
Signal Transduction of Transient Receptor Potential TRPM8 Channels: Role of PIP5K, Gq-Proteins, and c-Jun


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
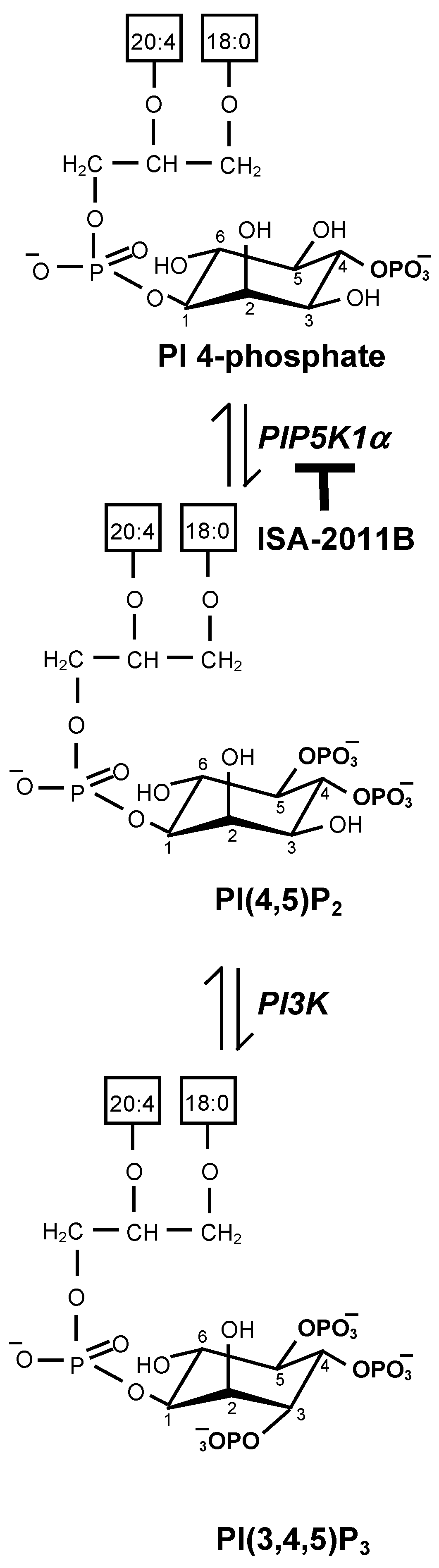
2.1. Biosynthesis of Phosphatidylinositol 4,5-Bisphosphate
2.2. Pharmacological Inhibition of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the TRPM8 Channel
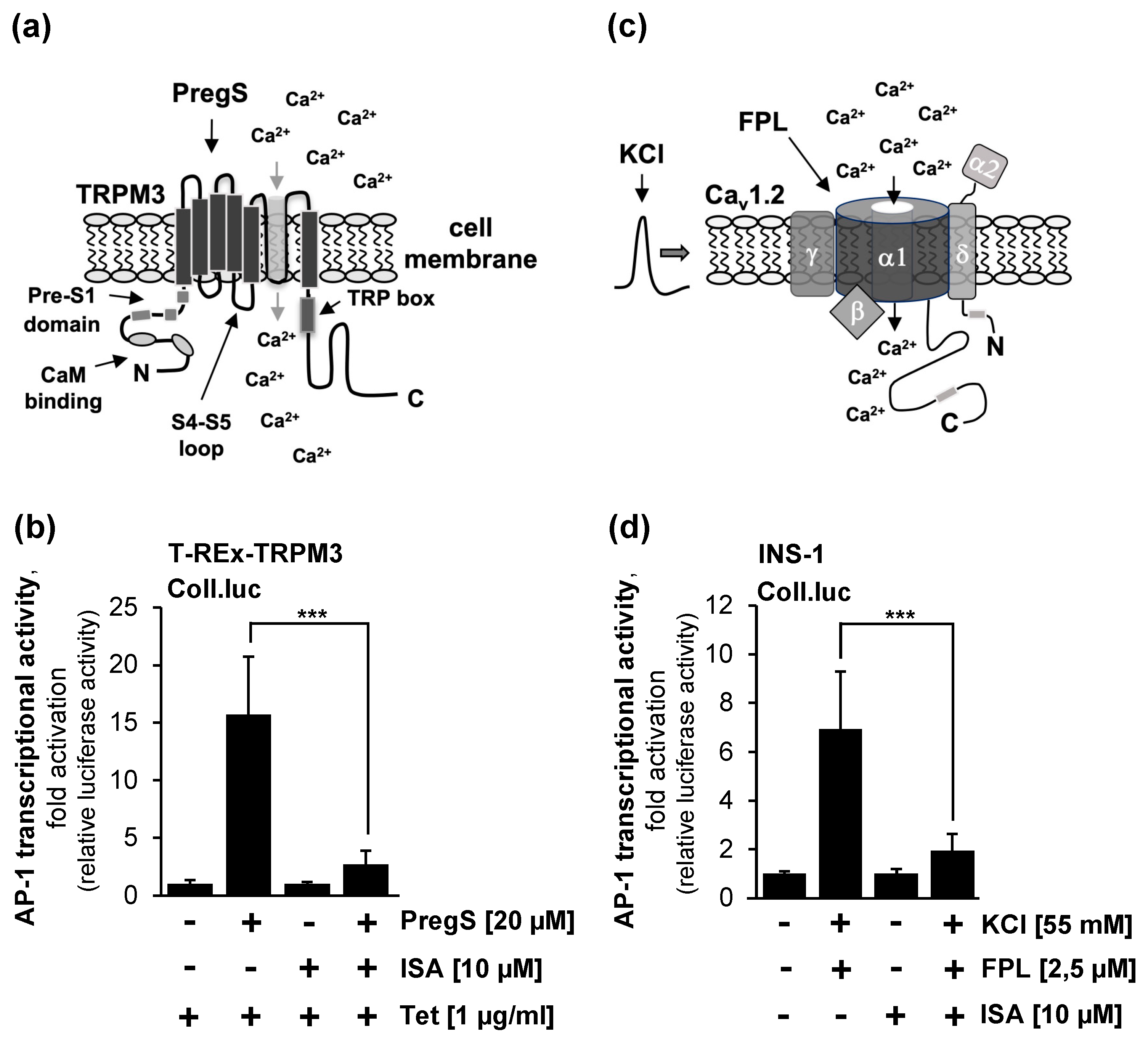
2.3. Pharmacological Inhibition of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the TRPM3 Channel
2.4. Pharmacological Inhibition of Phosphatidylinositol 4,5-Bisphosphate Biosynthesis Interferes with Signaling via the Voltage-Gated Cav1.2. Ca2+ Channel
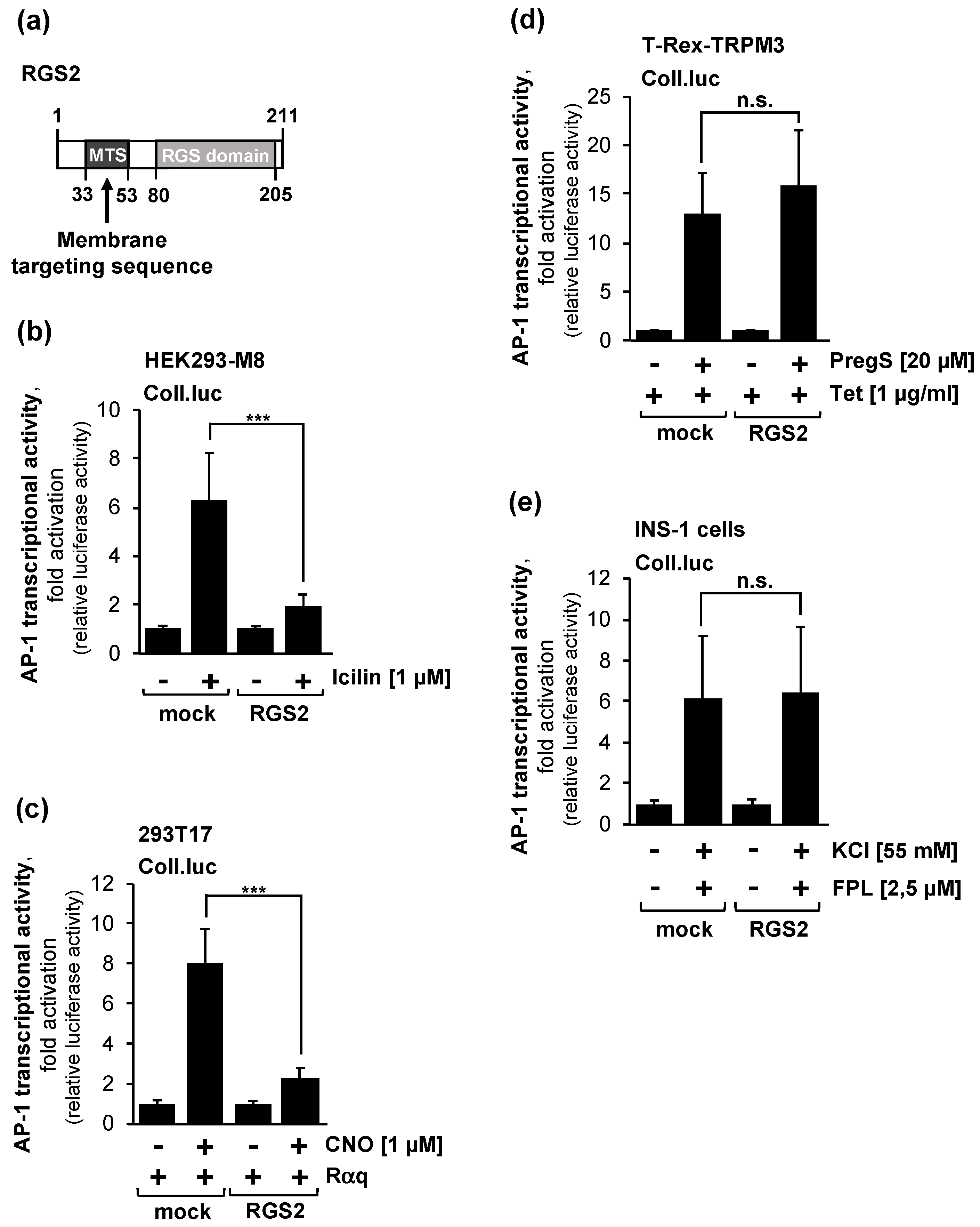
2.5. Overexpression of Regulator of G-Protein Signaling-2 (RGS2) Blocks the Activation of AP-1 after Stimulation of TRPM8 Channels
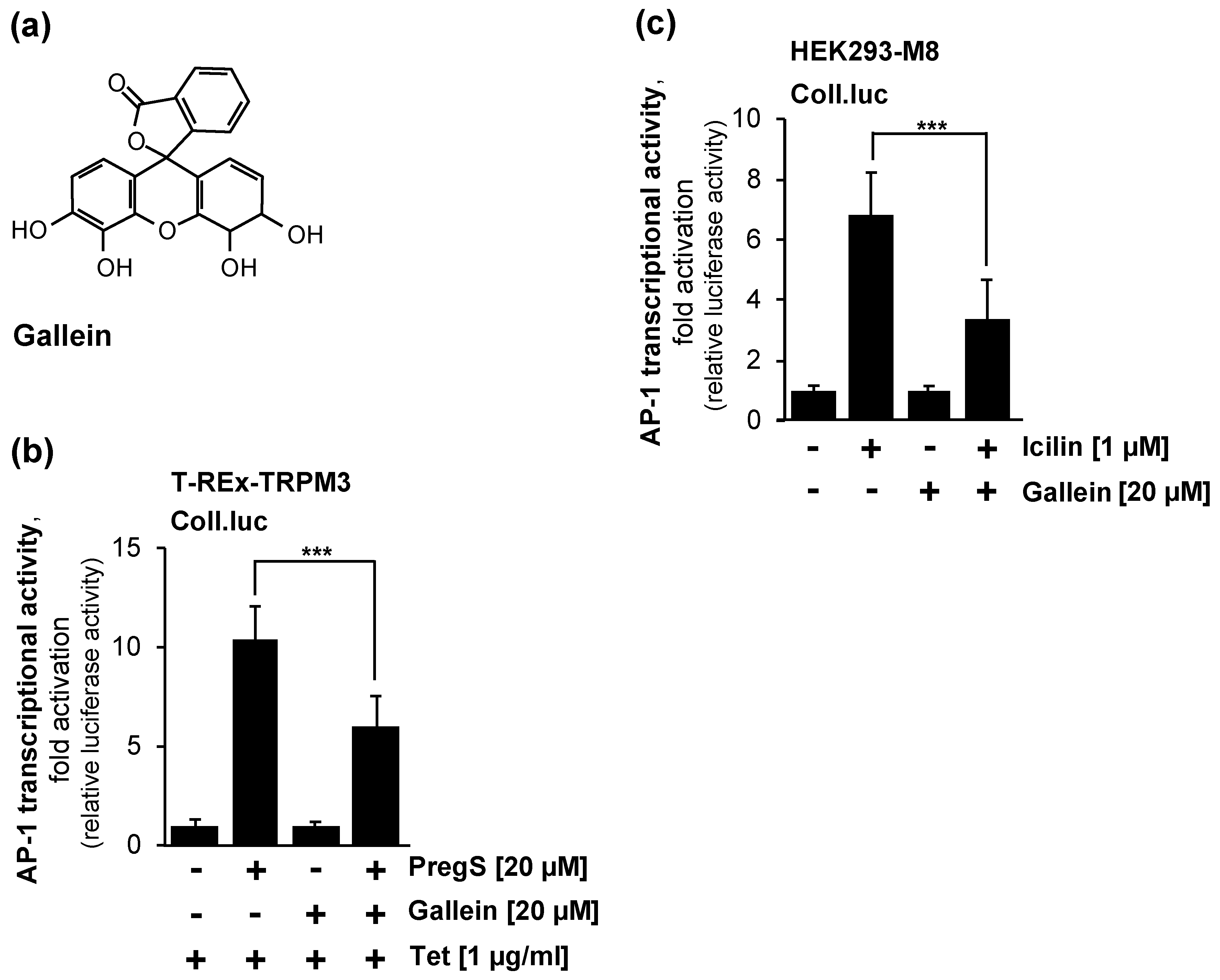
2.6. Pharmacological Inhibition of Gβγ Interferes with Signaling via the TRPM3 and TRPM8 Channels
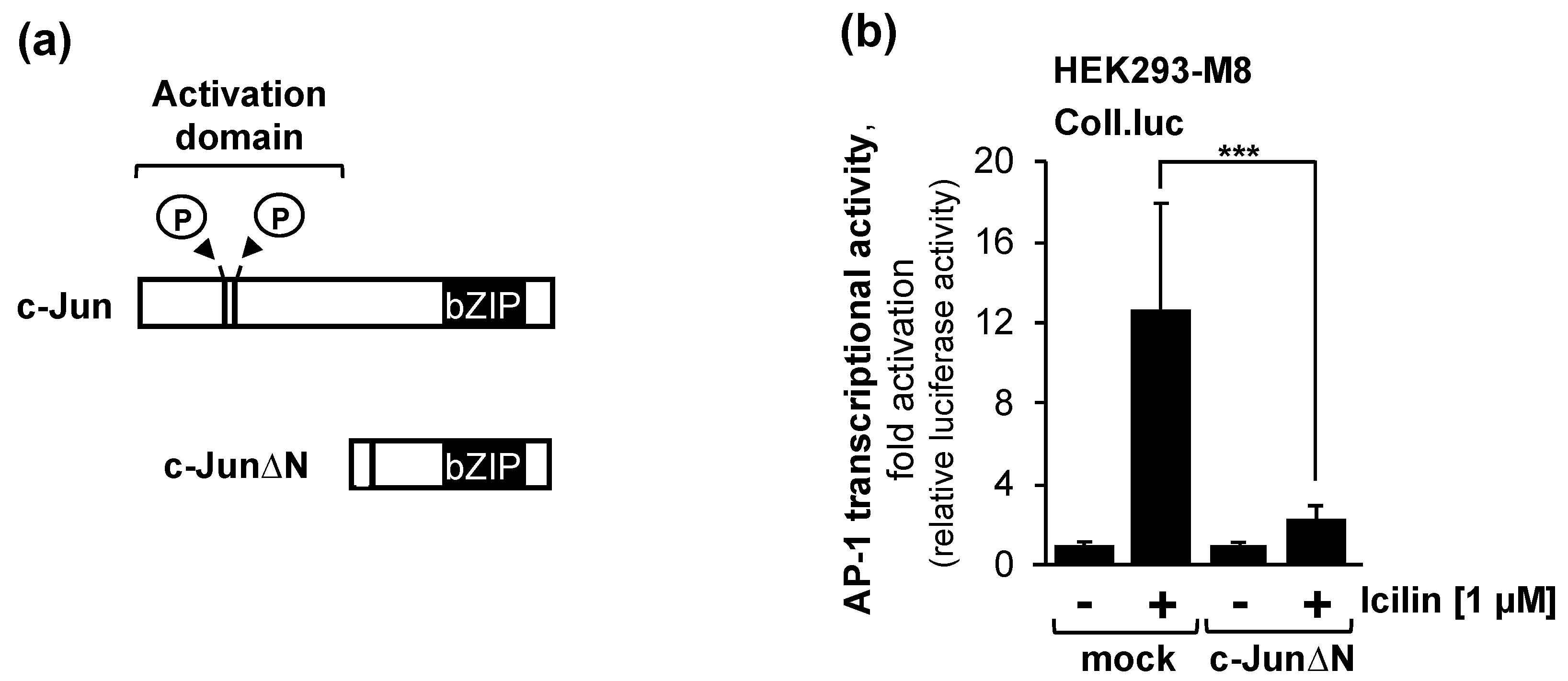
2.7. The Transcription Factor c-Jun or c-Jun-Dimeriziation Proteins Is Essential for the Activation of AP-1 after Stimulation of TRPM8 Channels with Icilin
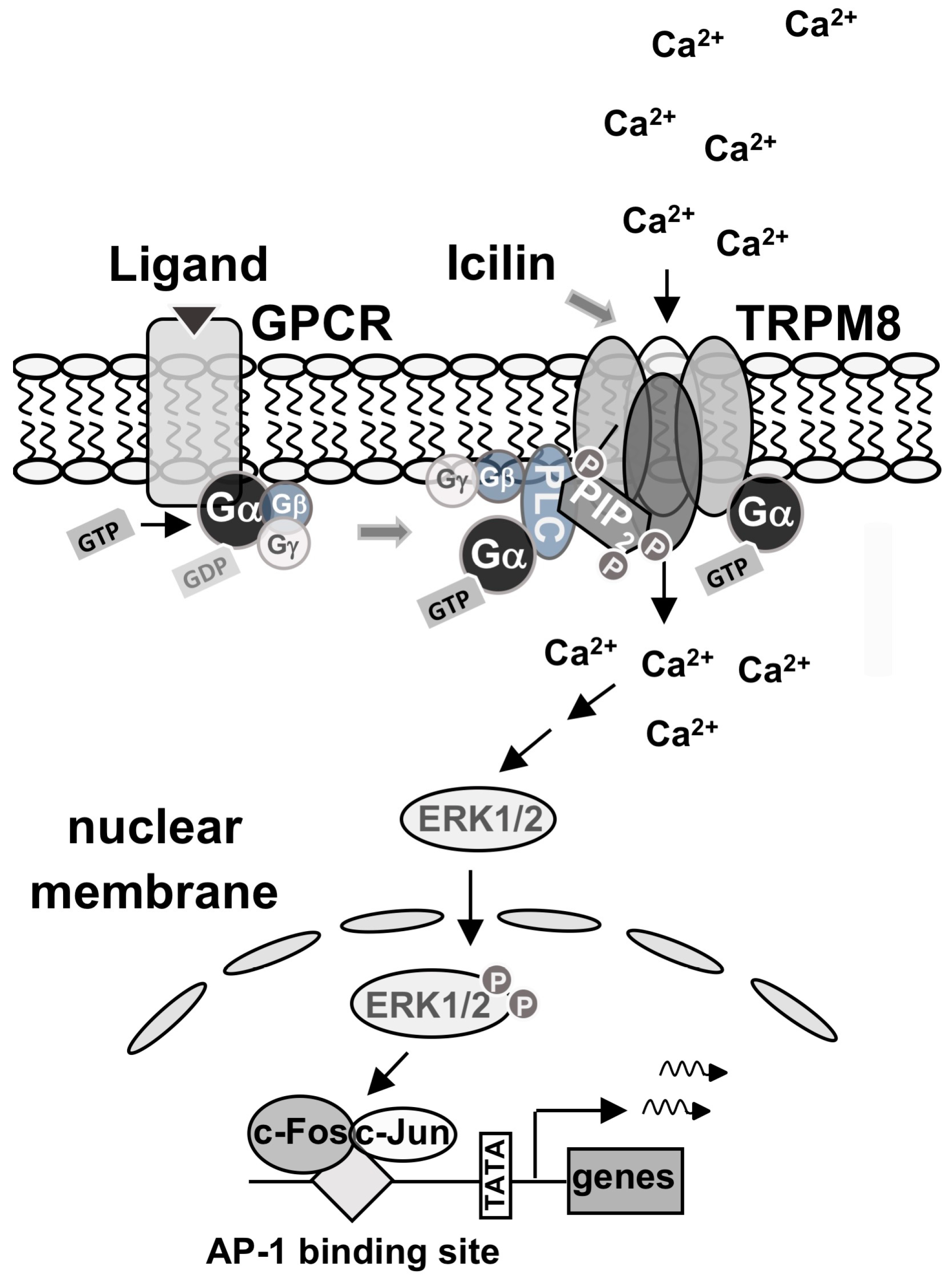
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Lentiviral Gene Transfer
4.3. Reporter Gene Assay
4.4. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKemy, D.D.; Neuhausser, W.M.; Julius, D. Identification of a cold receptor reveals a general role of TRP channels in thermosensatio. Nature 2002, 416, 52–58. [Google Scholar] [CrossRef]
- Peier, A.M.; Moqrich, A.; Hergarden, A.C.; Reeve, A.J.; Andersson, D.A.; Story, G.M.; Earley, T.J.; Dragoni, I.; McIntyre, P.; Bevan, S.; et al. A TRP channel that senses cold stimuli and menthol. Cell 2002, 108, 705–715. