


Molecular Insight into the Processes and Mechanisms of N2 Adsorption and Accumulation at the Hydrophobic Solid/Liquid Interface

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
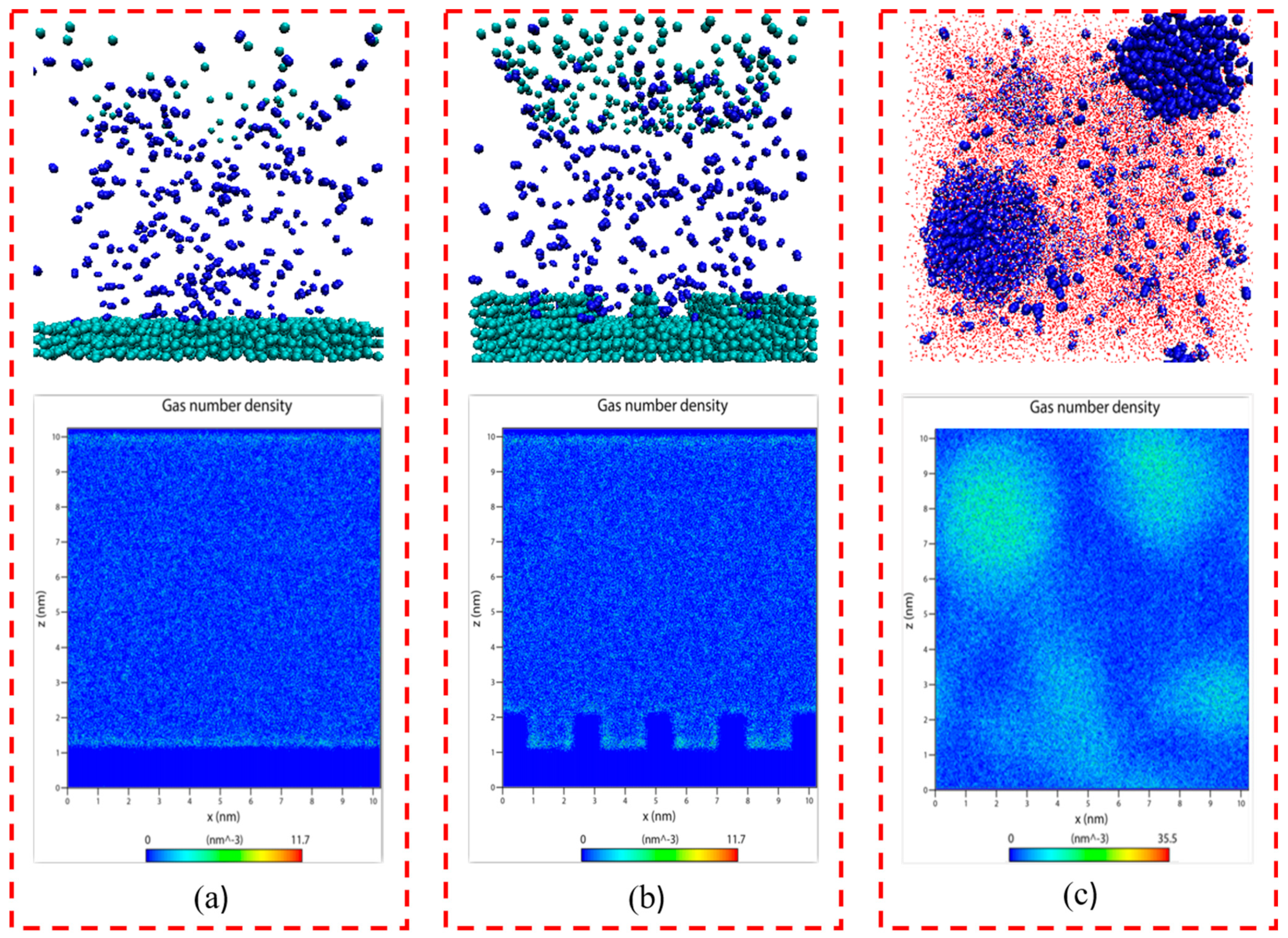
2.1. Gas–Solid and Gas–Liquid Systems
2.2. Gas–Solid–Liquid System
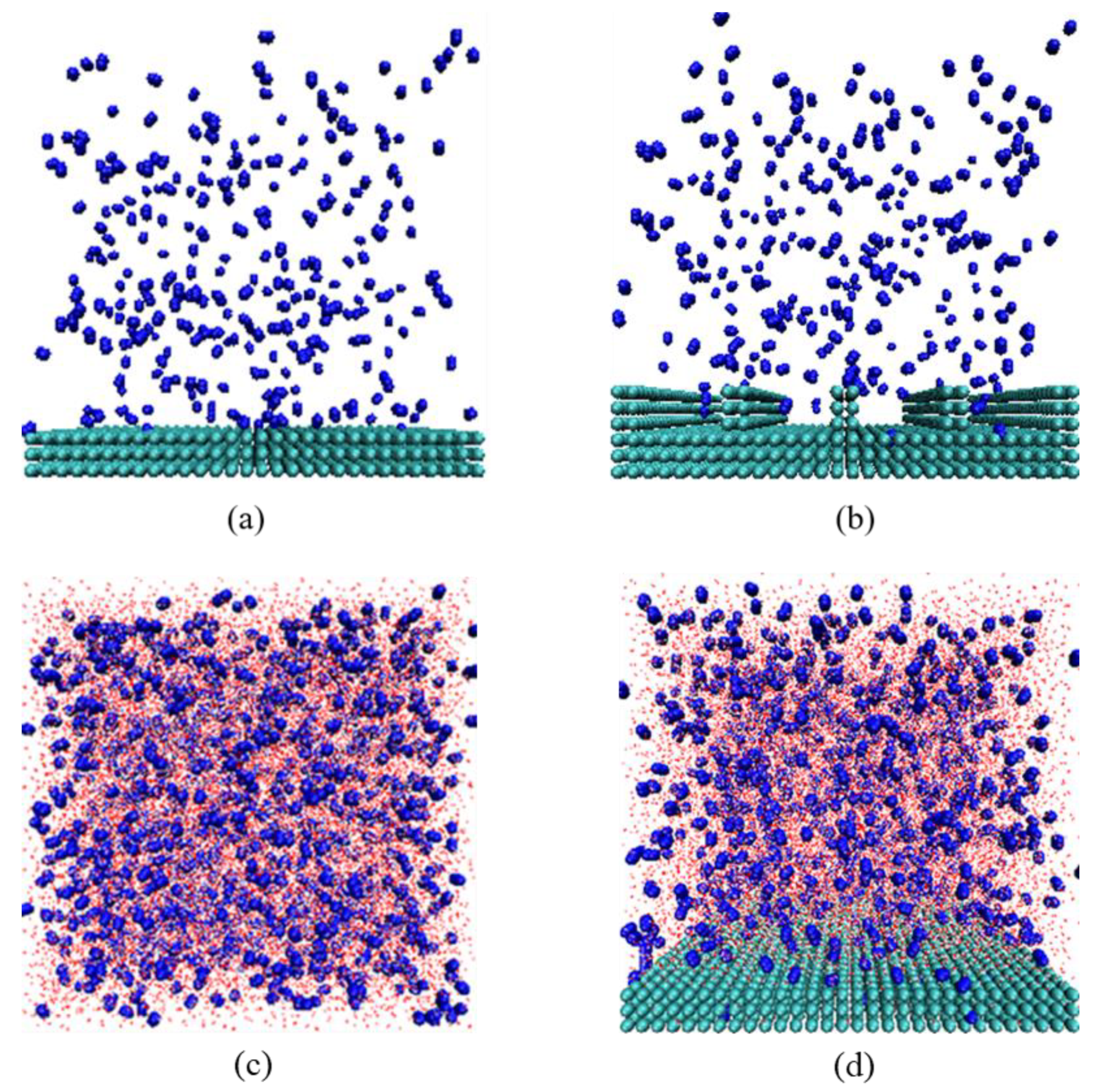
2.2.1. N2 Molecular Adsorption Process and Adsorption Configuration
2.2.2. Formation of a Loosely Arranged Water Molecular Layer at the Solid–Liquid Interface
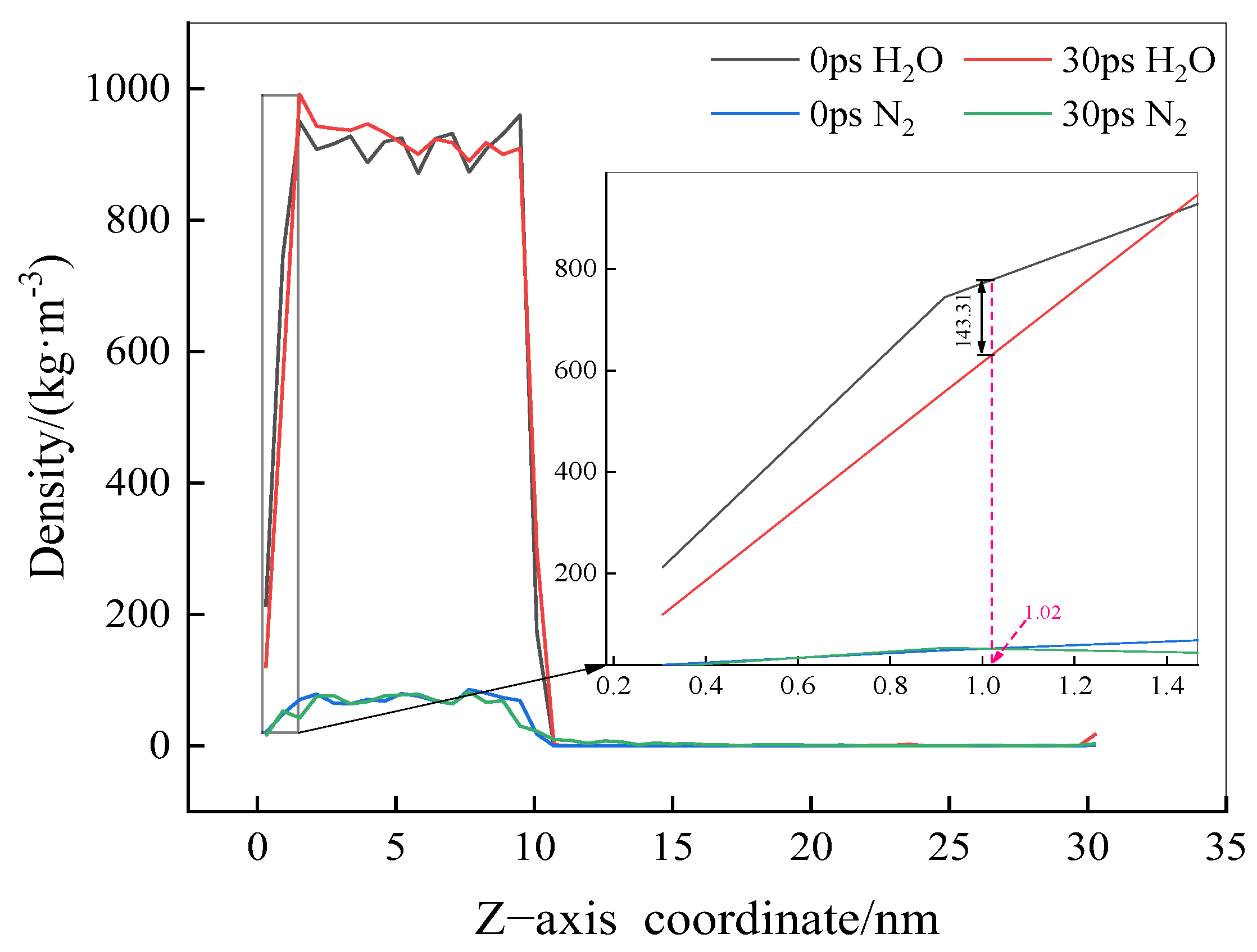
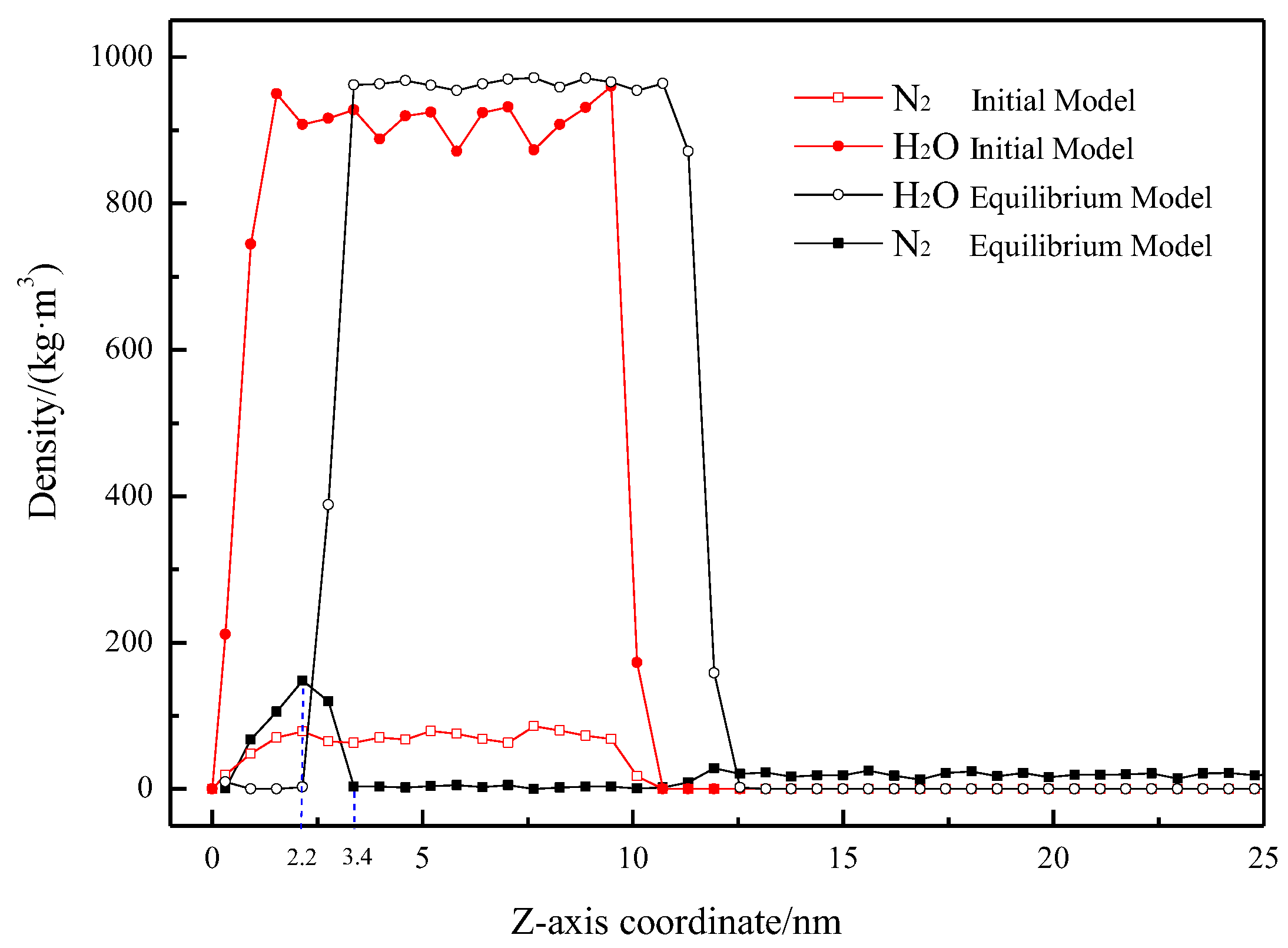
2.2.3. Density Distribution
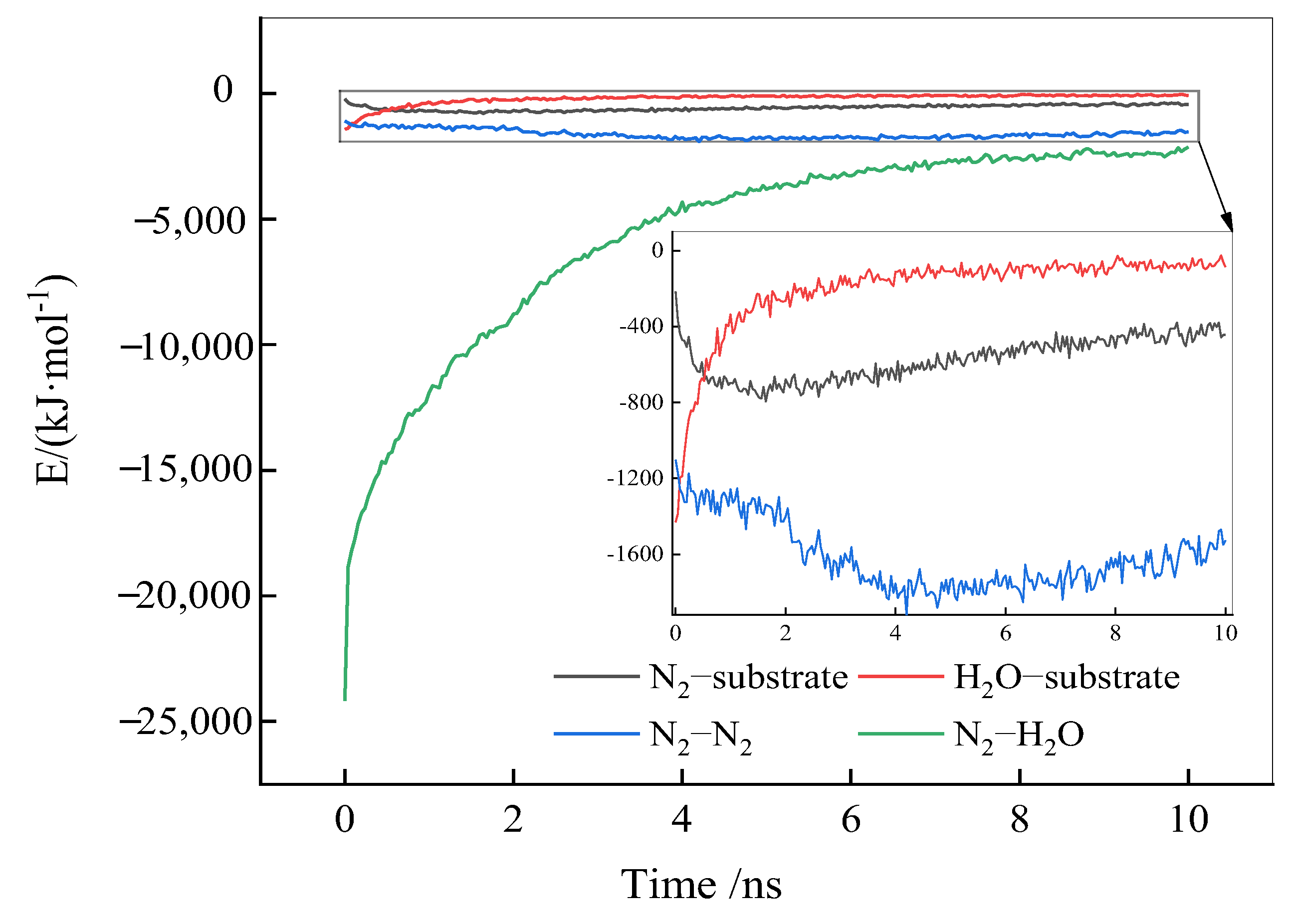
2.2.4. Interaction Energies
3. Models and Simulations
3.1. Models
3.2. Simulation Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Busse, A.; Sandham, N.; McHale, G.; Newton, M. Change in drag, apparent slip and optimum air layer thickness for laminar flow over an idealised superhydrophobic surface. J. Fluid. Mech. 2013, 727, 488–508. [Google Scholar] [CrossRef]
