.8 | 6.3 | 6.7 |
| 7 | 2 | III | a | >10,000 | >10,000 | 51.0 | 29.5 | 1.1 | 1.0 |
| 8 | 3 | III | a | >10,000 | >10,000 | 40.7 | 22.9 | 2.0 | 2.0 |
| 9 | 4 | III | a | >10,000 | >10,000 | 22.1 | 7.3 | 2.5 | 1.3 |
| 10 | 2 | IV | a | >10,000 | >10,000 | 360 | 422 | 1.0 | 1.4 |
| 11 | 3 | IV | a | >10,000 | >10,000 | 302 | 441 | 2.4 | 4.1 |
| 12 | 4 | IV | a | >10,000 | >10,000 | 104 | 140 | 1.9 | 2.7 |
| 13 | 2 | I | b | >10,000 | >10,000 | 23.7 | 6.2 | 1.0 | 1.9 |
| 14 | 3 | I | b | >10,000 | >10,000 | 40.1 | 13.6 | 1.0 | 3.4 |
| 15 | 4 | I | b | >10,000 | >10,000 | 29.0 | 42.9 | 1.2 | 2.3 |
| 16 | 2 | II | b | >10,000 | >10,000 | 8.2 | 51.9 | 1.0 | 1.4 |
| 17 | 3 | II | b | >10,000 | >10,000 | 16.1 | 60.8 | 2.6 | 2.1 |
| 18 | 4 | II | b | >10,000 | >10,000 | 28.5 | 72.1 | 1.9 | 3.2 |
| 19 | 2 | III | b | >10,000 | >10,000 | 84.7 | 5.8 | 1.4 | 2.2 |
| 20 | 3 | III | b | >10,000 | >10,000 | 72.0 | 22.4 | 2.3 | 2.4 |
| 21 | 4 | III | b | >10,000 | >10,000 | 50.3 | 15.9 | 3.9 | 4.1 |
| 22 | 2 | IV | b | >10,000 | >10,000 | 409 | 229 | 2.8 | 2.7 |
| 23 | 3 | IV | b | >10,000 | >10,000 | 242 | 304 | 1.6 | 1.8 |
| 24 | 4 | IV | b | >10,000 | >10,000 | 205 | 99.5 | 1.4 | 2.5 |
| 25 | 2 | I | c | >10,000 | >10,000 | 113 | 84.7 | 5.9 | 21.1 |
| 26 | 3 | I | c | >10,000 | >10,000 | 153 | 115 | 5.5 | 17.3 |
| 27 | 4 | I | c | >10,000 | >10,000 | 145 | 92.1 | 8.1 | 90.5 |
| 28 | 2 | II | c | >10,000 | >10,000 | 135 | 103 | 16.2 | 32.0 |
| 29 | 3 | II | c | >10,000 | >10,000 | 149 | 91.5 | 34.3 | 38.7 |
| 30 | 4 | II | c | >10,000 | >10,000 | 124 | 82.4 | 7.7 | 25.5 |
| 31 | 2 | III | c | >10,000 | >10,000 | 115 | 90.7 | 3.0 | 11.4 |
| 32 | 3 | III | c | >10,000 | >10,000 | 89.6 | 71.5 | 3.6 | 7.9 |
| 33 | 4 | III | c | >10,000 | >10,000 | 74.6 | 46.8 | 9.9 | 31.7 |
| 34 | 2 | IV | c | >10,000 | >10,000 | 666 | 851 | 5.7 | 14.3 |
| 35 | 3 | IV | c | >10,000 | >10,000 | 523 | 335 | 10.4 | 14.3 |
| 36 | 4 | IV | c | >10,000 | >10,000 | 332 | 298 | 19.0 | 31.7 |
| AAZ | 250.0 | 12.0 | 25.0 | 5.7 | - | - | |||
| Ver | - | - | - | - | 1.2 | 3.0 | |||
| EC50 µM 1 | |||
|---|---|---|---|
| Cmpd | MDR1 | MRP1 | BCRP |
| 13 | 14.45 ± 0.25 | 52 ± 10 | NA |
| 27 | 3.99 ± 0.11 | NA | NA |
| 32 | 4.53 ± 0.17 | NA | NA |
| 33 | 3.02 ± 0.09 | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braconi, L.; Riganti, C.; Parenti, A.; Cecchi, M.; Nocentini, A.; Bartolucci, G.; Menicatti, M.; Contino, M.; Colabufo, N.A.; Manetti, D.; et al. Dual Inhibitors of P-gp and Carbonic Anhydrase XII (hCA XII) against Tumor Multidrug Resistance with Piperazine Scaffold. Molecules 2024, 29, 3290. https://doi.org/10.3390/molecules29143290
Braconi L, Riganti C, Parenti A, Cecchi M, Nocentini A, Bartolucci G, Menicatti M, Contino M, Colabufo NA, Manetti D, et al. Dual Inhibitors of P-gp and Carbonic Anhydrase XII (hCA XII) against Tumor Multidrug Resistance with Piperazine Scaffold. Molecules. 2024; 29(14):3290. https://doi.org/10.3390/molecules29143290
Chicago/Turabian StyleBraconi, Laura, Chiara Riganti, Astrid Parenti, Marta Cecchi, Alessio Nocentini, Gianluca Bartolucci, Marta Menicatti, Marialessandra Contino, Nicola Antonio Colabufo, Dina Manetti, and et al. 2024. "Dual Inhibitors of P-gp and Carbonic Anhydrase XII (hCA XII) against Tumor Multidrug Resistance with Piperazine Scaffold" Molecules 29, no. 14: 3290. https://doi.org/10.3390/molecules29143290
APA StyleBraconi, L., Riganti, C., Parenti, A., Cecchi, M., Nocentini, A., Bartolucci, G., Menicatti, M., Contino, M., Colabufo, N. A., Manetti, D., Romanelli, M. N., Supuran, C. T., & Teodori, E. (2024). Dual Inhibitors of P-gp and Carbonic Anhydrase XII (hCA XII) against Tumor Multidrug Resistance with Piperazine Scaffold. Molecules, 29(14), 3290. https://doi.org/10.3390/molecules29143290