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, N. TRPM8 in health and disease: Cold sensing and beyond. Adv. Exp. Med. Biol. 2011, 704, 185–208. [Google Scholar] [PubMed]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stuck, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Colburn, R.W.; Lubin, M.L.; Stone, D.J., Jr.; Wang, Y.; Lawrence, D.; D’Andrea, M.R.; Brandt, M.R.; Liu, Y.; Flores, C.M.; Qin, N. Attenuated cold sensitivity in TRPM8 null mice. Neuron 2007, 54, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Dhaka, A.; Murray, A.N.; Mathur, J.; Earley, T.J.; Petrus, M.J.; Patapoutian, A. TRPM8 is required for cold sensation in mice. Neuron 2007, 54, 371–378. [Google Scholar] [CrossRef]
- Fleig, A.; Chubanov, V. TRPM8. In Mammalian Transient Receptor Potential (TRP) Cation Channels; Handbook of Experimental Pharmacology; Nilius, B., Flockerzi, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; Volume 222, pp. 547–579. [Google Scholar]
- Hantute-Ghesquier, A.; Haustrate, A.; Prevarskaya, N.; Lehen´kyi, V. TRPM family channels in cancer. Pharmaceuticals 2018, 11, 58. [Google Scholar] [CrossRef]
- Izquierdo, C.; Martín-Martínez, M.; Gómez-Monterrey, I.; González-Muñiz, R. TRPM8 channels: Advances in structural studies and pharmacological modulation. Int. J. Mol. Sci. 2021, 22, 8502. [Google Scholar] [CrossRef]
- Ramachandran, R.; Hyun, E.; Zhao, L.; Lapointe, T.K.; Chapman, K.; Hirota, C.L.; Ghosh, S.; McKemy, D.D.; Vergnolle, N.; Beck, P.L.; et al. TRPM8 activation attenuates inflammatory responses in mouse models of colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 7476–7481. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.I.; Liu, B.; Jabba, S.V.; Achanta, S.; Morris, J.B.; Jordt, S.-E. Transient receptor potential cation channel subfamily M member 8 channels mediate the anti-inflammatory effects of eucalyptol. Brit. J. Pharmacol. 2017, 174, 867–879. [Google Scholar] [CrossRef]
- Ulrich, M.; Wissenbach, U.; Thiel, G. The super-cooling compound icilin stimulates c-Fos and Egr-1 expression and activity involving TRPM8 channel activation, Ca2+ ion influx and activation of the ternary complex factor Elk-1. Biochem. Pharmacol. 2020, 177, 113936. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Backes, T.M.; Welck, J.; Steinhausen, S.; Fischer, A.