
Characterization of Excited-State Electronic Structure in Diblock π-Conjugated Oligomers with Adjustable Linker Electronic Coupling


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
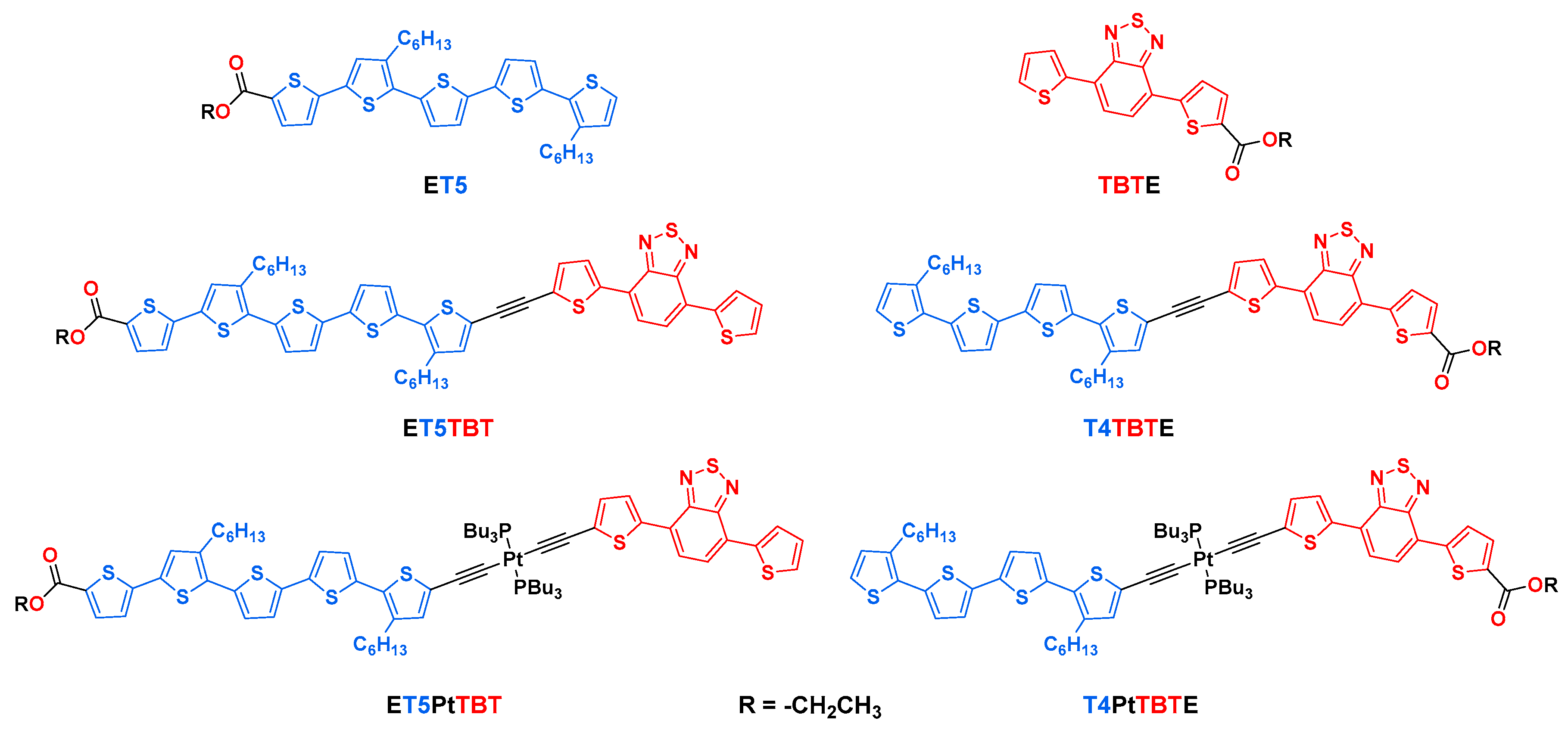
2.1. Structure and Synthesis
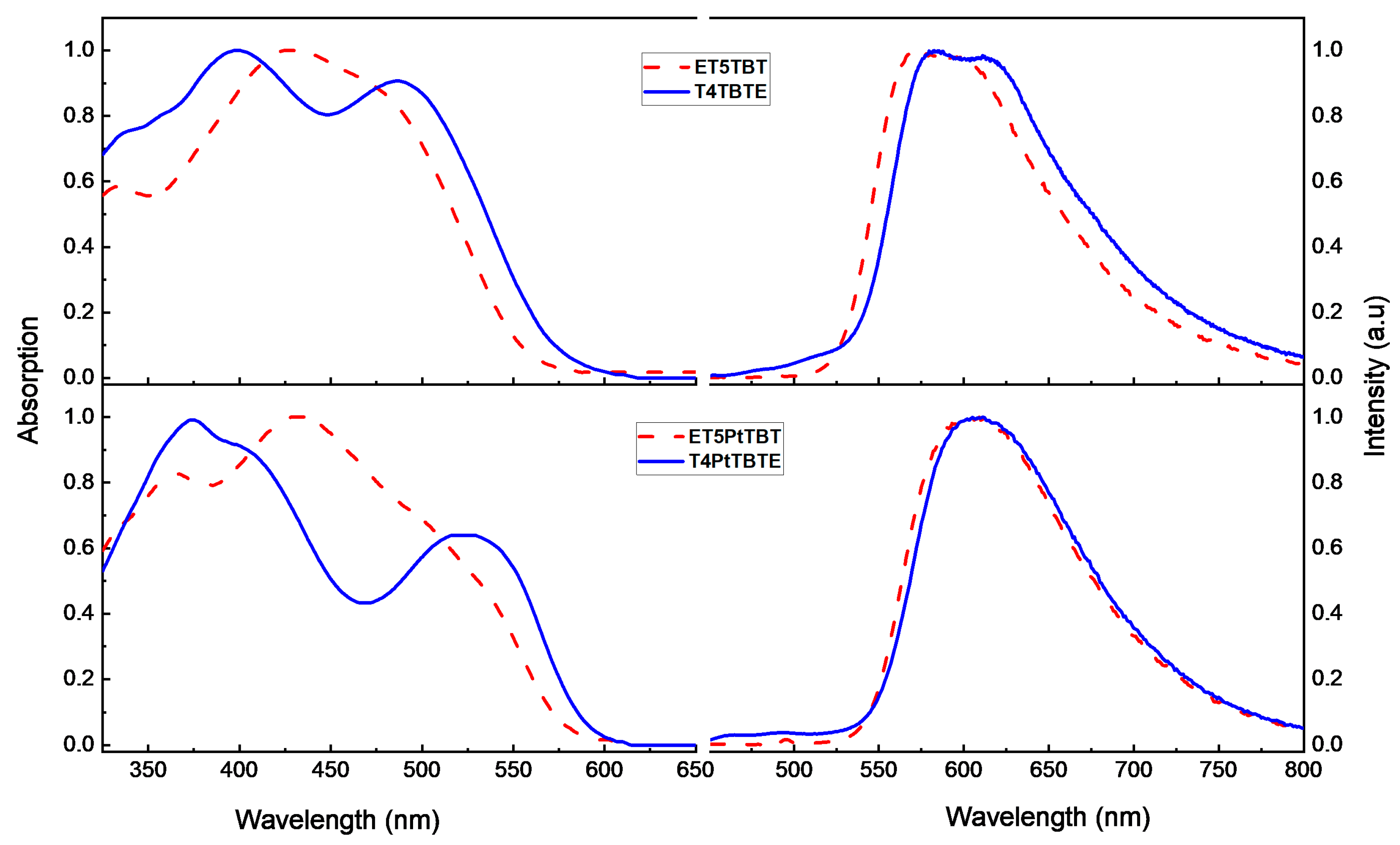
2.2. Photophysical Characterization
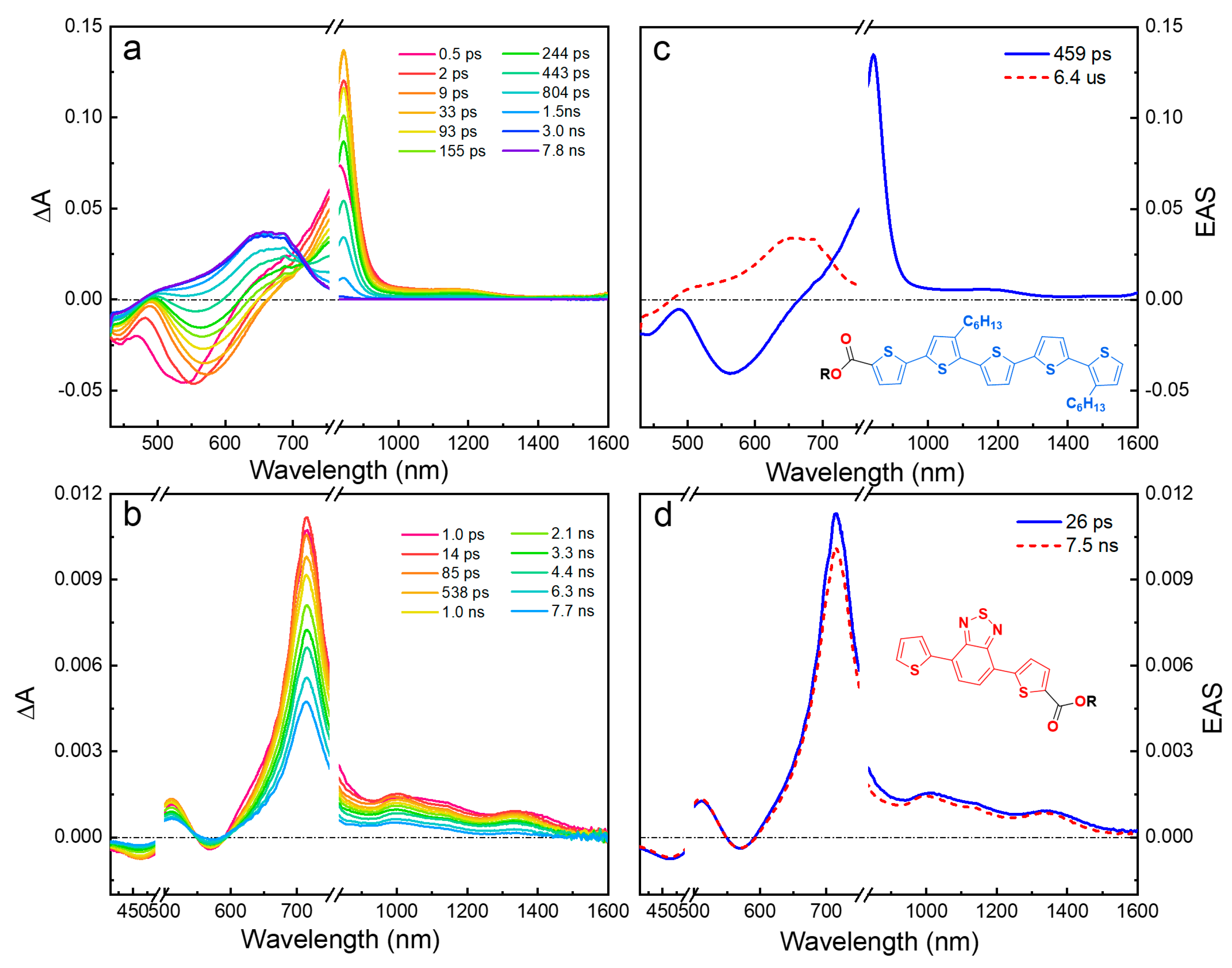
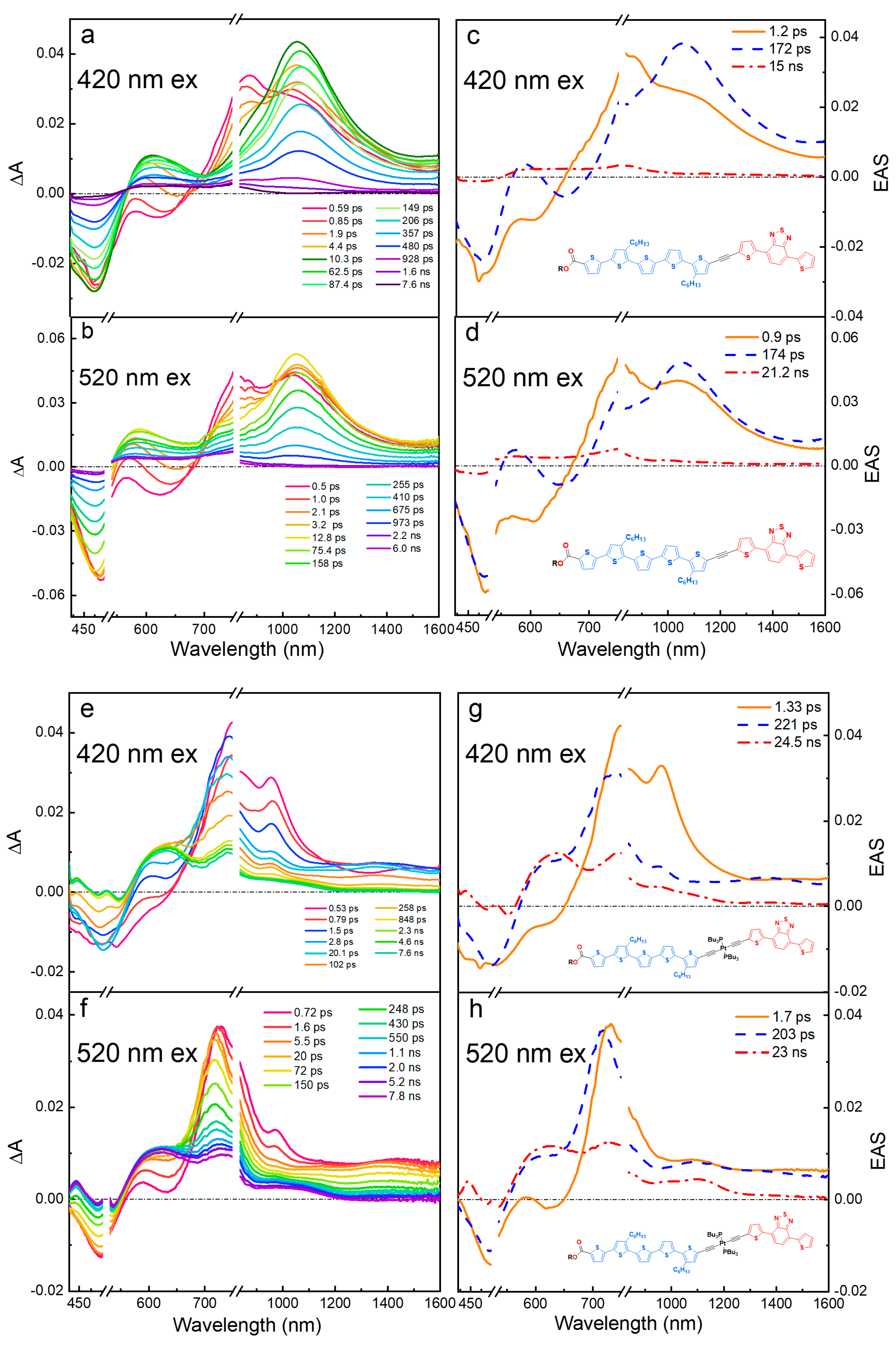
2.3. Ultrafast Transient Absorption Spectroscopy
2.3.1. Wavelength Photoselection and Effect of Linker Structure
2.3.2. Effect of Solvent Polarity on Excited-State Structure and Dynamics
3. Discussion
4. Experimental Methods
4.1. Synthesis and Characterization
4.2. Photophysical Analysis
4.3. Electrochemistry
4.4. Femtosecond Transient Absorption Spectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reynolds, J.R.; Thompson, B.C.; Skotheim, T.A. Handbook of Conducting Polymers, Two-Volume Set; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar]
- Mullen, K.; Wegner, G. Electronic Materials: The Oligomer Approach; Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Burroughes, J.H.; Bradley, D.D.; Brown, A.; Marks, R.; Mackay, K.; Friend, R.H.; Burns, P.L.; Holmes, A.B. Light-Emitting Diodes Based on Conjugated Polymers. Nature 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Facchetti, A. Π-Conjugated Polymers for Organic Electronics and Photovoltaic Cell Applications. Chem. Mater. 2011, 23, 733–758. [Google Scholar] [CrossRef]
- Zhu, D.; Ji, D.; Li, L.; Hu, W. Recent Progress in Polymer-Based Infrared Photodetectors. J. Mater. Chem. C 2022, 10, 13312–13323. [Google Scholar] [CrossRef]
