

Methane Formation Induced via Face-to-Face Orientation of Cyclic Fe Porphyrin Dimer in Photocatalytic CO2 Reduction


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
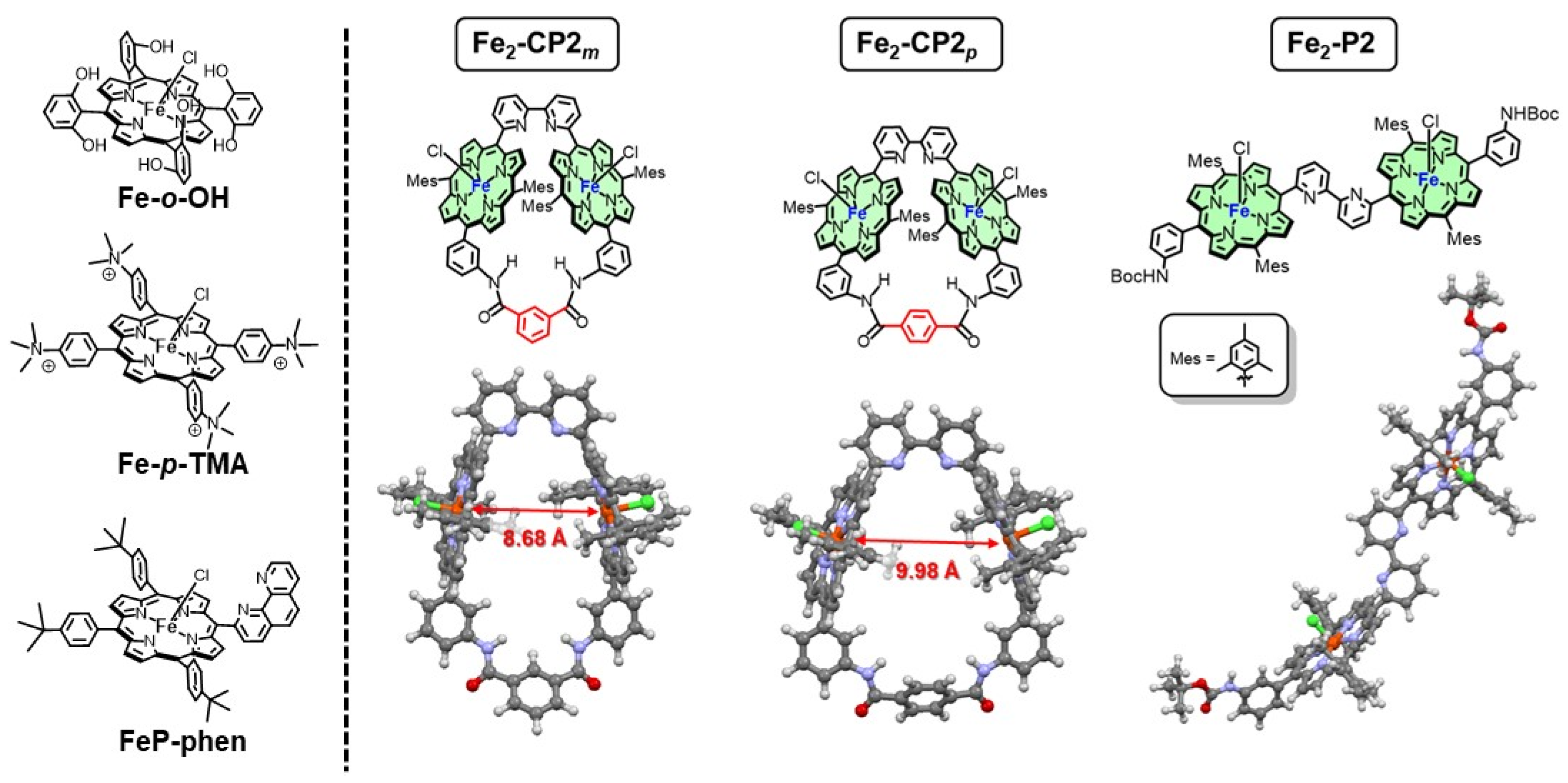
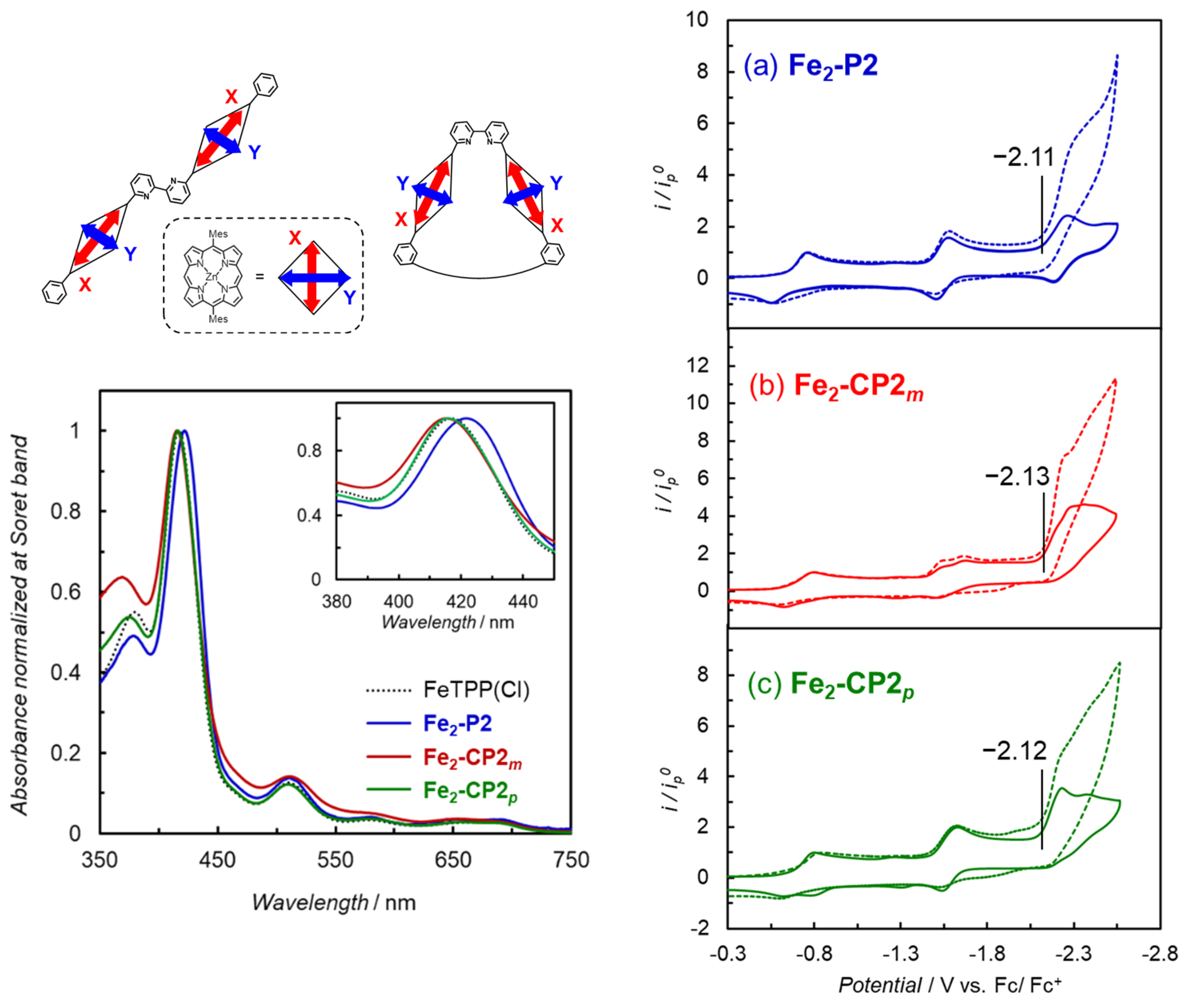
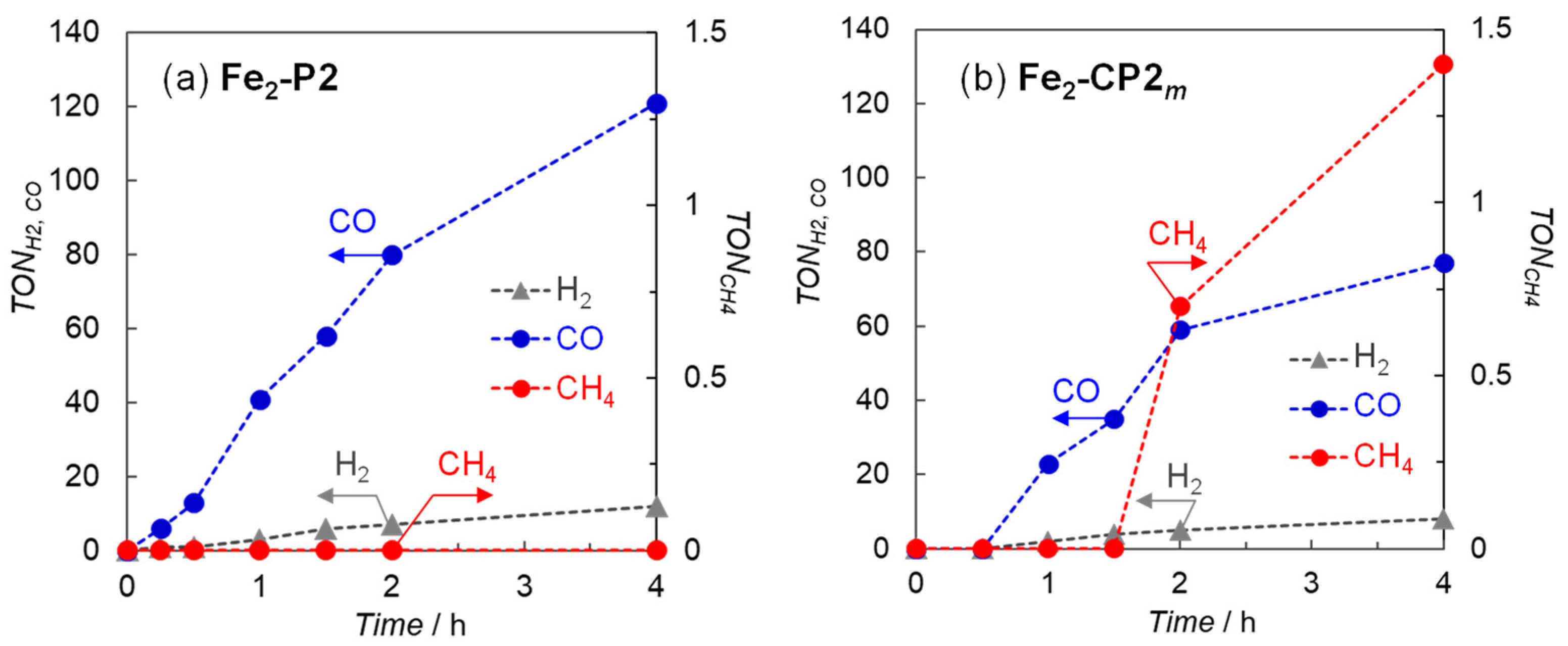
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure
3.2. Synthesis of Fb2-CP2m
3.3. Synthesis of Fe2-CP2m
3.4. Synthesis of Fb2-CP2p
3.5. Synthesis of Fe2-CP2p
3.6. Synthesis of Fe2-P2
3.7. Synthesis of FeP-phen
3.8. Photocatalytic CO2 Reduction
3.9. 13CO2-Labeling Experiment
3.10. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sato, S.; Arai, T.; Morikawa, T.; Uemura, K.; Suzuki, T.M.; Tanaka, H.; Kajino, T. Selective CO2 Conversion to Formate Conjugated with H2O Oxidation Utilizing Semiconductor/Complex Hybrid Photocatalysts. J. Am. Chem. Soc. 2011, 133, 15240. [Google Scholar] [CrossRef]
- Sahara, G.; Kumagai, H.; Maeda, K.; Kaeffer, N.; Artero, V.; Higashi, M.; Abe, R.; Ishitani, O. Photoelectrochemical Reduction of CO2 Coupled to Water Oxidation Using a Photocathode with a Ru(II)–Re(I) Complex Photocatalyst and a CoOx/TaON Photoanode. J. Am. Chem. Soc. 2016, 138, 14152. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48, 2996. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90. [Google Scholar] [CrossRef]
- Mohamed, E.A.; Zahran, Z.N.; Naruta, Y. Efficient electrocatalytic CO2 reduction with a molecular cofacial iron porphyrin dimer. Chem. Commun. 2015, 51, 16900. [Google Scholar] [CrossRef]