-L.; Langfermann, D.S.; Ulrich, M.; Wissenbach, U.; Rössler, O.G. Pharmacological inhibition of TRPM8-induced gene transcription. Biochem. Pharmacol. 2019, 170, 113678. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Calmodulin regulates transient receptor potential TRPM3 and TRPM8-induced gene transcription. Int. J. Mol. Sci. 2023, 24, 7902. [Google Scholar] [CrossRef]
- Thiel, G.; Rössler, O.G. Expression of the C-terminal domain of phospholipase Cβ3 inhibits signaling via Gαq-coupled receptors and transient receptor potential channels. Int. J. Mol. Sci. 2022, 23, 9590. [Google Scholar] [CrossRef] [PubMed]
- Rohacs, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nature Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef]
- Liu, B.; Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 1674–1681. [Google Scholar] [CrossRef]
- Daniels, R.L.; Takashima, Y.; McKemy, D.D. Activity of the neuronal cold sensor TRPM8 is regulated by phospholipase C via the phospholipid phosphoinositol 4,5-bisphosphate. J. Biol. Chem. 2009, 284, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Yudin, Y.; Lukacs, V.; Cao, C.; Rohács, T. Decrease in phosphatidylinositol 4,5-bisphosphate levels mediates desensitization of the cold sensor TRPM8 channels. J. Physiol. 2011, 589, 6007–6027. [Google Scholar] [CrossRef]
- Hammond, G.R.V.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef]
- Tóth, B.I.; Konrad, M.; Ghosh, D.; Mohr, F.; Halaszovich, C.R.; Leitner, M.G.; Vriens, J.; Oberwinkler, J.; Voets, T. Regulation of the transient receptor potential channel TRPM3 by phosphoinositides. J. Gen. Physiol. 2015, 146, 51–63. [Google Scholar] [CrossRef]
- Zhang, X.; Mak, S.; Li, L.; Parra, A.; Denlinger, B.; Belmonte, C.; McNaughton, P.A. Direct inhibition of the cold-activated TRPM8 ion channel by Gαq. Nat. Cell Biol. 2012, 14, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Klasen, K.; Hollatz, D.; Zielke, S.; Gisselmann, G.; Hatt, H.; Wetzel, C.H. The TRPM8 ion channel comprises direct Gq protein-activating capacity. Pflügers Arch.