- Xu, T.; Yu, L. How to Design Low Bandgap Polymers for Highly Efficient Organic Solar Cells. Mater.Today 2014, 17, 11–15. [Google Scholar] [CrossRef]
- Cheng, Y.-J.; Yang, S.-H.; Hsu, C.-S. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chem. Rev. 2009, 109, 5868–5923. [Google Scholar] [CrossRef]
- Dillon, R.J.; Pan, Z.; Jiang, J.; Winkel, R.W.; Papanikolas, J.M.; Schanze, K.S. Ultrafast Energy Transfer in Fully Conjugated Thiophene-Benzothiadiazole Capped Poly (Phenylene Ethynylene) Molecular Wires. J. Phys. Chem. C 2020, 124, 18920–18929. [Google Scholar] [CrossRef]
- Scarongella, M.; Laktionov, A.; Rothlisberger, U.; Banerji, N. Charge Transfer Relaxation in Donor–Acceptor Type Conjugated Materials. J. Mater. Chem. C 2013, 1, 2308–2319. [Google Scholar] [CrossRef]
- Hwang, I.; Beaupré, S.; Leclerc, M.; Scholes, G.D. Ultrafast Relaxation of Charge-Transfer Excitons in Low-Bandgap Conjugated Copolymers. Chem. Sci. 2012, 3, 2270–2277. [Google Scholar] [CrossRef]
- Ivanov, S.A.; Piryatinski, A.; Nanda, J.; Tretiak, S.; Zavadil, K.R.; Wallace, W.O.; Werder, D.; Klimov, V.I. Type-Ii Core/Shell Cds/Znse Nanocrystals: Synthesis, Electronic Structures, and Spectroscopic Properties. J. Am. Chem. Soc. 2007, 129, 11708–11719. [Google Scholar] [CrossRef]
- Beljonne, D.; Pourtois, G.; Silva, C.; Hennebicq, E.; Herz, L.; Friend, R.; Scholes, G.; Setayesh, S.; Müllen, K.; Brédas, J. Interchain vs. Intrachain Energy Transfer in Acceptor-Capped Conjugated Polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 10982–10987. [Google Scholar] [CrossRef]
