
The Dependence of Hydrophobic Interactions on the Shape of Solute Surface

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
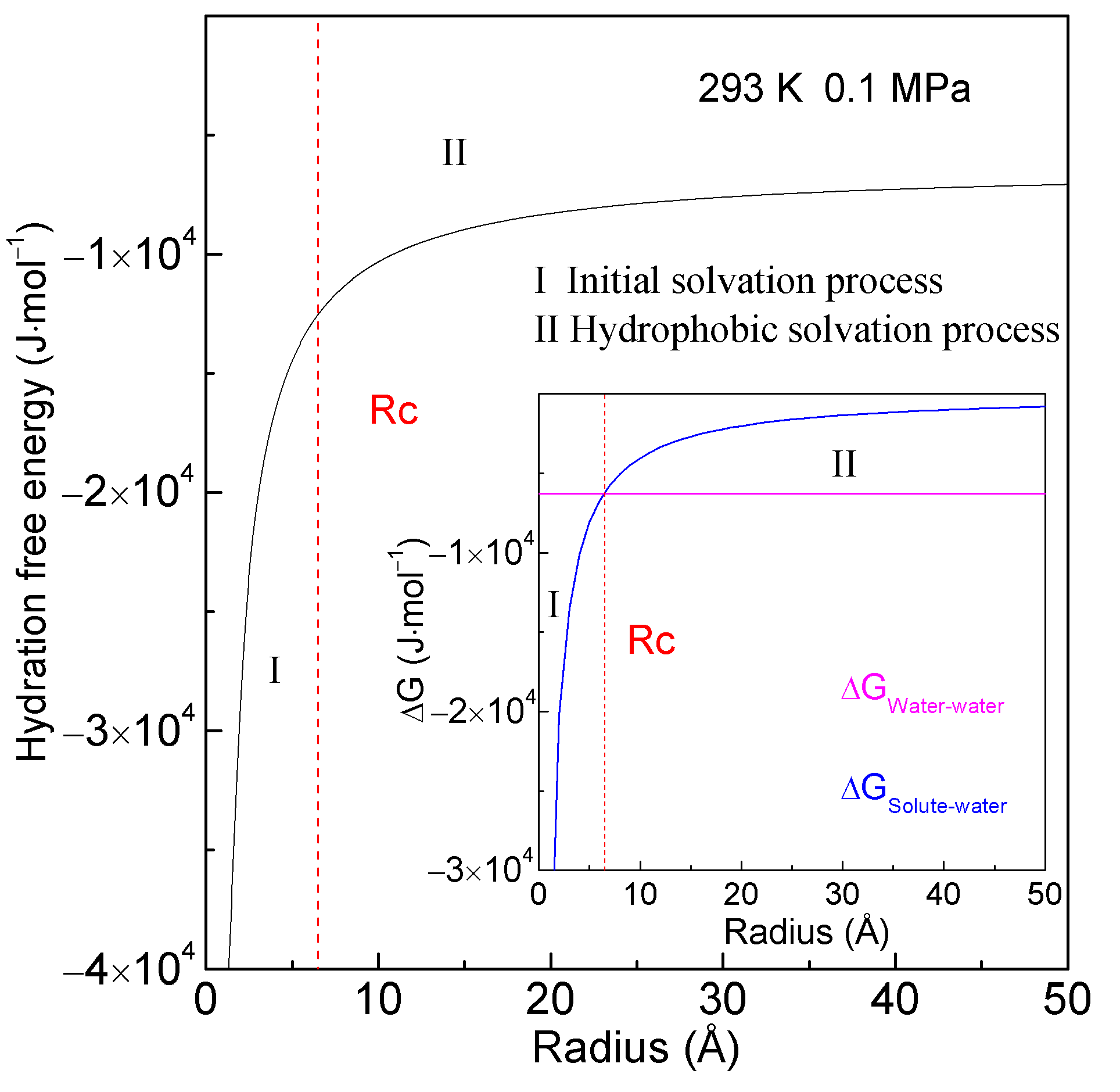
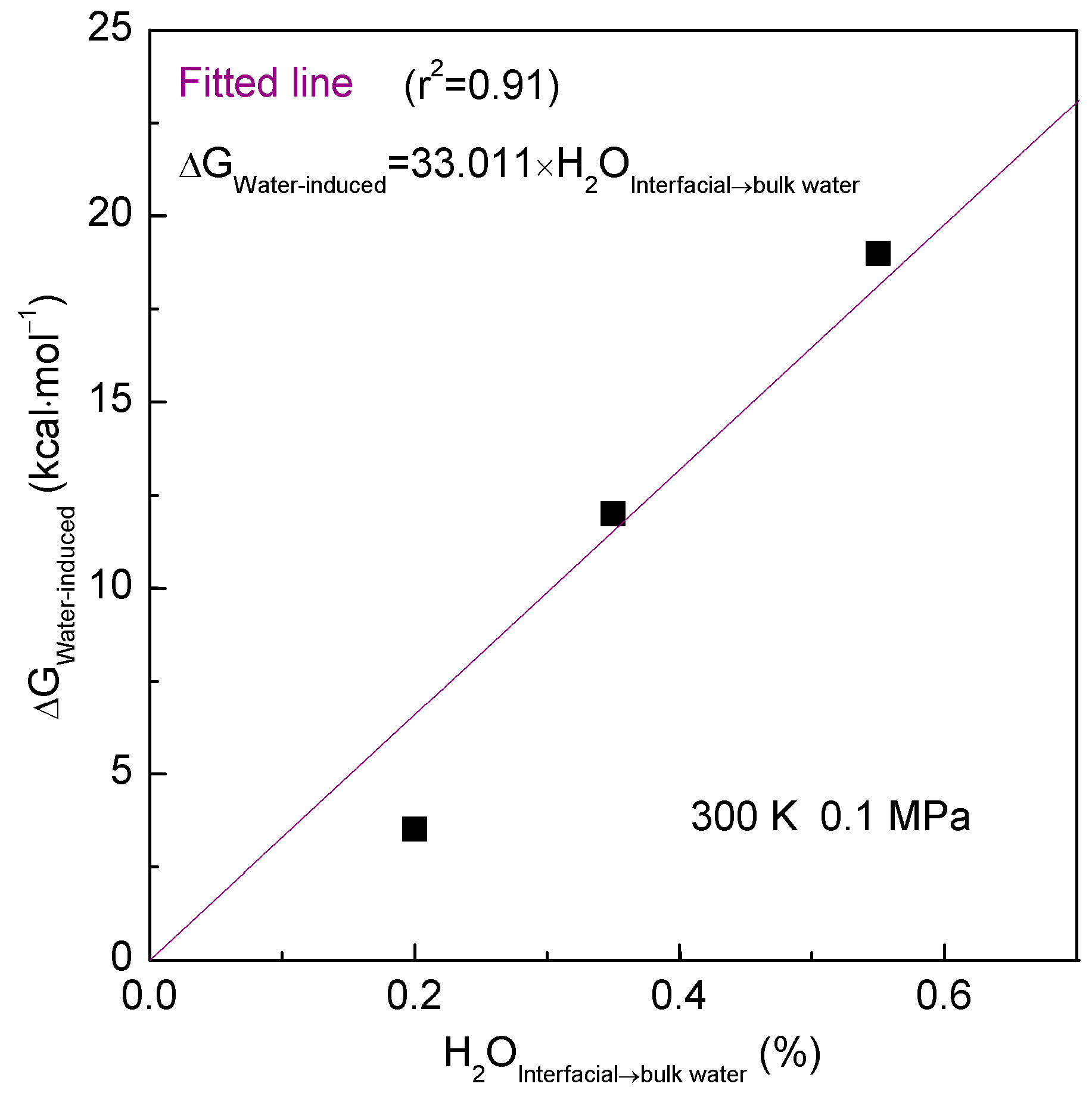