—Eur. J. Physiol. 2012, 463, 779–797. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Welck, J.; Wissenbach, U.; Rössler, O.G. Dihydrotestosterone activates AP-1 in prostate cancer cells. Int. J. Biochem. Cell Biol. 2019, 110, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Van den Bout, I.; Divecha, N. PIP5K-driven Ptd(4,5)P2 synthesis: Regulation and cellular functions. J. Cell Sci. 2009, 122, 3837–3850. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef]
- Semenas, J.; Hedblom, A.; Miftakhova, R.R.; Sarwar, M.; Larsson, R.; Shcherbina, L.; Johansson, M.E.; Härkönen, P.; Sterner, O.; Persson, J.L. The role of PI3K/AKT-related PIP5K1α and the discovery of its selective inhibitor for treatment of advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3689–E3698. [Google Scholar] [CrossRef] [PubMed]
- Kunkl, M.; Porciello, N.; Mastrogiovanni, M.; Capuano, C.; Lucantoni, F.; Moretti, C.; Persson, J.L.; Galandrini, R.; Buzzetti, R.; Tuosto, L. ISA-2011B, a phosphatidylinositol 4-phosphate 5-kinase α inhibitor, impairs CD28-dependent costimulatory and pro-inflammatory signals in human T lymphocytes. Front. Immunol. 2017, 8, 502. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rössler, O.G. Hyperforin activates gene transcription involving transient receptor potential C6 channels. Biochem. Pharmacol. 2017, 129, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Rössler, O.G.; Thiel, G. Pregnenolone sulfate activates basic region leucine zipper transcription factors in insulinoma cells: Role of voltage-gated Ca2+ channels and transient receptor potential melastatin 3 channels. Mol. Pharmacol. 2011, 80, 1179–1189. [Google Scholar] [CrossRef]
- Backes, T.M.; Rössler, O.G.; Hui, X.; Grötzinger, C.; Lipp, P.; Thiel, G. Stimulation of TRPV1 channels activates the AP-1 transcription factor. Biochem. Pharmacol. 2018, 150, 160–169. [Google Scholar] [CrossRef]
