
Strategies for Accessing cis-1-Amino-2-Indanol


Abstract
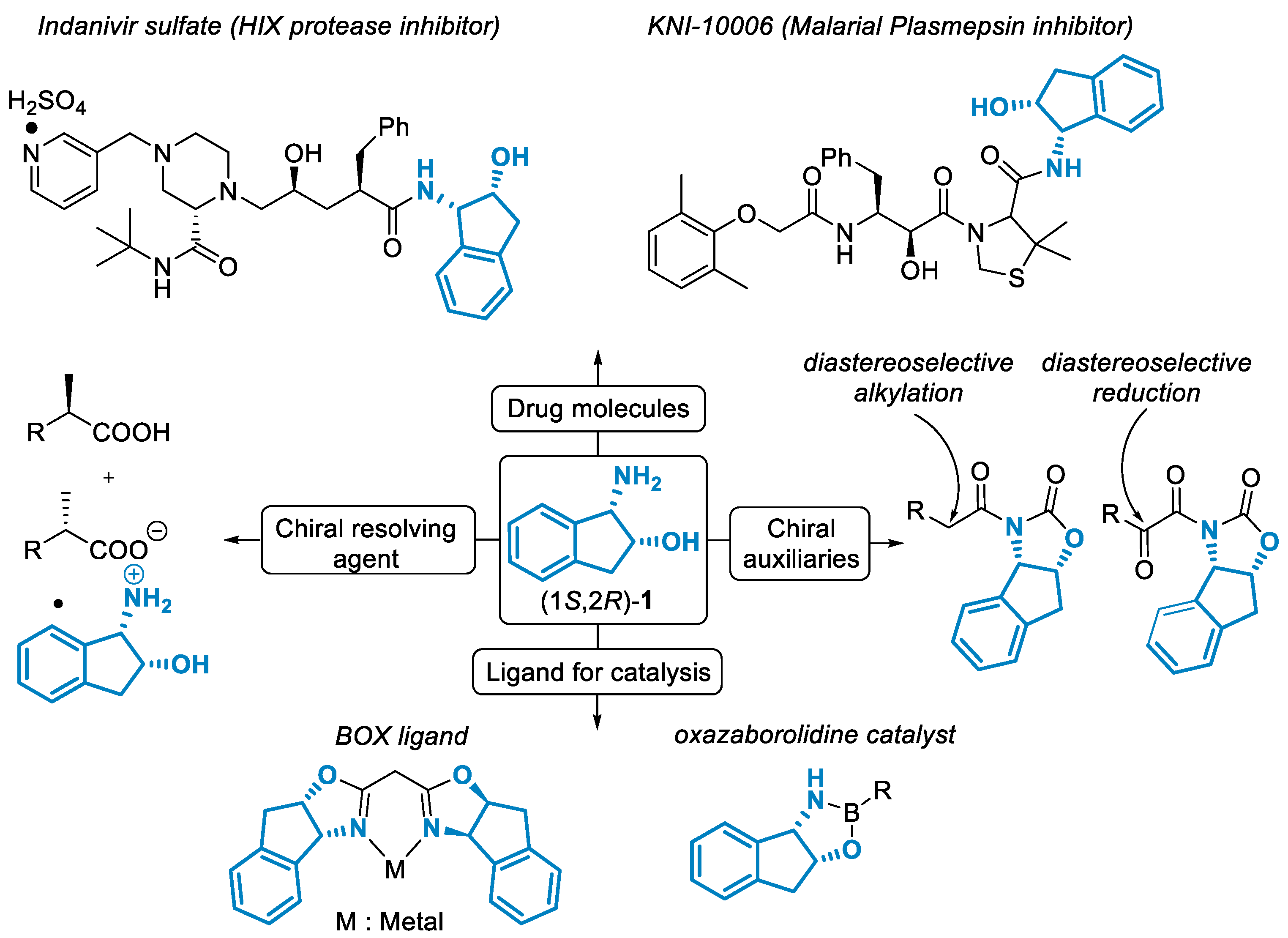
1. Introduction
2. Syntheses from Indane Skeleton
