

Chalcogen-Bond-Assisted Formation of the N→C Dative Bonds in the Complexes between Chalcogenadiazoles/Chalcogenatriazoles and Fullerene C60


Abstract
1. Introduction
2. Results and Discussion
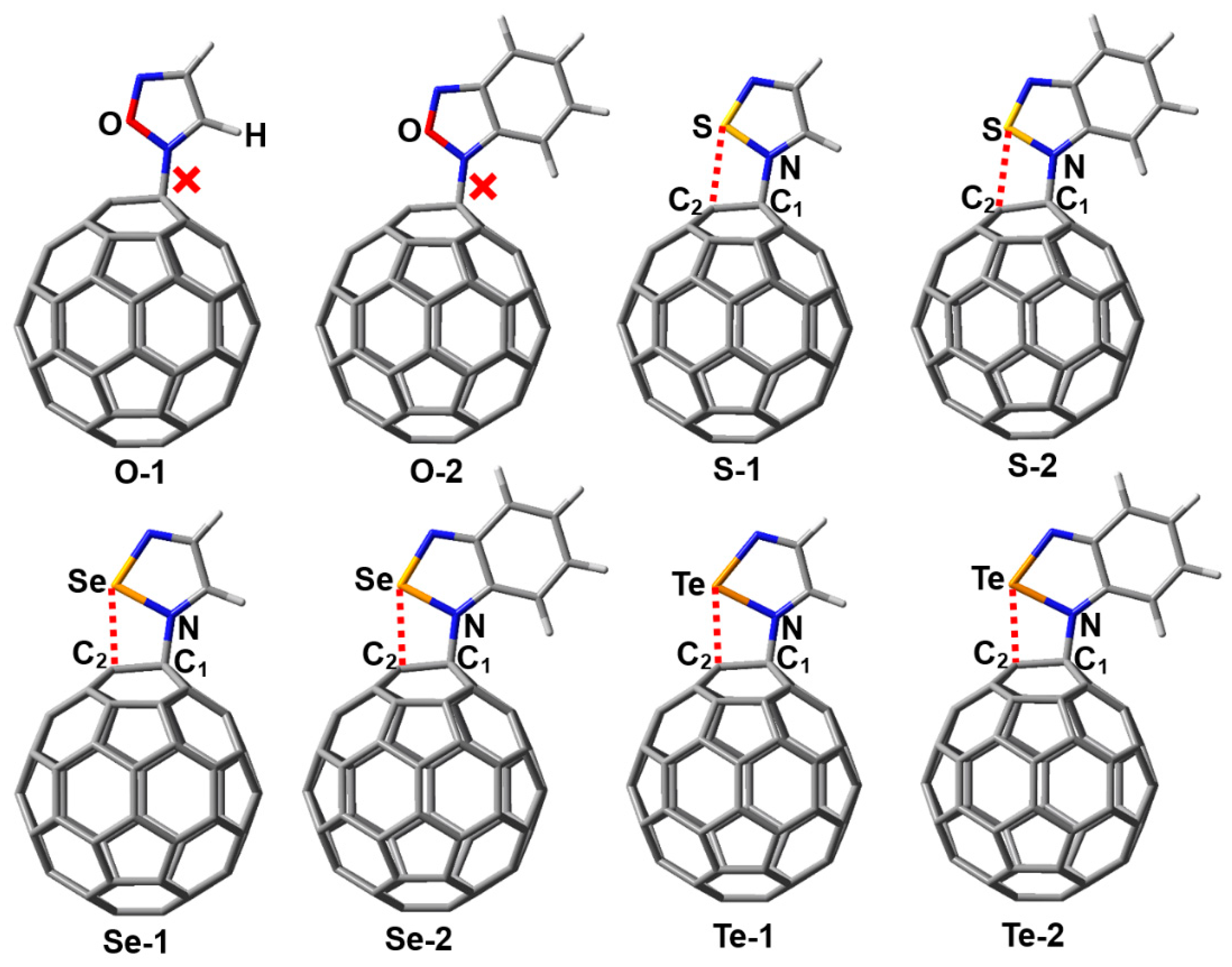
2.1. Complexes between Chalcogenadiazoles and Fullerene C60
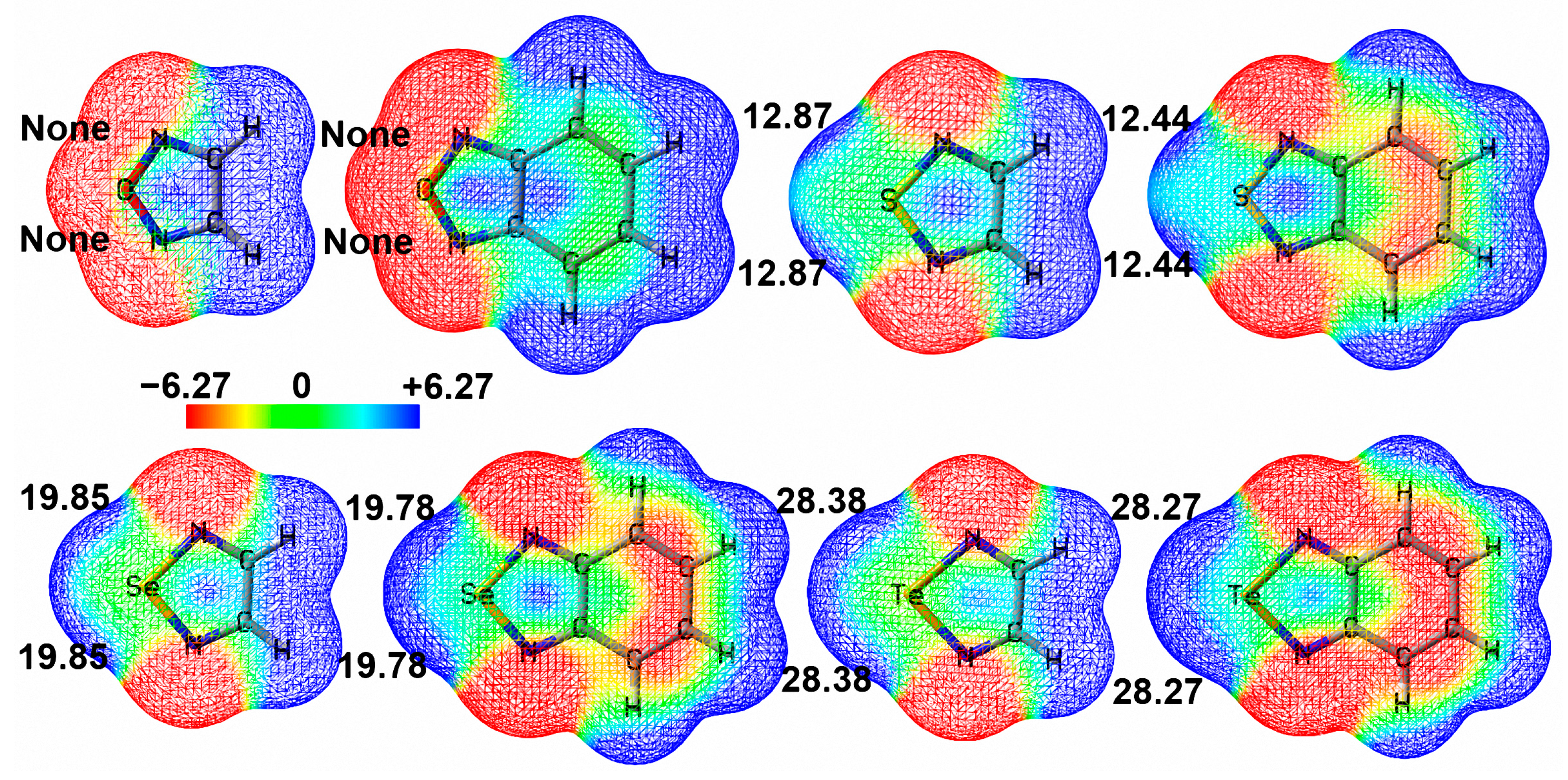
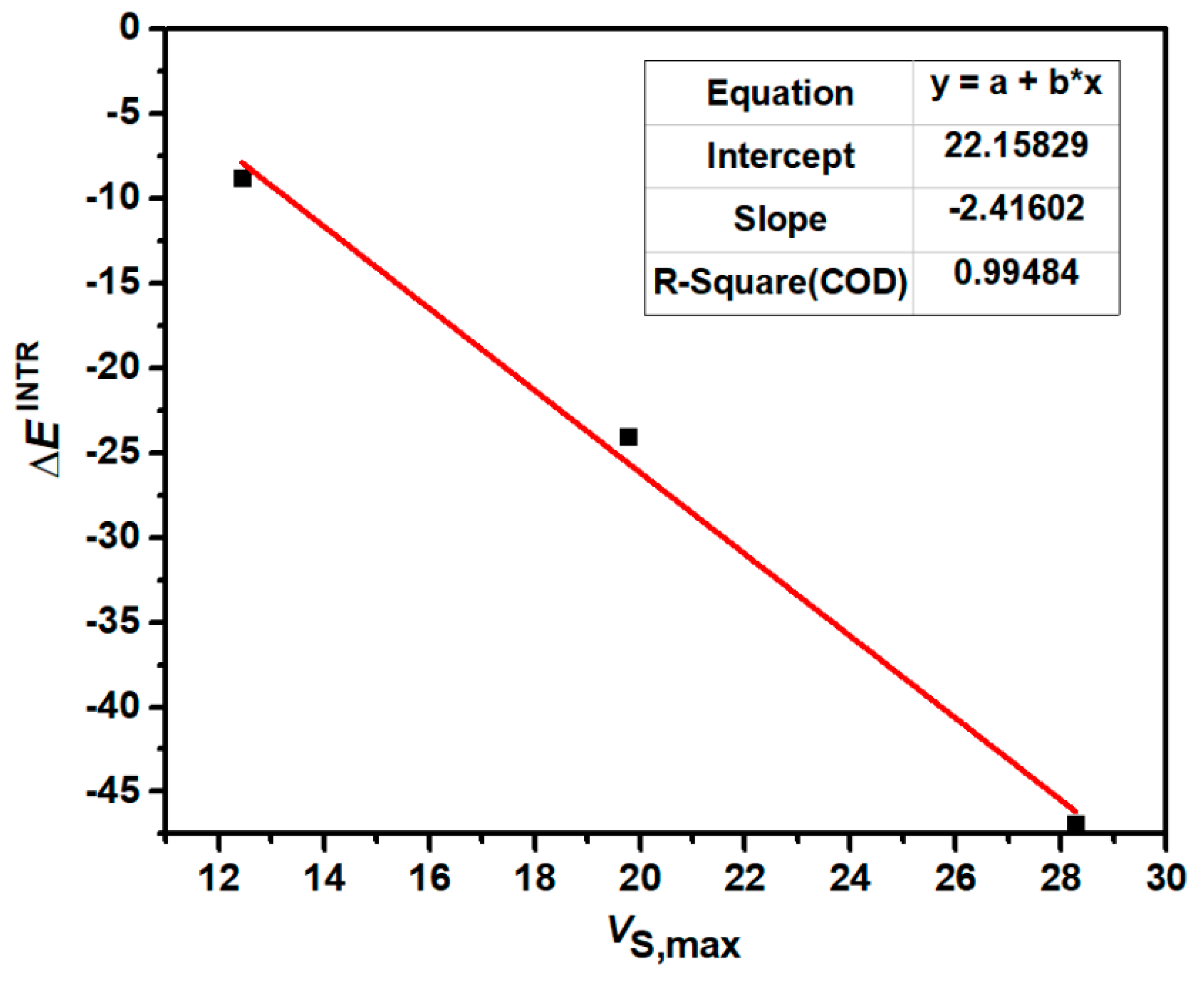
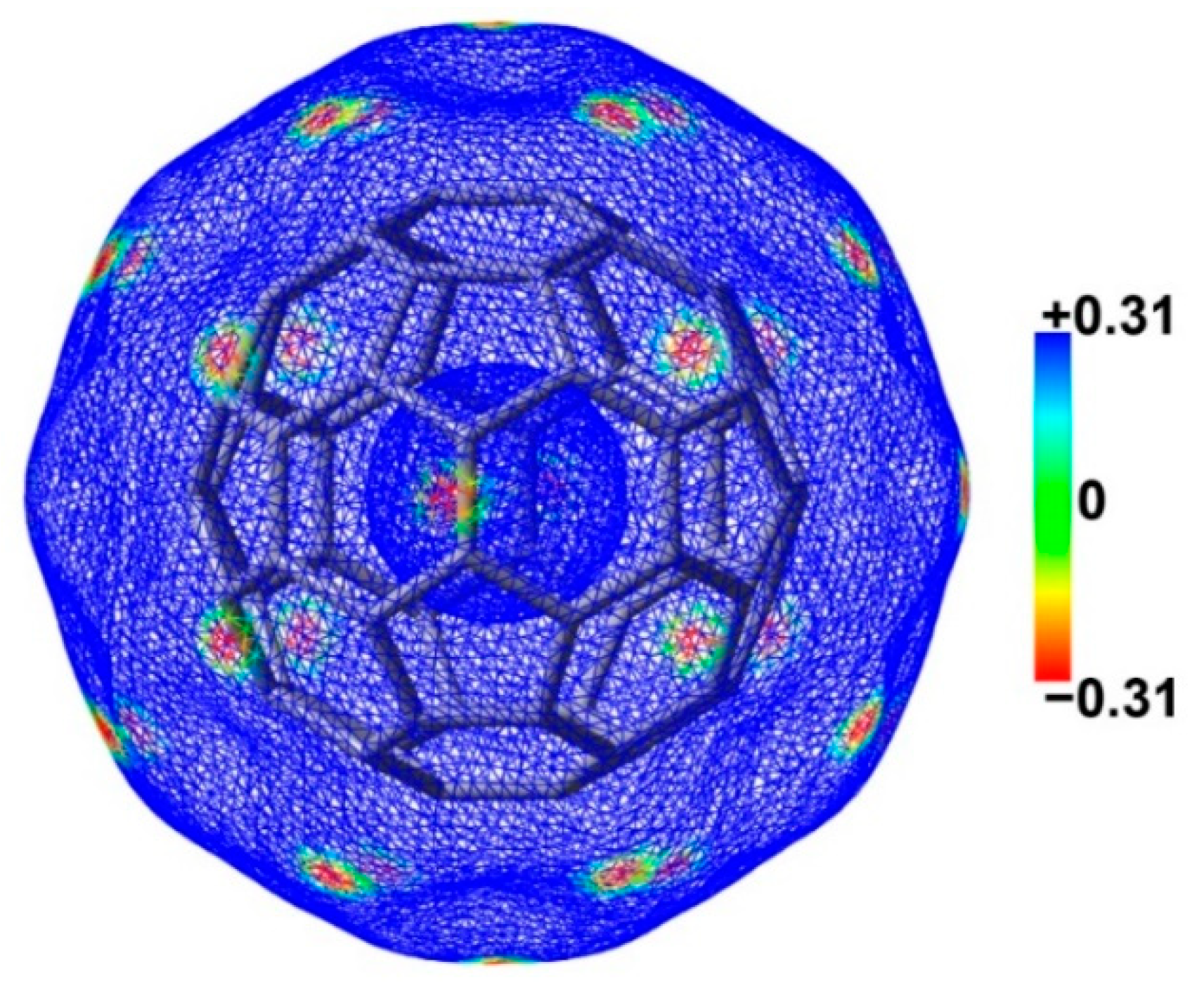
2.2. Molecular Electrostatic Potential Maps of Chalcogenadiazoles
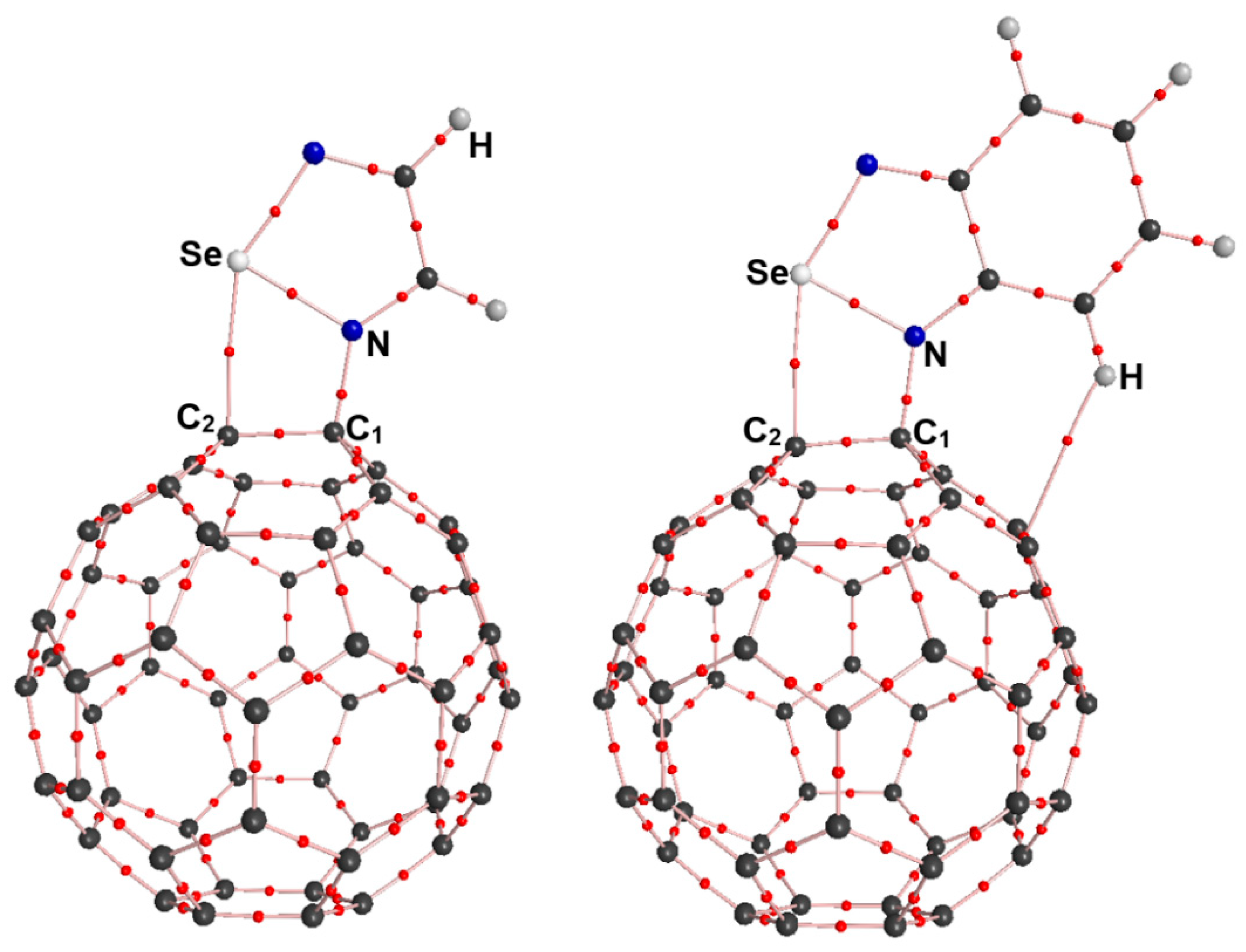
2.3. AIM Analyses
2.4. Complexes between Chalcogenatriazoles and Fullerene C60
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Minkin, V.I. Glossary of Terms Used in Theoretical Organic Chemistry (IUPAC Recommendations 1999). Pure Appl. Chem. 1999, 71, 1919–1981. [Google Scholar] [CrossRef]
- Haaland, A. Covalent versus Dative Bonds to Main Group Metals, a Useful Distinction. Angew. Chem. Int. Ed. Engl. 1989, 28, 992–1007. [Google Scholar] [CrossRef]
- Lepetit, C.; Maraval, V.; Canac, Y.; Chauvin, R. On the Nature of the Dative Bond: Coordination to Metals and Beyond. The Carbon Case. Coord. Chem. Rev. 2016, 308, 59–75. [Google Scholar] [CrossRef]
- Zhao, L.; Hermann, M.; Holzmann, N.; Frenking, G. Dative Bonding in Main Group Compounds. Coord. Chem. Rev. 2017, 344, 163–204. [Google Scholar] [CrossRef]