- Loviscach, L.; Backes, T.M.; Langfermann, D.S.; Ulrich, M.; Thiel, G. Zn2+ ions inhibit gene transcription following stimulation of the Ca2+ channels Cav1.2 and TRPM3. Metallomics 2020, 12, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A.; Keim, A.; Thiel, G. Regulation of immediate-early gene transcription following activation of Gαq-coupled designer receptors. J. Cell. Biochem. 2013, 114, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Backes, T.M.; Guethlein, L.A.; Rössler, O.G. Chromatin-embedded reporter genes: Quantification of stimulus-induced gene transcription. Gene 2021, 787, 145645. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Hofmann, T.; Philipp, S.E.; Stab, J.; Chen, X.; Benoit, M.; Xue, F.; Janssens, A.; Kerselaers, S.; et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron 2011, 70, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G.; Rubil, S.; Lesch, A.; Guethlein, L.A.; Rössler, O.G. Transient receptor potential TRPM3 channels: Pharmacology, signaling, and biological functions. Pharmacol. Res. 2017, 124, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Cabanas, H.; Muraki, K.; Balinas, C.; Eaton-Fitch, N.; Staines, D.; Marshall-Gradisnik, S. Validation of impaired transient receptor potential melastatin 3 ion channel activity in natural killer cells from chronic fatigue syndrome/myalgic encephalomyelitis patients. Mol. Med. 2019, 25, 14. [Google Scholar] [CrossRef] [PubMed]
- Roelens, R.; Peigneur, A.N.F.; Voets, T.; Vriens, J. Neurodevelopmental disorders caused by variants in TRPM3. Biochim. Biophys. Acta—Mol. Cell Res. 2024, 1871, 119709. [Google Scholar] [CrossRef] [PubMed]
- Badheka, D.; Borbiro, I.; Rohacs, T. Transient receptor potential melastatin 3 is a phosphoinositide-dependent ion channel. J. Gen. Physiol. 2015, 146, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Suh, B.-C.; Leal, K.; Hille, B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron 2010, 67, 224–238. [Google Scholar] [CrossRef]
- Kehrl, J.H.; Sinnarajah, S. RGS2: A multifunctional regulator of G-protein signaling. Int. J. Biochem. Cell Biol. 2002, 34, 432–438. [Google Scholar] [CrossRef]