- Kim, B.G.; Ma, X.; Chen, C.; Ie, Y.; Coir, E.W.; Hashemi, H.; Aso, Y.; Green, P.F.; Kieffer, J.; Kim, J. Energy Level Modulation of Homo, Lumo, and Band-Gap in Conjugated Polymers for Organic Photovoltaic Applications. Adv. Funct. Mater. 2013, 23, 439–445. [Google Scholar] [CrossRef]
- Müllen, K.; Pisula, W. Donor–Acceptor Polymers. J. Am. Chem. Soc. 2015, 137, 9503–9505. [Google Scholar] [CrossRef] [PubMed]
- Friend, R.H.; Bradley, D.D.C.; Townsend, P.D. Photo-Excitation in Conjugated Polymers. J. Phys. D Appl. Phys. 1987, 20, 1367. [Google Scholar] [CrossRef]
- Samuel, I.D.W.; Raksi, F.; Bradley, D.D.C.; Friend, R.H.; Burn, P.L.; Holmes, A.B.; Murata, H.; Tsutsui, T.; Saito, S. Femtosecond Transient Absorption Measurements in Poly(Arylenevinylene)S. Synth. Met. 1993, 55, 15–21. [Google Scholar] [CrossRef]
- Grebner, D.; Helbig, M.; Rentsch, S. Size-Dependent Properties of Oligothiophenes by Picosecond Time-Resolved Spectroscopy. J. Phys. Chem. 1995, 99, 16991–16998. [Google Scholar] [CrossRef]
- Moses, D.; Dogariu, A.; Heeger, A.J. Ultrafast Photoinduced Charge Generation in Conjugated Polymers. Chem. Phys. Lett. 2000, 316, 356–360. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, D.; Jeoung, S.C.; Han, J.-Y.; Jang, M.-S.; Shim, H.-K. Ultrafast Energy-Transfer Dynamics between Block Copolymer and Π-Conjugated Polymer Chains in Blended Polymeric Systems. Chem. Mater. 2001, 13, 2666–2674. [Google Scholar] [CrossRef]
- Lanzani, G.; Cerullo, G.; Polli, D.; Gambetta, A.; Zavelani-Rossi, M.; Gadermaier, C. Photophysics of Conjugated Polymers: The Contribution of Ultrafast Spectroscopy. Phys. Status Solidi 2004, 201, 1116–1131. [Google Scholar] [CrossRef]