- Zhang, C.; Gotico, P.; Guillot, R.; Dragoe, D.; Leibl, W.; Halime, Z.; Aukauloo, A. Bio-Inspired Bimetallic Cooperativity through a Hydrogen Bonding Spacer in CO2 Reduction. Angew. Chem. Int. Ed. 2023, 62, e202214665. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Savéant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Schmidt, L.C.; Bonin, J.; Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74. [Google Scholar] [CrossRef]
- Rao, H.; Lim, C.-H.; Bonin, J.; Miyake, G.M.; Robert, M. Visible-Light-Driven Conversion of CO2 to CH4 with an Organic Sensitizer and an Iron Porphyrin Catalyst. J. Am. Chem. Soc. 2018, 140, 17830. [Google Scholar] [CrossRef]
- Chen, J.Y.; Li, M.; Liao, R.Z. Mechanistic Insights into Photochemical CO2 Reduction to CH4 by a Molecular Iron–Porphyrin Catalyst. Inorg. Chem. 2023, 62, 9400. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kuramochi, Y.; Ito, S.; Kinbara, Y.; Satake, A. Metal-templated synthesis of rigid and conformationally restricted cyclic bisporphyrins: Specific retention times on a cyanopropyl-modified silica gel column. Org. Biomol. Chem. 2021, 19, 3159. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, Y.; Koike, K.; Morimoto, T.; Ishitani, O. Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor. J. Catal. 2013, 304, 22. [Google Scholar] [CrossRef]
- Aranzaes, J.R.; Daniel, M.-C.; Astruc, D. Metallocenes as references for the determination of redox potentials by cyclic voltammetry—Permethylated iron and cobalt sandwich complexes, inhibition by polyamine dendrimers, and the role of hydroxy-containing ferrocenes. Can. J. Chem. 2006, 84, 288. [Google Scholar] [CrossRef]
- Hammouche, M.; Lexa, D.; Momenteau, M.; Savéant, J.-M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113, 8455. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron (0) porphyrins. Synergistic effect of Lewis acid cations. J. Phys. Chem. 1996, 100, 19981. [Google Scholar] [CrossRef]