- Dembla, S.; Behrendt, M.; Mohr, F.; Goecke, C.; Sondermann, J.; Schneider, F.M.; Schmidt, M.; Stab, J.; Enzeroth, R.; Leitner, M.G.; et al. Ant-nociceptive action of peripheral mu-opioid receptors by G-beta-gamma protein-mediated inhibition of TRPM3 channels. eLife 2017, 6, e26280. [Google Scholar] [CrossRef] [PubMed]
- Quallo, T.; Alkhatib, O.; Gentry, C.; Andersson, D.A.; Bevan, S. G protein βγ subunits inhibit TRPM3 ion channel in sensory neurons. eLife 2017, 6, e26138. [Google Scholar] [CrossRef] [PubMed]
- Herlitze, S.; Garcia, D.E.; Mackie, K.; Hille, B.; Scheuer, T.; Catterall, W.A. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 1996, 380, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-protein betagamma subunits. Nature 1996, 380, 255–258. [Google Scholar] [CrossRef]
- Chiu, R.; Boyle, W.J.; Meek, J.; Smeal, T.; Hunter, T.; Karin, M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell 1988, 54, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Rohacs, T. Phosphoinositide regulation of TRP channels: A functional overview in the structural era. Annu. Rev. Physiol. 2024, 86, 329–355. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Carnevale, V.; Gabrielle, M.; Gianti, E.; Rohacs, T. Computational and functional studies of the PI(4,5)P2 binding site of the TRPM3 ion channel reveal interactions with other regulators. J. Biol. Chem. 2022, 298, 102547. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; MacKinnon, R. Structural and functional analyses of a GPCR-inhibited ion channel TRPM3. Neuron 2023, 111, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Wills, R.C.; Goulden, B.D.; Hammond, G.R.V. Genetically encoded lipid biosensors. Mol. Biol. Cell 2018, 29, 1526–1532. [Google Scholar] [CrossRef]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.-C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Falasca, M.; Logan, S.K.; Lehto, V.P.; Baccante, G.; Lemmon, M.A.; Schlessinger, J. Activation of phospholipase Cγ by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998, 17, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.K.; Kim, O.