- Nandi, A.; Kozuch, S. History and Future of Dative Bonds. Chem. Eur. J. 2020, 26, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A. The Chemistry of the Fullerenes; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Lamanec, M.; Lo, R.; Nachtigallová, D.; Bakandritsos, A.; Mohammadi, E.; Dračínský, M.; Zbořil, R.; Hobza, P.; Wang, W. The Existence of a N→C Dative Bond in the C60–Piperidine Complex. Angew. Chem. Int. Ed. 2021, 60, 1942–1950. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Lamanec, M.; Wang, W.; Manna, D.; Bakandritsos, A.; Dračínský, M.; Zbořil, R.; Nachtigallová, D.; Hobza, P. Structure-Directed Formation of the Dative/Covalent Bonds in Complexes with C70···Piperidine. Phys. Chem. Chem. Phys. 2021, 23, 4365–4375. [Google Scholar] [CrossRef]
- Lo, R.; Manna, D.; Lamanec, M.; Wang, W.; Bakandritsos, A.; Dračínský, M.; Zbořil, R.; Nachtigallová, D.; Hobza, P. Addition Reaction between Piperidine and C60 to Form 1,4-Disubstituted C60 Proceeds through van der Waals and Dative Bond Complexes: Theoretical and Experimental Study. J. Am. Chem. Soc. 2021, 143, 10930–10939. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Manna, D.; Hobza, P. Cyclo[n]carbons Form Strong N→C Dative/Covalent Bonds with Piperidine. J. Phys. Chem. A 2021, 125, 2923–2931. [Google Scholar] [CrossRef]