- Anxolabéhère-Mallart, E.; Bonin, J.; Fave, C.; Robert, M. Small-molecule activation with iron porphyrins using electrons, photons and protons: Some recent advances and future strategies. Dalton Trans. 2019, 48, 5869. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction mechanisms of catalytic photochemical CO2 reduction using Re (I) and Ru (II) complexes. Coord. Chem. Rev. 2018, 373, 333. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Kamiya, M.; Ishida, H. Photocatalytic CO2 Reduction in N,N-Dimethylacetamide/Water as an Alternative Solvent System. Inorg. Chem. 2014, 53, 332. [Google Scholar] [CrossRef]
- Pearson, R.M.; Lim, C.-H.; McCarthy, B.G.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Suzuki, Y.; Asai, S.; Suzuki, T.; Iwama, H.; Asano, M.S.; Satake, A. Significance of the connecting position between Zn(II) porphyrin and Re(I) bipyridine tricarbonyl complex units in dyads for room-temperature phosphorescence and photocatalytic CO2 reduction: Unexpected enhancement by triethanolamine in catalytic activity. Chem. Sci. 2023, 14, 8743. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, B.G.; Pearson, R.M.; Lim, C.-H.; Sartor, S.M.; Damrauer, N.H.; Miyake, G.M. Structure–property relationships for tailoring phenoxazines as reducing photoredox catalysts. J. Am. Chem. Soc. 2018, 140, 5088. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.-H.; Ilic, S.; Alherz, A.; Worrell, B.T.; Bacon, S.S.; Hynes, J.T.; Glusac, K.D.; Musgrave, C.B. Benzimidazoles as Metal-Free and Recyclable Hydrides for CO2 Reduction to Formate. J. Am. Chem. Soc. 2018, 141, 272. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Xu, J.; Idros, U.M.; Katsuhira, J.; Fuki, M.; Hayashi, M.; Yamanaka, M.; Kobori, Y.; Matsubara, R. Metal-free reduction of CO2 to formate using a photochemical organohydride-catalyst recycling strategy. Nat. Chem. 2023, 15, 794. [Google Scholar] [CrossRef] [PubMed]