-H.; Hur, J.; Yu, S.H.; Lamichhane, S.; Lee, J.W.; Ojha, U.; Hong, J.H.; Lee, C.S.; Cha, J.-Y.; et al. Increased intracellular Ca2+ concentrations prevent membrane localization of PH domains through the formation of Ca2+-phosphoinositides. Proc. Natl. Acad. Sci. USA 2017, 114, 11926–11931. [Google Scholar] [CrossRef] [PubMed]
- Kadamur, G.; Ross, E.M. Intrinsinc pleckstrin homology (PH) domain motion in phospholipase C-β exposes a Gβγ protein binding site. J. Biol. Chem. 2016, 291, 11394–11406. [Google Scholar] [CrossRef] [PubMed]
- Szmanska, E.; Sobota, A.; Czurylo, E.; Kwiatkowska, K. Expression of PI(4,5)P2-binding proteins lowers the PI(4,5)P2 level and inhibits FcγRIIA-mediated cell spreading and phagocytosis. Eur. J. Immunol. 2008, 38, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Schoeber, J.P.; Topala, C.N.; Wang, X.; Diepens, R.J.; Lambers, T.T.; Hoenderop, J.G.; Bindels, R.J. RGS2 inhibits the epithelial Ca2+ channel TRPV6. J. Biol. Chem. 2006, 281, 29669–29674. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, M.; Gruss, F.; Enzeroth, R.; Dembla, S.; Zhao, S.; Crassous, P.-A.; Mohr, F.; Nys, M.; Louros, N.; Gallardo, R.; et al. The structural basis for an on-off switch controlling Gβγ-mediated inhibition of TRPM3 channels. Proc. Nat. Acad. Sci. USA 2020, 117, 29090–29100. [Google Scholar] [CrossRef] [PubMed]
- Rohács, T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv. Biol. Reg. 2013, 53, 341–355. [Google Scholar] [CrossRef]
- Falzone, M.E.; MacKinnon, R. The mechanism of Gαq regulation of PLCβ3-catalyzed PIP2 hydrolysis. Proc. Natl. Acad. Sci. USA 2023, 120, e2315011120. [Google Scholar] [CrossRef]
- Falzone, M.E.; MacKinnon, R. Gβγ activates PIP2 hydrolysis by recruiting and orienting PLCβ on the membrane surface. Proc. Natl. Acad. Sci. USA 2023, 120, e2301121120. [Google Scholar] [CrossRef]
- Stephens, L.; Jackson, T.R.; Hawkins, P.T. Activation of phosphatidylinositol 4,5-bisphosphate supply by agonists and non-hydrolysable GTP analogues. Biochem. J. 1993, 296, 481–488. [Google Scholar] [CrossRef]