- Baldwin, R.; Rose, G. How the hydrophobic factor drives protein folding. Proc. Natl. Acad. Sci. USA 2016, 113, 12462–12466. [Google Scholar] [CrossRef] [PubMed]
- Farhang, F.; Nguyen, A.V.; Sewell, K.B. Fundamental Investigation of the Effects of Hydrophobic Fumed Silica on the Formation of Carbon Dioxide Gas Hydrates. Energy Fuels 2014, 28, 7025–7037. [Google Scholar] [CrossRef]
- Xu, W.; Lu, Z.; Sun, X.; Jiang, L.; Duan, X. Superwetting Electrodes for Gas-Involving Electrocatalysis. Acc. Chem. Res. 2018, 51, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, X.; Liu, S.; Hao, J.; Shao, Z.; Yi, B. Study on hydrophobicity loss of the gas diffusion layer in PEMFCs by electrochemical oxidation. RSC Adv. 2014, 4, 3852–3856. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Sun, J.; Zhang, Z.; Li, G.; Fang, H.; Xiao, X.; Zeng, X.; Hu, J. Detection of novel gaseous states at the highly oriented pyrolytic graphite-water interface. Langmuir 2007, 23, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Hampton, M.A.; Nguyen, A.V. Nanobubbles do not sit alone at the solid-liquid interface. Langmuir 2013, 29, 6123–6130. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, I.; Sivan, U. Three-Dimensional Characterization of Layers of Condensed Gas Molecules Forming Universally on Hydrophobic Surfaces. J. Am. Chem. Soc. 2018, 33, 10473–10481. [Google Scholar] [CrossRef]
- Theodorakis, P.; Che, Z. Surface Nanobubbles: Theory, Simulation, and Experiment. A Review. Adv. Colloid. Interface Sci. 2019, 272, 101995. [Google Scholar] [CrossRef]
- Zhang, X.; Maeda, N. Interfacial gaseous states on crystalline surfaces. J. Phys. Chem. C 2011, 115, 736–743. [Google Scholar] [CrossRef]
- Weijs, J.; Snoeijer, J.; Lohse, D. Formation of surface nanobubbles and the universality of their contact angles: A molecular dynamics approach. Phys. Rev. Lett. 2012, 108, 104501. [Google Scholar] [CrossRef] [PubMed]