2.1. Thermodynamic Analysis
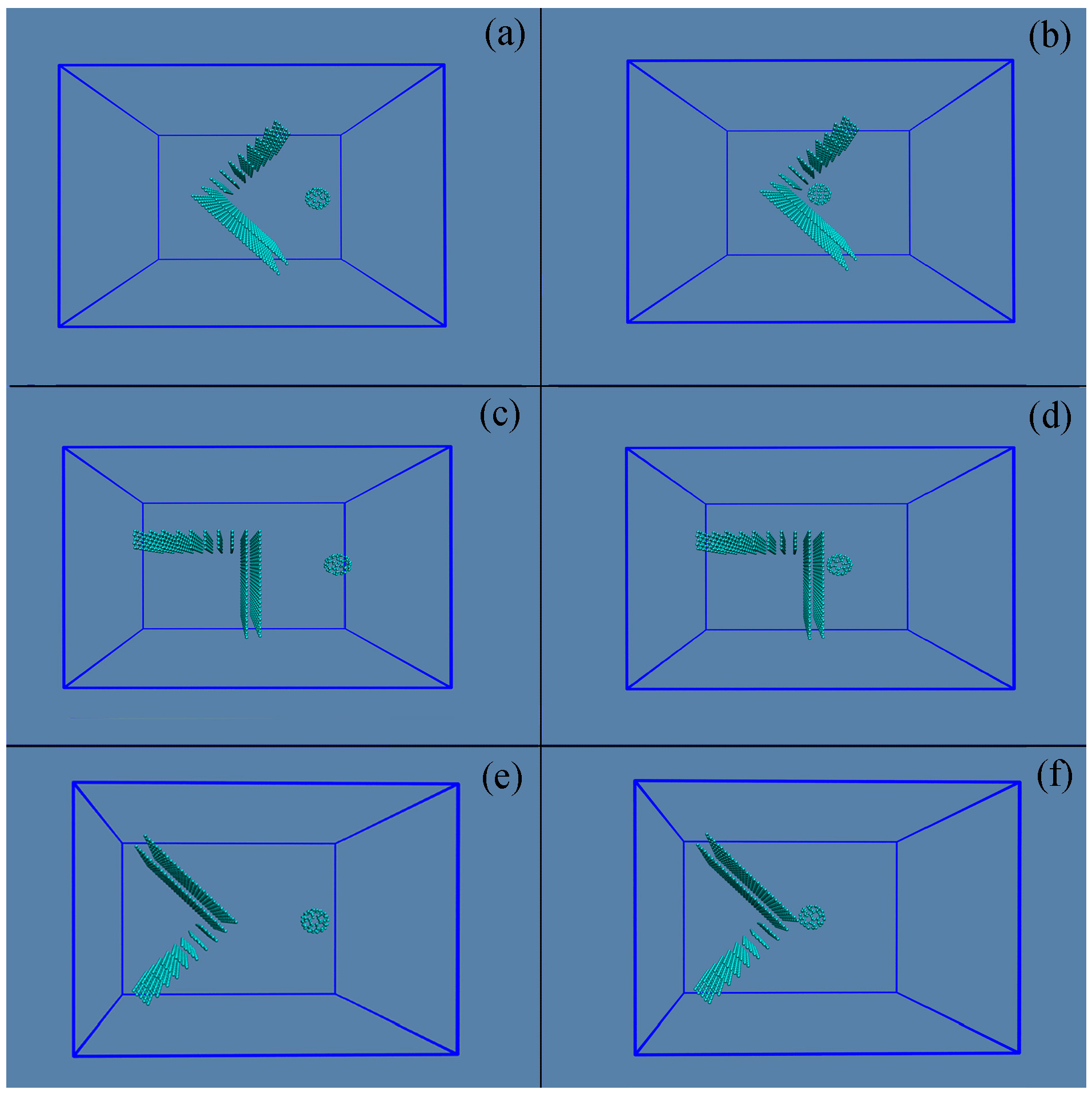
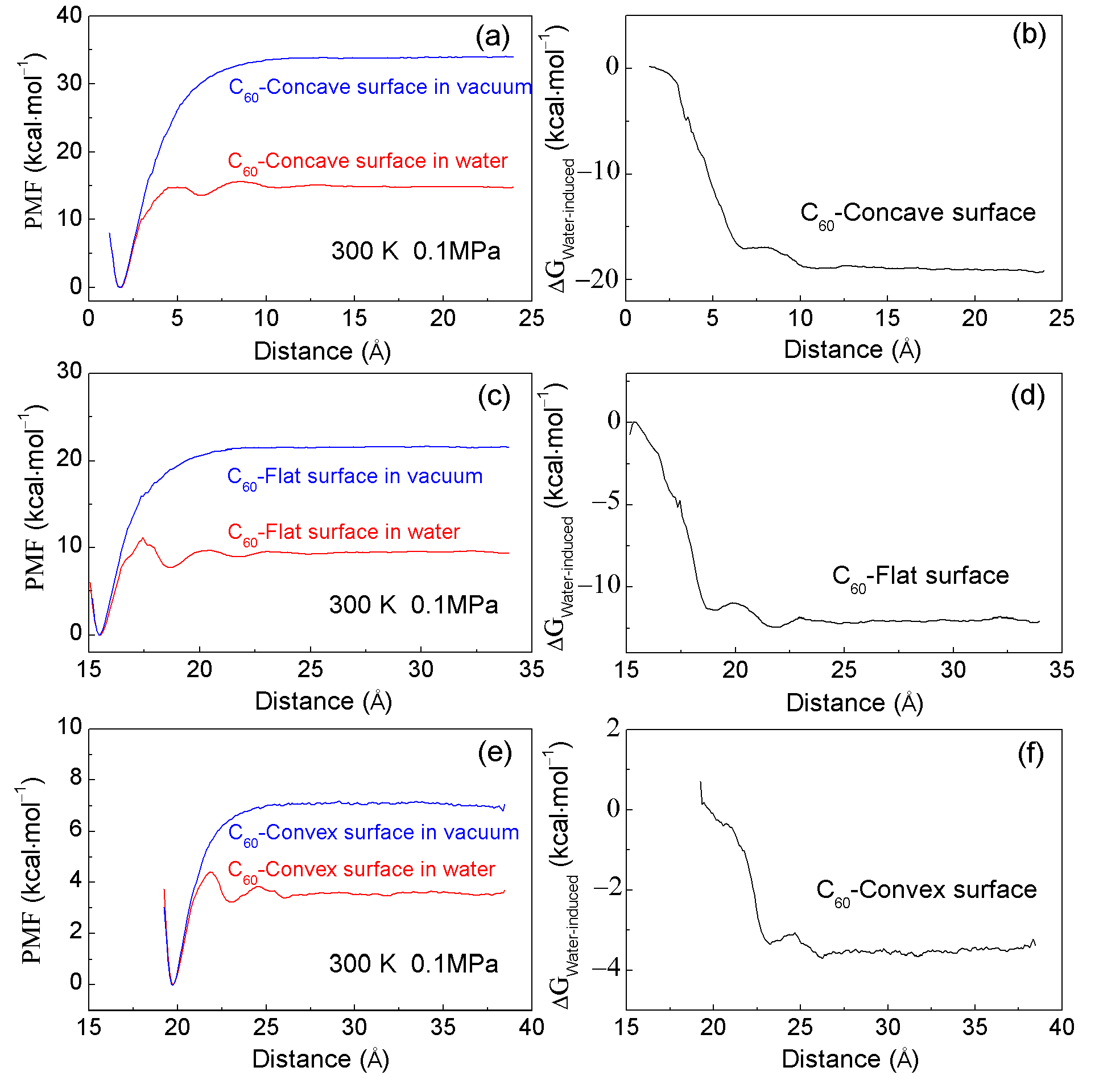
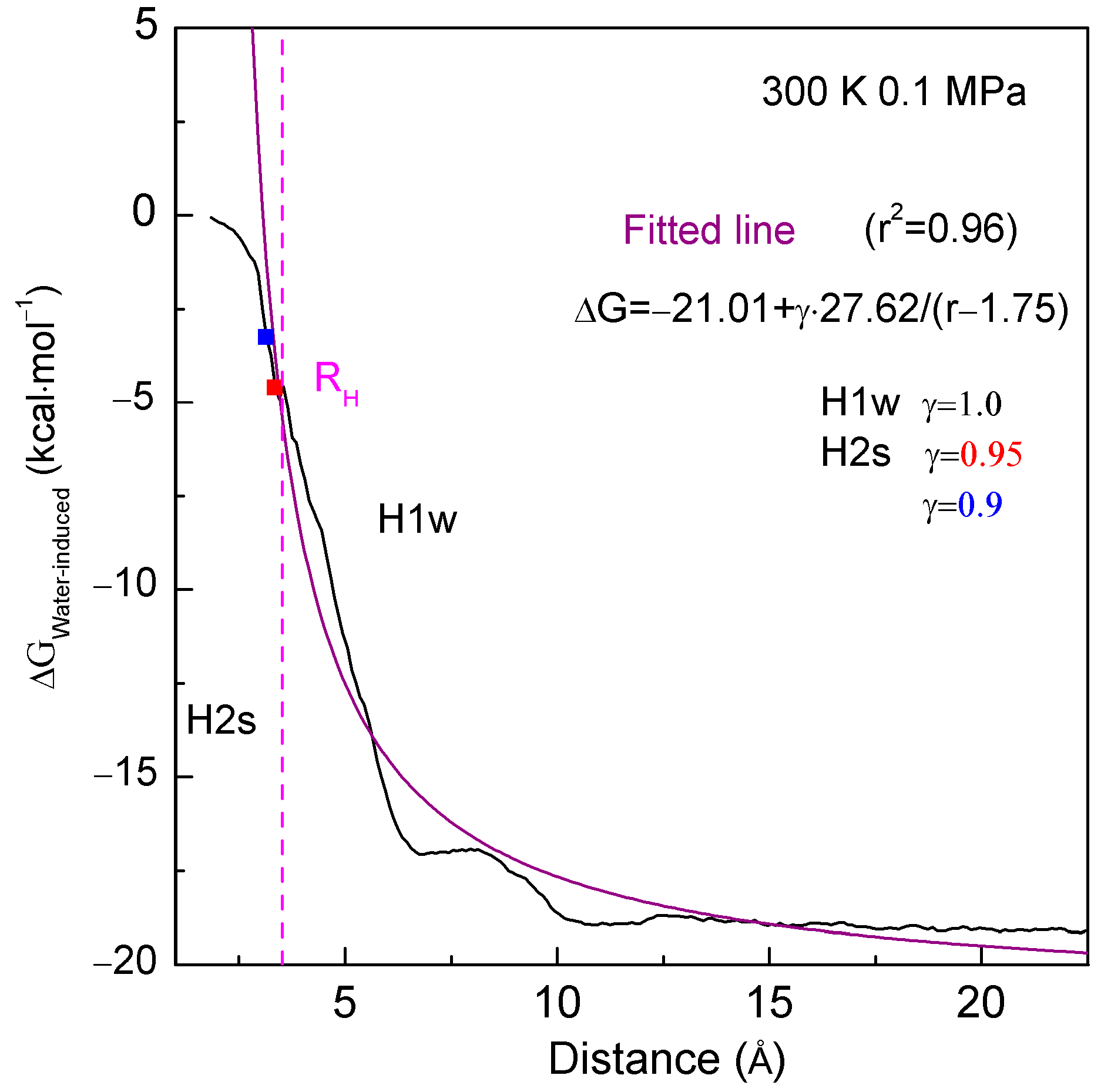
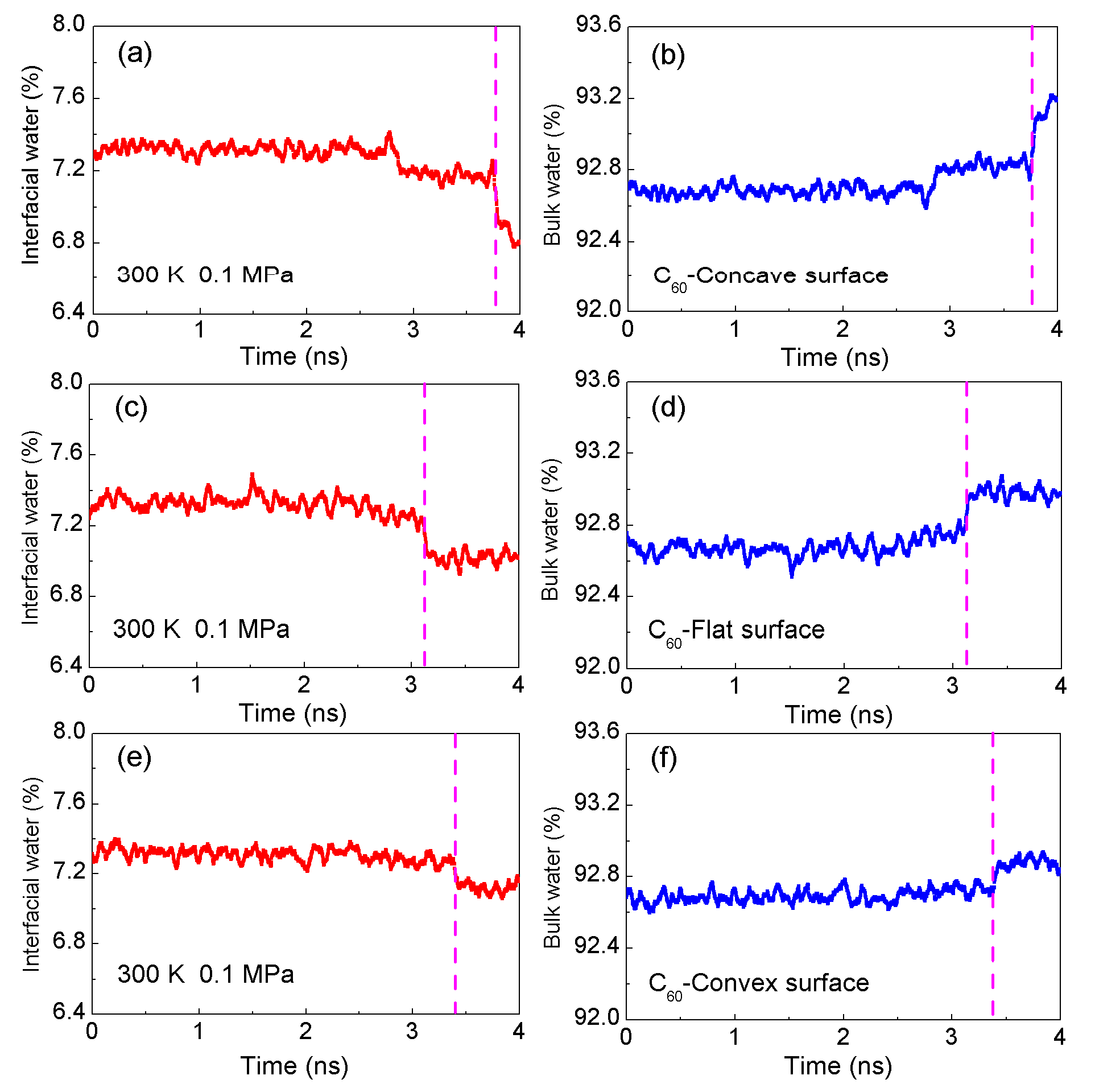
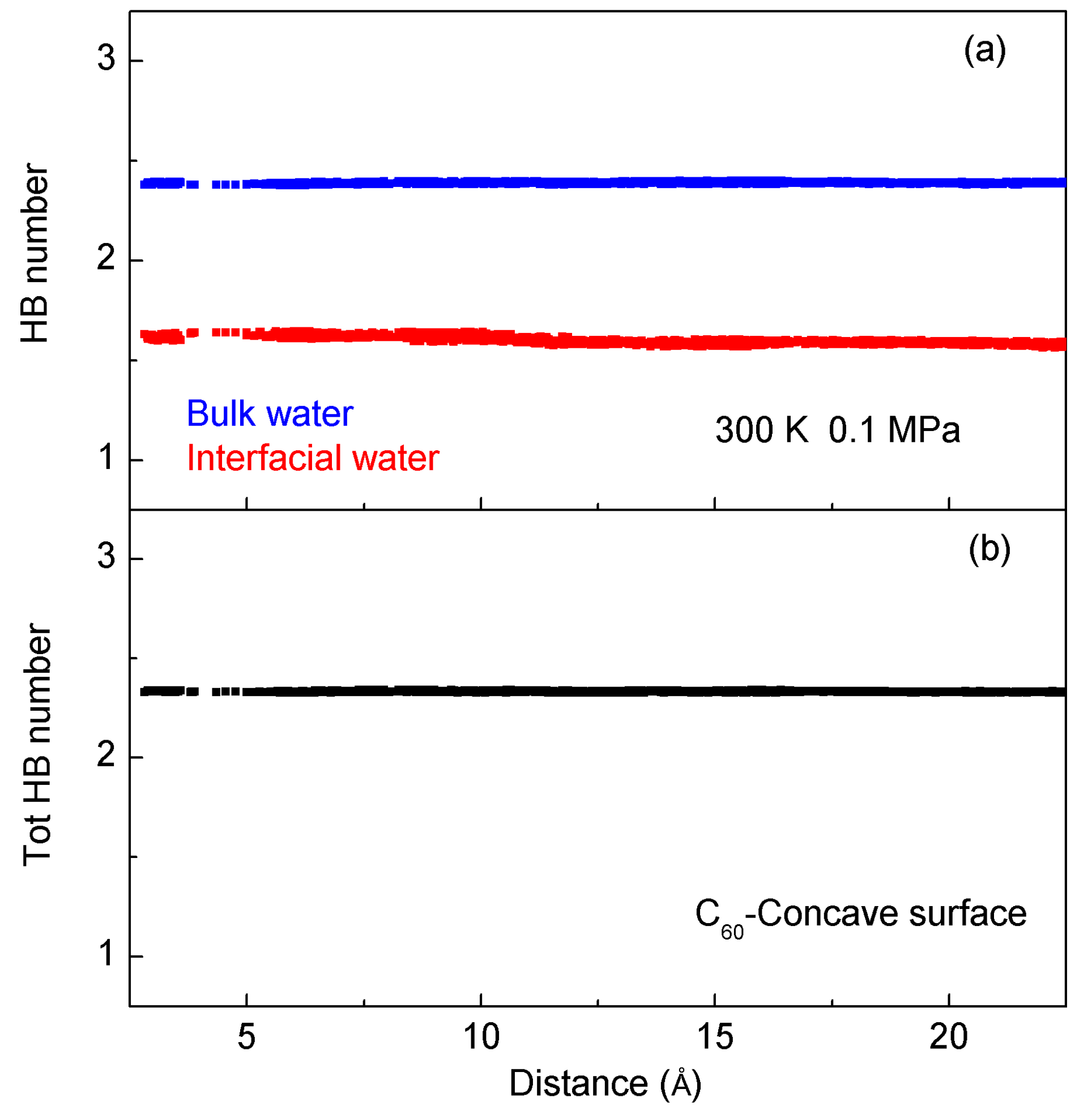