- Barneda, D.; Janardan, V.; Niewczas, I.; Collins, D.M.; Cosulich, S.; Clark, J.; Stephens, L.R.; Hawkins, P.T. Acyl chain selection couples the consumption and synthesis of phosphoinositides. EMBO J. 2022, 41, e110038. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, U.; Nyako, M.; Woturski, W.; Zaitseva, I.; McLaughlin, S. Diffusion coefficient of fluorescent phosphatidylinositol 4,5-bisphosphate in the plasma membrane of cells. Mol. Biol. Cell 2008, 19, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Bödding, M.; Wissenbach, U.; Flockerzi, V. Characterization of TRPM8 as a pharmacophore receptor. Cell Calcium 2007, 42, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Naylor, J.; Milligan, C.J.; Zeng, F.; Jones, C.; Beech, D.J. Production of a specific extracellular inhibitor of TRPM3 channels. Brit. J. Pharmacol. 2008, 155, 567–573. [Google Scholar] [CrossRef]
- Hohmeier, H.E.; Mulder, H.; Chen, G.; Henkel-Rieger, R.; Prentki, M.; Newgard, C.B. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin release. Diabetes 2000, 49, 424–430. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, G.; Rössler, O.G. Signal Transduction of Transient Receptor Potential TRPM8 Channels: Role of PIP5K, Gq-Proteins, and c-Jun. Molecules 2024, 29, 2602. https://doi.org/10.3390/molecules29112602
Thiel G, Rössler OG. Signal Transduction of Transient Receptor Potential TRPM8 Channels: Role of PIP5K, Gq-Proteins, and c-Jun. Molecules. 2024; 29(11):2602. https://doi.org/10.3390/molecules29112602
Chicago/Turabian StyleThiel, Gerald, and Oliver G. Rössler. 2024. "Signal Transduction of Transient Receptor Potential TRPM8 Channels: Role of PIP5K, Gq-Proteins, and c-Jun" Molecules 29, no. 11: 2602. https://doi.org/10.3390/molecules29112602
APA StyleThiel, G., & Rössler, O. G. (2024). Signal Transduction of Transient Receptor Potential TRPM8 Channels: Role of PIP5K, Gq-Proteins, and c-Jun. Molecules, 29(11), 2602. https://doi.org/10.3390/molecules29112602