- Nandi, A.; Martin, J.M.L. Heavy-Atom Tunneling in the Covalent/Dative Bond Complexation of Cyclo [18]carbon–Piperidine. J. Phys. Chem. B 2022, 126, 1799–1804. [Google Scholar] [CrossRef]
- Lo, R.; Manna, D.; Hobza, P. Tuning the P–C Dative/Covalent Bond Formation in R3P–C60 Complexes by Changing the R Group. Chem. Commun. 2021, 57, 3363–3366. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Manna, D.; Hobza, P. P-Doped Graphene–C60 Nanocomposite: A Donor–Acceptor Complex with a P–C Dative Bond. Chem. Commun. 2022, 58, 1045–1048. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.-C.C.; Rappé, A.K.; Berryman, O.B.; Ho, S.P. Hydrogen Bond Enhanced Halogen Bonds: A Synergistic Interaction in Chemistry and Biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Fanfrlík, J.; Holub, J.; Růžičková, Z.; Řezáč, J.; Lane, P.D.; Wann, D.A.; Hnyk, D.; Růžička, A.; Hobza, P. Competition between Halogen, Hydrogen and Dihydrogen Bonding in Brominated Carboranes. ChemPhysChem 2016, 17, 3373–3376. [Google Scholar] [CrossRef]
- Brammer, L. Halogen Bonding, Chalcogen Bonding, Pnictogen Bonding, Tetrel Bonding: Origins, Current Status and Discussion. Faraday Discuss. 2017, 203, 485–507. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Noncovalent Bonds through Sigma and Pi-Hole Located on the Same Molecule. Guiding Principles and Comparisons. Molecules 2021, 26, 1740. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond. Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Kolb, S.; Oliver, G.A.; Werz, D.B. Chemistry Evolves, Terms Evolve, but Phenomena Do Not Evolve: From Chalcogen–Chalcogen Interactions to Chalcogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 22306–22310. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. A 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Lo, R.; Manna, D.; Miriyala, V.M.; Nachtigallová, D.; Hobza, P. Trends in the Stability of Covalent Dative Bonds with Variable Solvent Polarity Depend on the Charge Transfer in the Lewis Electron-Pair System. Phys. Chem. Chem. Phys. 2023, 25, 25961–25964. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C–H–O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Characterization of a Dihydrogen Bond on the Basis of the Electron Density. J. Phys. Chem. A 1998, 102, 1873–1878. [Google Scholar] [CrossRef]
- Wang, W.; Wong, N.-B.; Zheng, W.; Tian, A. Theoretical Study on the Blueshifting Halogen Bond. J. Phys. Chem. A 2004, 108, 1799–1805. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Li, X.; Zheng, S.; Meng, L. Ab Initio and AIM Studies on Typical π-Type and Pseudo-π-Type Halogen Bonds: Comparison with Hydrogen Bonds. Int. J. Quantum Chem. 2011, 111, 3725–3740. [Google Scholar] [CrossRef]
- Grabowski, S.J. QTAIM Characteristics of Halogen Bond and Related Interactions. J. Phys. Chem. A 2012, 116, 1838–1845. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6169. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The Calculation of Small Molecular Interactions by the Difference of Separate Total Energies. Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Anderson, L.N.; Aquino, F.W.; Raeber, A.E.; Chen, X.; Wong, B.M. Halogen Bonding Interactions: Revised Benchmarks and a New Assessment of Exchange vs Dispersion. J. Chem. Theory Comput. 2018, 14, 180–190. [Google Scholar] [CrossRef]
- de Azevedo Santos, L.; Ramalho, T.C.; Hamlin, T.A.; Bickelhaupt, F.M. Chalcogen Bonds: Hierarchical Ab Initio Benchmark and Density Functional Theory Performance Study. J. Comput. Chem. 2021, 42, 688–698. [Google Scholar] [CrossRef]
- Wang, W.; Sun, T.; Zhang, Y.; Wang, Y.B. The Benzene⋯Naphthalene Complex: A more Challenging System than the Benzene Dimer for newly Developed Computational Methods. J. Chem. Phys. 2015, 143, 114312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, Y.; Wang, Y.B. Highly Accurate Benchmark Calculations of the Interaction Energies in the Complexes C6H6···C6X6 (X = F, Cl, Br, and I). Int. J. Quantum Chem. 2017, 117, e25345. [Google Scholar] [CrossRef]