- Takeda, N.; Asaoka, S.; Miller, J.R. Nature and Energies of Electrons and Holes in a Conjugated Polymer, Polyfluorene. J. Am. Chem. Soc. 2006, 128, 16073–16082. [Google Scholar] [CrossRef]
- Bolinger, J.C.; Traub, M.C.; Brazard, J.; Adachi, T.; Barbara, P.F.; Vanden Bout, D.A. Conformation and Energy Transfer in Single Conjugated Polymers. Acc. Chem. Res. 2012, 45, 1992–2001. [Google Scholar] [CrossRef]
- Reid, O.G.; Pensack, R.D.; Song, Y.; Scholes, G.D.; Rumbles, G. Charge Photogeneration in Neat Conjugated Polymers. Chem. Mater. 2014, 26, 561–575. [Google Scholar] [CrossRef]
- Voss, M.G.; Scholes, D.T.; Challa, J.R.; Schwartz, B.J. Ultrafast Transient Absorption Spectroscopy of Doped P3ht Films: Distinguishing Free and Trapped Polarons. Faraday Discuss. 2019, 216, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Dimitriev, O.P. Dynamics of Excitons in Conjugated Molecules and Organic Semiconductor Systems. Chem. Rev. 2022, 122, 8487–8593. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.; Song, Y.; O’Connor, L.; Wang, X.; Dailing, E.A.; Bragg, A.E.; Ayzner, A.L. Exciton Transfer Between Extended Electronic States in Conjugated Inter-Polyelectrolyte Complexes. ACS Appl. Mater. Interfaces 2024, 16, 19995–20010. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Huang, Y.-S.; Huettner, S.; Sommer, M.; Brinkmann, M.; Mulherin, R.; Niedzialek, D.; Beljonne, D.; Clark, J.; Huck, W.T. Control of Intrachain Charge Transfer in Model Systems for Block Copolymer Photovoltaic Materials. J. Am. Chem. Soc. 2013, 135, 5074–5083. [Google Scholar] [CrossRef] [PubMed]