- Sendner, C.; Horinek, D.; Bocquet, L.; Netz, R. Interfacial water at hydrophobic and hydrophilic surfaces: Slip, viscosity, and diffusion. Langmuir 2009, 25, 10768. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Zhang, L.; Guo, J.; Liu, S.; Li, B. Adsorption and accumulation mechanism of N2 on groove-type rough surfaces: A molecular simulation study. J. Mol. Liq. 2022, 366, 120260. [Google Scholar] [CrossRef]
- Grein, F. CH4–N2, NH3–N2, H2O–N2 and HF–N2 complexes: Ab initio studies and comparisons—Transition to hydrogen bonding. Theor. Chem. Acc. 2020, 139, 166. [Google Scholar] [CrossRef]
- Mezger, M.; Schöder, S.; Reichert, H.; Schröder, H.; Okasinski, J.; Honkimäki, V.; Ralston, J.; Bilgram, J.; Roth, R.; Dosch, H. Water and ice in contact with octadecyl-trichlorosilane functionalized surfaces: A high resolution X-ray reflectivity study. J. Chem. Phys. 2008, 128, 244705. [Google Scholar] [CrossRef]
- Mezger, M.; Sedlmeier, F.; Horinek, D.; Reichert, H.; Pontoni, D.; Dosch, H. On the origin of the hydrophobic water gap: An X-ray reflectivity and MD simulation study. J. Am. Chem. Soc. 2010, 132, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, X.; Shin, H.; Wang, J.; Tai, R.; Zhang, X.; Fang, H.; Xiao, W.; Wang, L.; Wang, C.; et al. Ultrahigh density of gas molecules confined in surface nanobubbles in ambient water. J. Am. Chem. Soc. 2020, 142, 5583–5593. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, C.; Zhang, J.; Xiao, S.; Zhang, Z.; Shen, Y.; Sun, B.; He, J. Displacement mechanism of oil in shale inorganic nanopores by supercritical carbon dioxide from molecular dynamics simulations. Energy Fuels 2017, 31, 738–746. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, Y.; Yan, Y.; Wang, Z.; Zhang, J. Molecular insight into the oil extraction and transport in CO2 flooding with reservoir depressurization. Int. J. Heat Mass Transf. 2020, 148, 119051. [Google Scholar] [CrossRef]