- Hedberg, K.; Hedberg, L.; Bethune, D.S.; Brown, C.A.; Dorn, H.C.; Johnson, R.D.; De Vries, M. Bond Lengths in Free Molecules of Buckminsterfullerene, C60, from Gas-Phase Electron Diffraction. Science 1991, 254, 410–412. [Google Scholar] [CrossRef]
- Biegler-König, F.; Schönbohm, J.; Bayles, D. AIM2000-A Program to Analyze and Visualize Atoms in Molecules. J. Comput. Chem. 2001, 22, 545–559. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative Analysis of Molecular Surface Based on Improved Marching Tetrahedra Algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | d(N···C1) | d(Ch···C2) | µ | q(N) | q(C1) | q(Ch) | q(C2) | ||
|---|---|---|---|---|---|---|---|---|---|
| S-1 | 1.504 | 2.569 | −4.52 | 20.85 | 8.14 | 0.081 | 1.121 | 0.351 | −0.507 |
| S-2 | 1.500 | 2.557 | −8.75 | 18.13 | 10.18 | 0.090 | 1.153 | 0.381 | −0.545 |
| Se-1 | 1.480 | 2.467 | −20.05 | 11.40 | 5.60 | 0.007 | 0.923 | 0.341 | −0.221 |
| Se-2 | 1.478 | 2.468 | −24.04 | 8.35 | 7.59 | −0.004 | 0.952 | 0.383 | −0.241 |
| Te-1 | 1.467 | 2.471 | −42.54 | −4.11 | 3.39 | −0.142 | 1.019 | 0.443 | −0.245 |
| Te-2 | 1.466 | 2.464 | −46.88 | −7.32 | 4.92 | −0.150 | 1.043 | 0.497 | −0.245 |
| Complex | ρb | ▽2ρb | λ1 | λ2 | λ3 | ε |
|---|---|---|---|---|---|---|
| S-1 | 0.0393 | 0.0659 | −0.0336 | −0.0257 | 0.1253 | 0.3091 |