- Tautz, R.; Da Como, E.; Wiebeler, C.; Soavi, G.; Dumsch, I.; Fröhlich, N.; Grancini, G.; Allard, S.; Scherf, U.; Cerullo, G. Charge Photogeneration in Donor–Acceptor Conjugated Materials: Influence of Excess Excitation Energy and Chain Length. J. Am. Chem. Soc. 2013, 135, 4282–4290. [Google Scholar] [CrossRef]
- Love, J.A.; Nagao, I.; Huang, Y.; Kuik, M.; Gupta, V.; Takacs, C.J.; Coughlin, J.E.; Qi, L.; van der Poll, T.S.; Kramer, E.J. Silaindacenodithiophene-Based Molecular Donor: Morphological Features and Use in the Fabrication of Compositionally Tolerant, High-Efficiency Bulk Heterojunction Solar Cells. J. Am. Chem. Soc. 2014, 136, 3597–3606. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Schanze, K.S. Fluorescent Charge-Transfer Excited States in Acceptor Derivatized Thiophene Oligomers. J. Phys. Chem. A 2020, 124, 7001–7013. [Google Scholar] [CrossRef]
- Jones, A.L.; Jiang, J.; Schanze, K.S. Excitation-Wavelength-Dependent Photoinduced Electron Transfer in a Π-Conjugated Diblock Oligomer. J. Am. Chem. Soc. 2020, 142, 12658–12668. [Google Scholar] [CrossRef] [PubMed]
- Gobeze, H.B.; Jagadesan, P.; Schanze, K.S. Photophysics and Charge Transfer in Oligo (Thiophene) Based Conjugated Diblock Oligomers. Phys. Chem. Chem. Phys. 2023, 25, 23685–23695. [Google Scholar] [CrossRef]
- Gobeze, H.B.; Martinez, D.; Schanze, K.S. Excited State Electronic Structure and Dynamics in Diblock Π-Conjugated Oligomers. J. Photochem. Photobiol. A 2023, 444, 114966. [Google Scholar] [CrossRef]
- Younus, M.; Valandro, S.; Gobeze, H.B.; Ahmed, S.; Schanze, K.S. Wavelength and Solvent Controlled Energy and Charge Transfer in Donor-Acceptor Substituted Platinum Acetylide Complexes. J. Photochem. Photobiol. A 2023, 435, 114303. [Google Scholar] [CrossRef]