- Salzmann, R.; Ziegler, C.J.; Godbout, N.; McMahon, M.T.; Suslick, K.S.; Oldfield, E. Carbonyl Complexes of Iron(II), Ruthenium(II), and Osmium(II) 5,10,15,20-Tetraphenylporphyrinates: A Comparative Investigation by X-ray Crystallography, Solid-State NMR Spectroscopy, and Density Functional Theory. J. Am. Chem. Soc. 1998, 120, 11323. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Fujisawa, Y.; Satake, A. Photocatalytic CO2 Reduction Mediated by Electron Transfer via the Excited Triplet State of Zn(II) Porphyrin. J. Am. Chem. Soc. 2020, 142, 705. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuramochi, Y.; Hashimoto, M.; Satake, A. Methane Formation Induced via Face-to-Face Orientation of Cyclic Fe Porphyrin Dimer in Photocatalytic CO2 Reduction. Molecules 2024, 29, 2453. https://doi.org/10.3390/molecules29112453
Kuramochi Y, Hashimoto M, Satake A. Methane Formation Induced via Face-to-Face Orientation of Cyclic Fe Porphyrin Dimer in Photocatalytic CO2 Reduction. Molecules. 2024; 29(11):2453. https://doi.org/10.3390/molecules29112453
Chicago/Turabian StyleKuramochi, Yusuke, Masaya Hashimoto, and Akiharu Satake. 2024. "Methane Formation Induced via Face-to-Face Orientation of Cyclic Fe Porphyrin Dimer in Photocatalytic CO2 Reduction" Molecules 29, no. 11: 2453. https://doi.org/10.3390/molecules29112453
APA StyleKuramochi, Y., Hashimoto, M., & Satake, A. (2024). Methane Formation Induced via Face-to-Face Orientation of Cyclic Fe Porphyrin Dimer in Photocatalytic CO2 Reduction. Molecules, 29(11), 2453. https://doi.org/10.3390/molecules29112453