2.2. MD Simulations
3. Method
3.1. Simulated Systems
3.2. PMF Calculations
4. Conclusions
- (1)
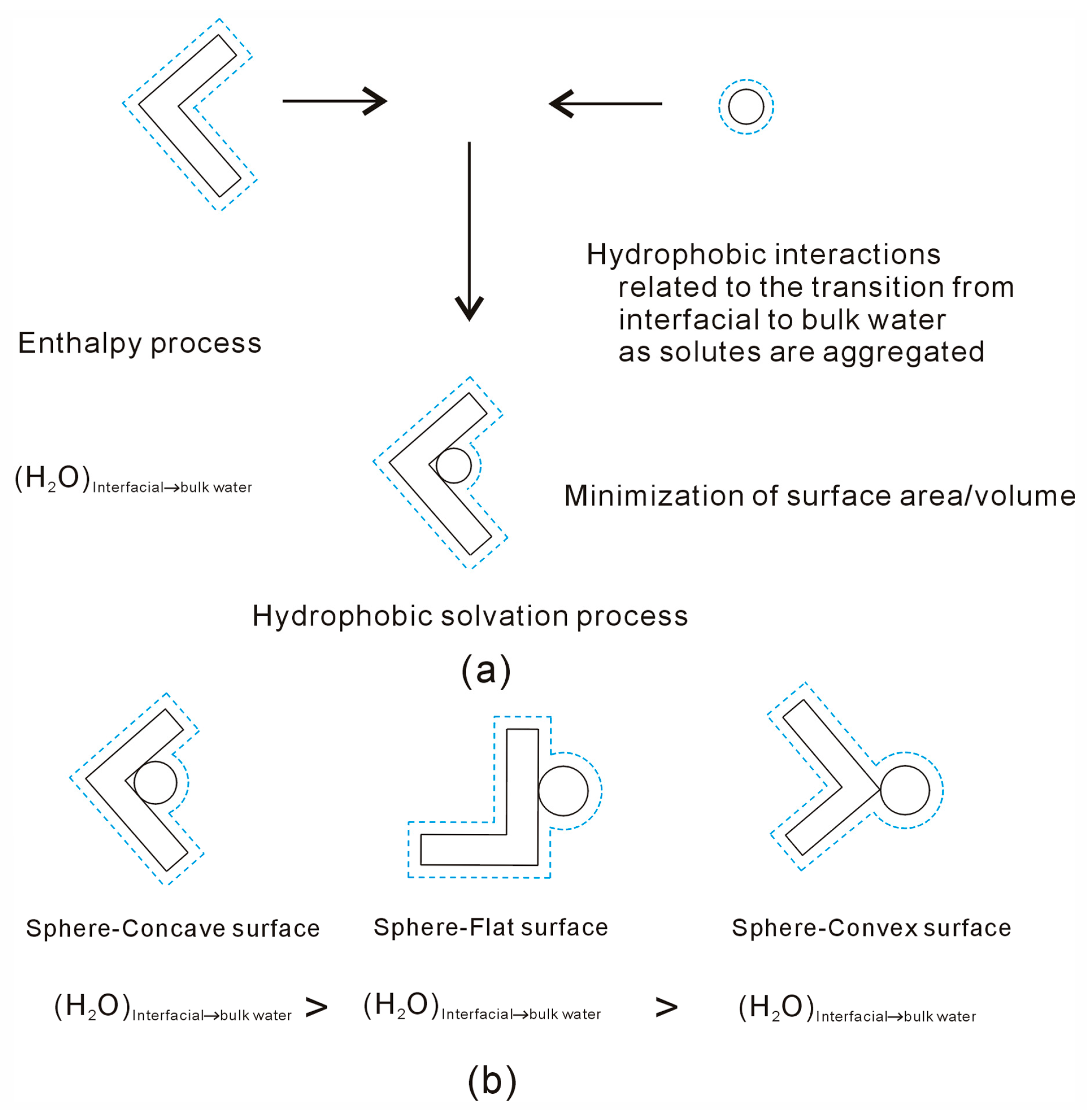
- Solutes primarily impact the hydrogen bonds of interfacial water, which may be weaker than those of bulk water. Due to hydrophobic interactions, the solutes may be attracted and tend to be aggregated to maximize the hydrogen bonding of water. As solutes associate in water, the strength of hydrophobic interactions may be closely related to the water molecules transformed from interfacial to bulk water.
- (2)
- It is inferred that hydrophobic interactions are influenced by the geometric shape of the solute. This dependence on the shape of the solute surface aligns with the directional nature of hydrophobic processes, suggesting that solutes dissolve in a manner that minimizes their surface area-to-volume ratio.