- Turlington, M.D.; Gobeze, H.B.; Younus, M.; Schanze, K.S. Excitation-Wavelength-Dependent Charge Injection and Hole Localization in Diblock Oligomers Anchored to Tio2. ACS Appl. Mater. Interfaces 2023, 15, 45399–45410. [Google Scholar] [CrossRef]
- Jones, S.C.; Coropceanu, V.; Barlow, S.; Kinnibrugh, T.; Timofeeva, T.; Brédas, J.-L.; Marder, S.R. Delocalization in Platinum−Alkynyl Systems: A Metal-Bridged Organic Mixed-Valence Compound. J. Am. Chem. Soc. 2004, 126, 11782–11783. [Google Scholar] [CrossRef]
- Haque, A.; Al-Balushi, R.A.; Al-Busaidi, I.J.; Khan, M.S.; Raithby, P.R. Rise of Conjugated Poly-Ynes and Poly(Metalla-Ynes): From Design through Synthesis to Structure–Property Relationships and Applications. Chem. Rev. 2018, 118, 8474–8597. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, H.-B. Linear Neutral Platinum–Acetylide Moiety: Beyond the Links. Chem. Commun. 2014, 50, 5171–5186. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.; Rettig, W.; Bonafoux, D.; Lapouyade, R. Photoinduced Intramolecular Charge Transfer in a Series of Differently Twisted Donor−Acceptor Biphenyls as Revealed by Fluorescence. J. Phys. Chem. A 1999, 103, 3388–3401. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.; Seger, R.; Mullen, K.M.; van Stokkum, I.H. Glotaran: A Java-Based Graphical User Interface for the R Package Timp. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef]
- Gish, M.K.; Jones, A.L.; Papanikolas, J.M.; Schanze, K.S. Role of Structure in Ultrafast Charge Separation and Recombination in Naphthalene Diimide End-Capped Thiophene Oligomers. J. Phys. Chem. C 2018, 122, 18802–18808. [Google Scholar] [CrossRef]