| S-2 | 0.0398 | 0.0683 | −0.0345 | −0.0259 | 0.1287 | 0.3305 |
| Se-1 | 0.0550 | 0.0493 | −0.0509 | −0.0491 | 0.1493 | 0.0370 |
| Se-2 | 0.0544 | 0.0517 | −0.0495 | −0.0489 | 0.1502 | 0.0136 |
| Te-1 | 0.0645 | 0.0302 | −0.0618 | −0.0544 | 0.1464 | 0.1355 |
| Te-2 | 0.0651 | 0.0313 | −0.0623 | −0.0559 | 0.1496 | 0.1138 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, W. Chalcogen-Bond-Assisted Formation of the N→C Dative Bonds in the Complexes between Chalcogenadiazoles/Chalcogenatriazoles and Fullerene C60. Molecules 2024, 29, 2685. https://doi.org/10.3390/molecules29112685
Zhang Y, Wang W. Chalcogen-Bond-Assisted Formation of the N→C Dative Bonds in the Complexes between Chalcogenadiazoles/Chalcogenatriazoles and Fullerene C60. Molecules. 2024; 29(11):2685. https://doi.org/10.3390/molecules29112685
Chicago/Turabian StyleZhang, Yu, and Weizhou Wang. 2024. "Chalcogen-Bond-Assisted Formation of the N→C Dative Bonds in the Complexes between Chalcogenadiazoles/Chalcogenatriazoles and Fullerene C60" Molecules 29, no. 11: 2685. https://doi.org/10.3390/molecules29112685
APA StyleZhang, Y., & Wang, W. (2024). Chalcogen-Bond-Assisted Formation of the N→C Dative Bonds in the Complexes between Chalcogenadiazoles/Chalcogenatriazoles and Fullerene C60. Molecules, 29(11), 2685. https://doi.org/10.3390/molecules29112685