- (3)
- Hydrophobic interactions exhibit a correlation with the geometric shape of dissolved solutes. This understanding can be applied to interpret the molecular packing parameter commonly utilized in studying the self-assembly behavior of amphiphilic molecules in aqueous solutions.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tanford, C. Hydrophobic free energy, micelle formation and the association of proteins with amphiphiles. J. Mol. Biol. 1972, 67, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 1976, 72, 1525–1568. [Google Scholar] [CrossRef]
- Nagarajan, R. Molecular packing parameter and surfactant self-assembly: The neglected role of the surfactant tail. Langmuir 2002, 18, 31–38. [Google Scholar] [CrossRef]
- Frank, H.S.; Evans, M.W. Free volume and entropy in condensed systems III. Entropy in binary liquid mixtures; partial molal entropy in dilute solutions; structure and thermodynamics in aqueous electrolytes. J. Chem. Phys. 1945, 13, 507–532. [Google Scholar] [CrossRef]
- Stillinger, F.H. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J. Solution Chem. 1973, 2, 141–158. [Google Scholar] [CrossRef]
- Huang, D.M.; Geissler, P.L.; Chandler, D. Scaling of hydrophobic solvation free energies. J. Phys. Chem. B 2001, 105, 6704–6709. [Google Scholar] [CrossRef]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at small and large length scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Huang, D.M.; Chandler, D. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc. Natl. Acad. Sci. USA 2000, 97, 8324–8327. [Google Scholar] [CrossRef]
- Davis, J.G.; Gierszal, K.P.; Wang, P.; Ben-Amotz, D. Water structural transformation at molecular hydrophobic interfaces. Nature 2012, 491, 582–585. [Google Scholar] [CrossRef]
- Rego, N.B.; Patel, A.J. Understanding hydrophobic effects: Insights from water density fluctuations. Annu. Rev. Condens. Matter Phys. 2022, 13, 303–324. [Google Scholar] [CrossRef]
- Grade, S. Sticky when dry. Nat. Chem. 2020, 12, 587–588. [Google Scholar] [CrossRef]
- Ben-Amotz, D. Water-mediated hydrophobic interactions. Annu. Rev. Phys. Chem. 2016, 67, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.W.; Sullivan, M.R.; Long, J.A.; Tang, D.; Nguyen, T.; Ben-Amotz, D.; Gibb, B.C.; Ashbaugh, H.S. Spontaneous drying of non-polar deep-cavity cavitand pockets in aqueous solution. Nat. Chem. 2020, 12, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G. Molecular binding under water’s influence. Nat. Chem. 2010, 2, 906–907. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q. The physical origin of hydrophobic effects. Chem. Phys. Lett. 2017, 672, 21–25. [Google Scholar] [CrossRef]
- Sun, Q.; Su, X.W.; Cheng, C.B. The dependence of hydrophobic interactions on the solute size. Chem. Phys. 2019, 516, 199–205. [Google Scholar] [CrossRef]
- Sun, Q. The Hydrophobic effects: Our current understanding. Molecules 2022, 27, 7009. [Google Scholar] [CrossRef] [PubMed]
- Hillyer, M.B.; Gibb, B.C. Molecular shape and the hydrophobic effect. Annu. Rev. Phys. Chem. 2016, 67, 307–329. [Google Scholar] [CrossRef]
- Rasaiah, J.C.; Garde, S.; Hummer, G. Water in nonpolar confinement: From nanotubes to proteins and beyond. Annu. Rev. Phys. Chem. 2008, 59, 713–740. [Google Scholar] [CrossRef]
- Auer, B.M.; Skinner, J.L. IR and Raman spectra of liquid water: Theory and interpretation. J. Chem. Phys. 2008, 128, 224511. [Google Scholar] [CrossRef]
- Bakker, H.J.; Skinner, J.L. Vibrational spectroscopy as a probe of structure and dynamics in liquid water. Chem. Rev. 2010, 110, 1498–1517. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q. Local statistical interpretation for water structure. Chem. Phys. Lett. 2013, 568, 90–94. [Google Scholar] [CrossRef]
- Stanley, H.E.; Teixeira, J. Interpretation of the unusual behavior of H2O and D2O at low temperatures: Tests of a percolation model. J. Chem. Phys. 1980, 73, 3404–3422. [Google Scholar] [CrossRef]
- Nilsson, A.; Pettersson, L.G.M. Perspective on the structure of liquid water. Chem. Phys. 2011, 389, 1–34. [Google Scholar] [CrossRef]
- Russo, J.; Tanaka, H. Understanding water’s anomalies with locally favoured structures. Nat. Commun. 2014, 5, 3556. [Google Scholar] [CrossRef]
- Hamm, P. Markov state model of the two-state behaviour of water. J. Chem. Phys. 2016, 145, 134501. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Tanaka, H. Microscopic structural descriptor of liquid water. J. Chem. Phys. 2018, 148, 124503. [Google Scholar] [CrossRef] [PubMed]
- Skinner, L.B.; Huang, C.; Schlesinger, D.; Pettersson, L.G.M.; Nilsson, A.; Benmore, C.J. Benchmark Oxygen-Oxygen pair-distribution function of ambient water from X-ray diffraction measurements with a wide Q-range. J. Chem. Phys. 2013, 138, 074506. [Google Scholar] [CrossRef]
- Smith, J.D.; Cappa, C.D.; Wilson, K.R.; Cohen, R.C.; Geissler, P.L.; Saykally, R.J. Unified description of temperature-dependent hydrogen-bond rearrangements in liquid water. Proc. Natl. Acad. Sci. USA 2005, 102, 14171–14174. [Google Scholar] [CrossRef]
- Hura, G.; Sorenson, J.M.; Glaeser, R.M.; Head-Gordon, T. A high-quality X-ray scattering experiment on liquid water at ambient conditions. J. Chem. Phys. 2000, 113, 9140. [Google Scholar] [CrossRef]
- Sun, Q.; Guo, Y. Vibrational sum frequency generation spectroscopy of the air/water interface. J. Mol. Liquids 2016, 213, 28–32. [Google Scholar] [CrossRef]
- Dorsey, N.E. Properties of Ordinary Water Substance; ACS Monograph No. 81; Reinhold Publishing Corp.: New York, NY, USA, 1940. [Google Scholar]