- Cekli, S.; Winkel, R.W.; Alarousu, E.; Mohammed, O.F.; Schanze, K.S. Triplet Excited State Properties in Variable Gap Π-Conjugated Donor–Acceptor–Donor Chromophores. Chem. Sci. 2016, 7, 3621–3631. [Google Scholar] [CrossRef]
- Kissling, G.P.; Ruhstaller, B.; Pernstich, K.P. Measuring Frontier Orbital Energy Levels of Oled Materials Using Cyclic Voltammetry in Solution. Org. Electron. 2023, 122, 106888. [Google Scholar] [CrossRef]
- Cardona, C.M.; Li, W.; Kaifer, A.E.; Stockdale, D.; Bazan, G.C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Adv. Mater. 2011, 23, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Valandro, S.R.; Li, Z.; Kim, S.; Schanze, K.S. Photoinduced Intramolecular Electron Transfer in Phenylene Ethynylene Naphthalimide Oligomers. J. Phys. Chem. A 2021, 125, 3863–3873. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Grumstrup, E.M.; Gilligan, A.T.; Papanikolas, J.M.; Schanze, K.S. Light-Harvesting Polymers: Ultrafast Energy Transfer in Polystyrene-Based Arrays of Π-Conjugated Chromophores. J. Phys. Chem. B 2014, 118, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.L.; Gish, M.K.; Zeman, C.J.I.V.; Papanikolas, J.M.; Schanze, K.S. Photoinduced Electron Transfer in Naphthalene Diimide End-Capped Thiophene Oligomers. J. Phys. Chem. A 2017, 121, 9579–9588. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Fujitsuka, M.; Majima, T. Photoaccelerated Hole Transfer in Oligothiophene Assemblies. J. Phys. Chem. C 2017, 121, 649–655. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, N.; Ho, C.-L.; Fu, Y.; Lau, W.-S.; Xie, Z.; Wang, L.; Wong, W.-Y. Synthesis, Characterization, Photophysical and Photovoltaic Properties of New Donor–Acceptor Platinum(II) Acetylide Complexes. J. Organomet. Chem. 2016, 812, 2–12. [Google Scholar] [CrossRef]