- Fang, T.; Wang, M.; Gao, Y.; Zhang, Y.; Yan, Y.; Zhang, J. Enhanced oil recovery with CO2/N2 slug in low permeability reservoir: Molecular dynamics simulation. Chem. Eng. Sci. 2019, 197, 204–211. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.; Smith, J.; Kasson, P.; Van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef] [PubMed]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Yu, H.; Chen, Z.; Xin, Y.; Chai, J.; Fu, L.Y.; Zhang, J.; Zhang, H. Simulation on oily contamination removal by ozone using molecular dynamics. Chemosphere 2022, 308, 136473. [Google Scholar] [CrossRef]
- Li, J.; Han, Y.; Qu, G.; Cheng, J.; Xue, C.; Gao, X.; Sun, T.; Ding, W. Molecular dynamics simulation of the aggregation behavior of N-Dodecyl-N, N-Dimethyl-3-Ammonio-1-Propanesulfonate/sodium dodecyl benzene sulfonate surfactant mixed system at oil/water interface. Colloids Surf. A Physicochem. Eng. Asp. 2017, 531, 73–80. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, B.; Su, D. Molecular Insight into the Processes and Mechanisms of N2 Adsorption and Accumulation at the Hydrophobic Solid/Liquid Interface. Molecules 2024, 29, 2711. https://doi.org/10.3390/molecules29112711
Li B, Su D. Molecular Insight into the Processes and Mechanisms of N2 Adsorption and Accumulation at the Hydrophobic Solid/Liquid Interface. Molecules. 2024; 29(11):2711. https://doi.org/10.3390/molecules29112711
Chicago/Turabian StyleLi, Bao, and Dan Su. 2024. "Molecular Insight into the Processes and Mechanisms of N2 Adsorption and Accumulation at the Hydrophobic Solid/Liquid Interface" Molecules 29, no. 11: 2711. https://doi.org/10.3390/molecules29112711
APA StyleLi, B., & Su, D. (2024). Molecular Insight into the Processes and Mechanisms of N2 Adsorption and Accumulation at the Hydrophobic Solid/Liquid Interface. Molecules, 29(11), 2711. https://doi.org/10.3390/molecules29112711