- Sun, Q.; Wang, W.; Cui, S. Directional nature of hydrophobic interactions: Implications for the mechanism of molecular recognition. Chem. Phys. 2021, 547, 111200. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Computer simulation of hydrophobic hydration forces on stacked plates at short range. J. Phys. Chem. 1995, 99, 2893–2899. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Molecular dynamics study of the dependence of water solvation free energy on solute curvature and surface area. J. Phys. Chem. 1995, 99, 2885–2892. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Giovanbattista, N.; Debenedetti, P.G.; Rossky, P.J. Hydration behavior under confinement by nanoscale surfaces with patterned hydrophobicity and hydrophilicity. J. Phys. Chem. C 2007, 111, 1323–1332. [Google Scholar] [CrossRef]
- Liu, P.; Huang, X.; Zhou, R.; Berne, B.J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature 2005, 437, 159–162. [Google Scholar] [CrossRef]
- ten Wolde, P.R.; Chandler, D. Drying induced hydrophobic polymer collapse. Proc. Natl. Acad. Sci. USA 2002, 99, 6539–6543. [Google Scholar] [CrossRef]
- Leibler, L.; Orland, H.; Wheeler, J.C. Theory of critical micelle concentration for solutions of block copolymers. J. Chem. Phys. 1983, 79, 3550. [Google Scholar] [CrossRef]
- Noolandi, J.; Hong, K.M. Theory of block copolymer micelles in solution. Macromolecules 1983, 16, 1443–1448. [Google Scholar] [CrossRef]
- Whitmore, D.; Noolandi, J. Theory of micelle formation in block copolymer-homopolymer blends. Macromolecules 1985, 18, 657–665. [Google Scholar] [CrossRef]
- Nagarajan, R. Constructing a molecular theory of self-assembly: Interplay of ideas from surfactants and block copolymers. Adv. Colloid Interfac. 2017, 244, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Monte Carlo free energy estimates using non-Boltzmann sampling: Application to the sub-critical Lennard-Jones fluid. Chem. Phys. Lett. 1974, 28, 578–581. [Google Scholar] [CrossRef]
- Torrie, G.M.; Valleau, J.P. Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. Mu1tidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995, 16, 1339–1350. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]
- Ashbaugh, H.S.; Weiss, K.; Williams, S.M.; Meng, B.; Surampudi, L.N. Temperature and pressure dependence of methane correlations and osmotic second virial coefficients in water. J. Phys. Chem. B 2015, 119, 6280–6294. [Google Scholar] [CrossRef]
- Zangi, R. Driving force for hydrophobic interaction at different length scales. J. Phys. Chem. B 2011, 115, 2303–2311. [Google Scholar] [CrossRef]
- Uddin, N.M.; Capaldi, F.M.; Farouk, B. Molecular dynamics simulations of carbon nanotube dispersions in water: Effects of nanotube length, diameter, chirality and surfactant structures. Comput. Mat. Sci. 2012, 53, 133–144. [Google Scholar] [CrossRef]
- Li, L.W.; Bedrov, D.; Smith, G.D. Water-induced interactions between carbon nanoparticles. J. Phys. Chem. B 2006, 110, 10509–10513. [Google Scholar] [CrossRef] [PubMed]
- Ball, P. Water as an active constituent in cell biology. Chem. Rev. 2008, 108, 74–108. [Google Scholar] [CrossRef]
- Ball, P. Water is an active matrix of life for cell and molecular biology. Proc. Natl. Acad. Sci. USA 2017, 114, 13327–13335. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.D.; Neilson, G.W.; Enderby, J.E. Ions in water: Characterizing the forces that control chemical processes and biological structure. Biophys. Chem. 2007, 128, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Cappa, C.D.; Smith, J.D.; Messer, B.M.; Cohen, R.C.; Saykally, R.J. Effects of cations on the hydrogen bond network of liquid water: New results from X-ray absorption spectroscopy of liquid microjets. J. Phys. Chem. B 2006, 110, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Omta, A.W.; Kropman, M.F.; Woutersen, S.; Bakker, H.J. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science 2003, 301, 347–349. [Google Scholar] [CrossRef]
- Moilanen, D.E.; Wong, D.; Rosenfeld, D.E.; Fenn, E.E.; Fayer, M.D. Ion-water hydrogen-bond switching observed with 2D IR vibrational echo chemical exchange spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Turton, D.A.; Hunger, J.; Hefter, G.; Buchner, R.; Wynne, K. Glasslike behavior in aqueous electrolyte solutions. J. Chem. Phys. 2008, 128, 161102. [Google Scholar] [CrossRef]
- Kumar, R.; Schmidt, J.R.; Skinner, J.L. Hydrogen bonding definitions and dynamics in liquid water. J. Chem. Phys. 2007, 126, 204107. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD-Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- GrossField, A. WHAM: An Implementation of the Weighted Histogram Analysis Method. Available online: http://membrane.urmc.rochester.edu/content/wham/version2.0.10 (accessed on 1 January 2020).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-Z.; Chen, Y.-N.; Sun, Q. The Dependence of Hydrophobic Interactions on the Shape of Solute Surface. Molecules 2024, 29, 2601. https://doi.org/10.3390/molecules29112601
Liu Y-Z, Chen Y-N, Sun Q. The Dependence of Hydrophobic Interactions on the Shape of Solute Surface. Molecules. 2024; 29(11):2601. https://doi.org/10.3390/molecules29112601
Chicago/Turabian StyleLiu, Yu-Zhen, Yan-Nan Chen, and Qiang Sun. 2024. "The Dependence of Hydrophobic Interactions on the Shape of Solute Surface" Molecules 29, no. 11: 2601. https://doi.org/10.3390/molecules29112601
APA StyleLiu, Y.-Z., Chen, Y.-N., & Sun, Q. (2024). The Dependence of Hydrophobic Interactions on the Shape of Solute Surface. Molecules, 29(11), 2601. https://doi.org/10.3390/molecules29112601