- Akkuratov, A.V.; Susarova, D.K.; Kozlov, O.V.; Chernyak, A.V.; Moskvin, Y.L.; Frolova, L.A.; Pshenichnikov, M.S.; Troshin, P.A. Design of (X-Dadad)N Type Copolymers for Efficient Bulk Heterojunction Organic Solar Cells. Macromolecules 2015, 48, 2013–2021. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gobeze, H.B.; Younus, M.; Turlington, M.D.; Ahmed, S.; Schanze, K.S. Characterization of Excited-State Electronic Structure in Diblock π-Conjugated Oligomers with Adjustable Linker Electronic Coupling. Molecules 2024, 29, 2678. https://doi.org/10.3390/molecules29112678
Gobeze HB, Younus M, Turlington MD, Ahmed S, Schanze KS. Characterization of Excited-State Electronic Structure in Diblock π-Conjugated Oligomers with Adjustable Linker Electronic Coupling. Molecules. 2024; 29(11):2678. https://doi.org/10.3390/molecules29112678
Chicago/Turabian StyleGobeze, Habtom B., Muhammed Younus, Michael D. Turlington, Sohel Ahmed, and Kirk S. Schanze. 2024. "Characterization of Excited-State Electronic Structure in Diblock π-Conjugated Oligomers with Adjustable Linker Electronic Coupling" Molecules 29, no. 11: 2678. https://doi.org/10.3390/molecules29112678
APA StyleGobeze, H. B., Younus, M., Turlington, M. D., Ahmed, S., & Schanze, K. S. (2024). Characterization of Excited-State Electronic Structure in Diblock π-Conjugated Oligomers with Adjustable Linker Electronic Coupling. Molecules, 29(11), 2678. https://doi.org/10.3390/molecules29112678