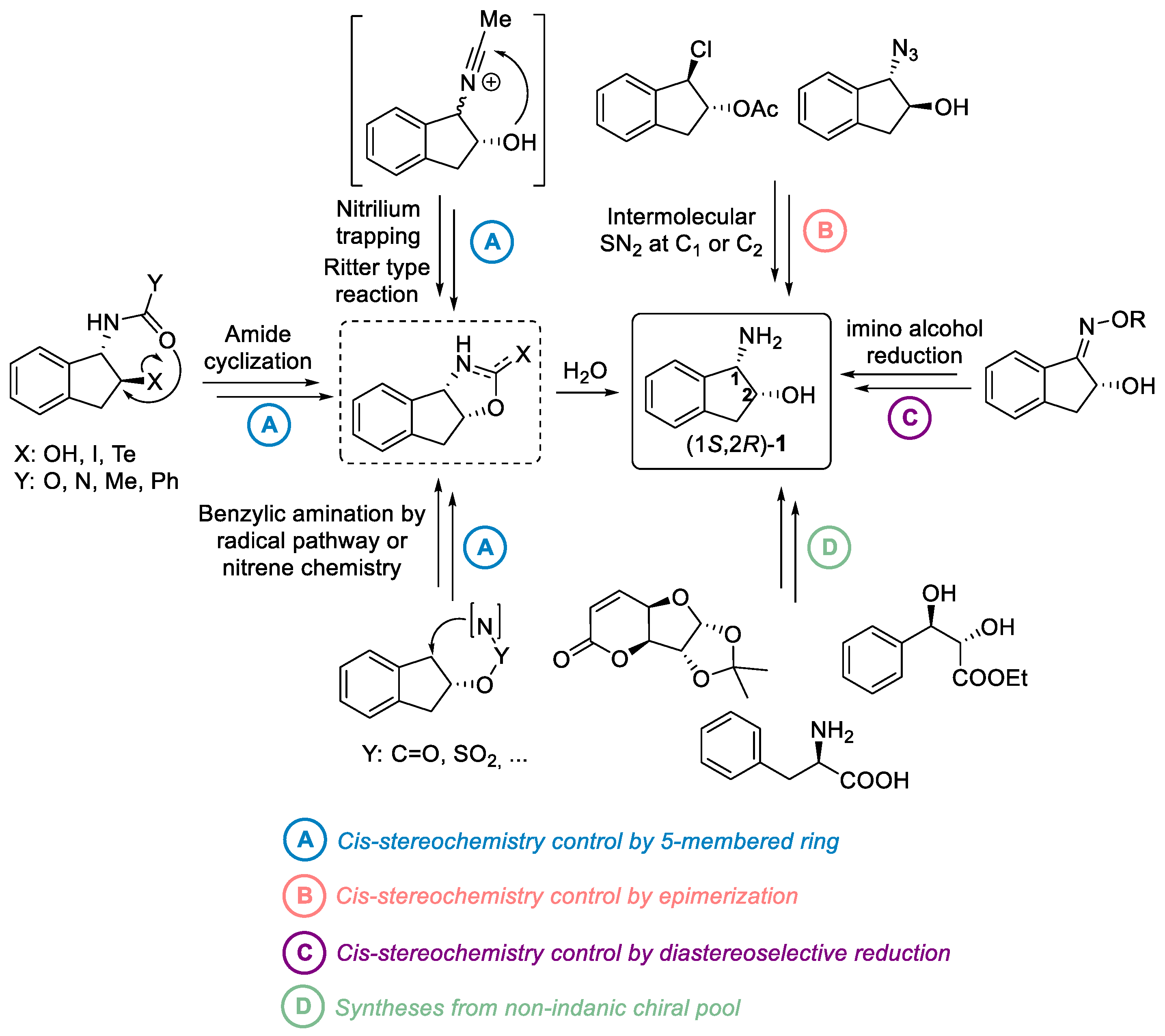
2.1. cis Stereochemistry Controlled by Intramolecular Formation of 5-Membered Ring
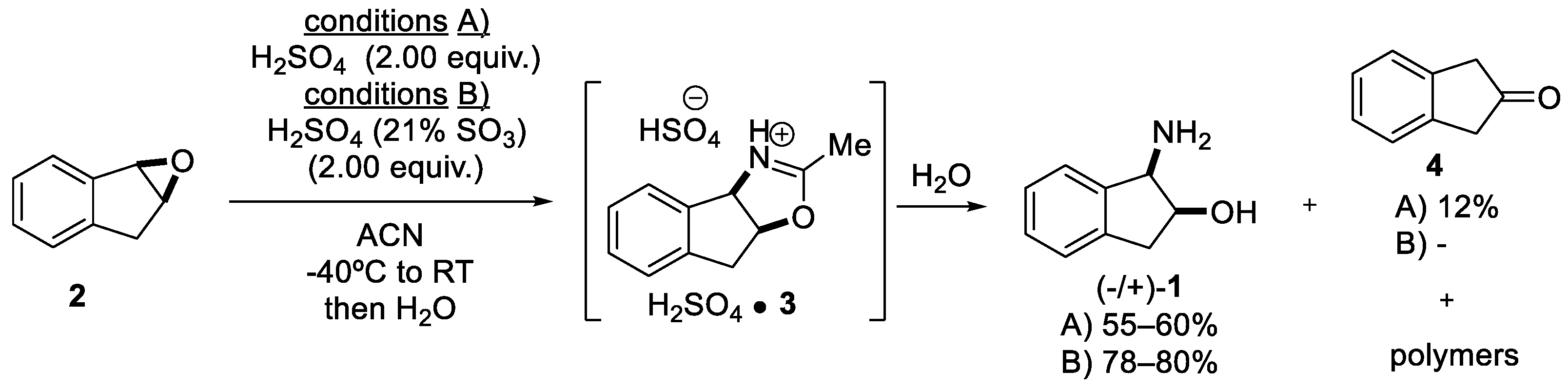
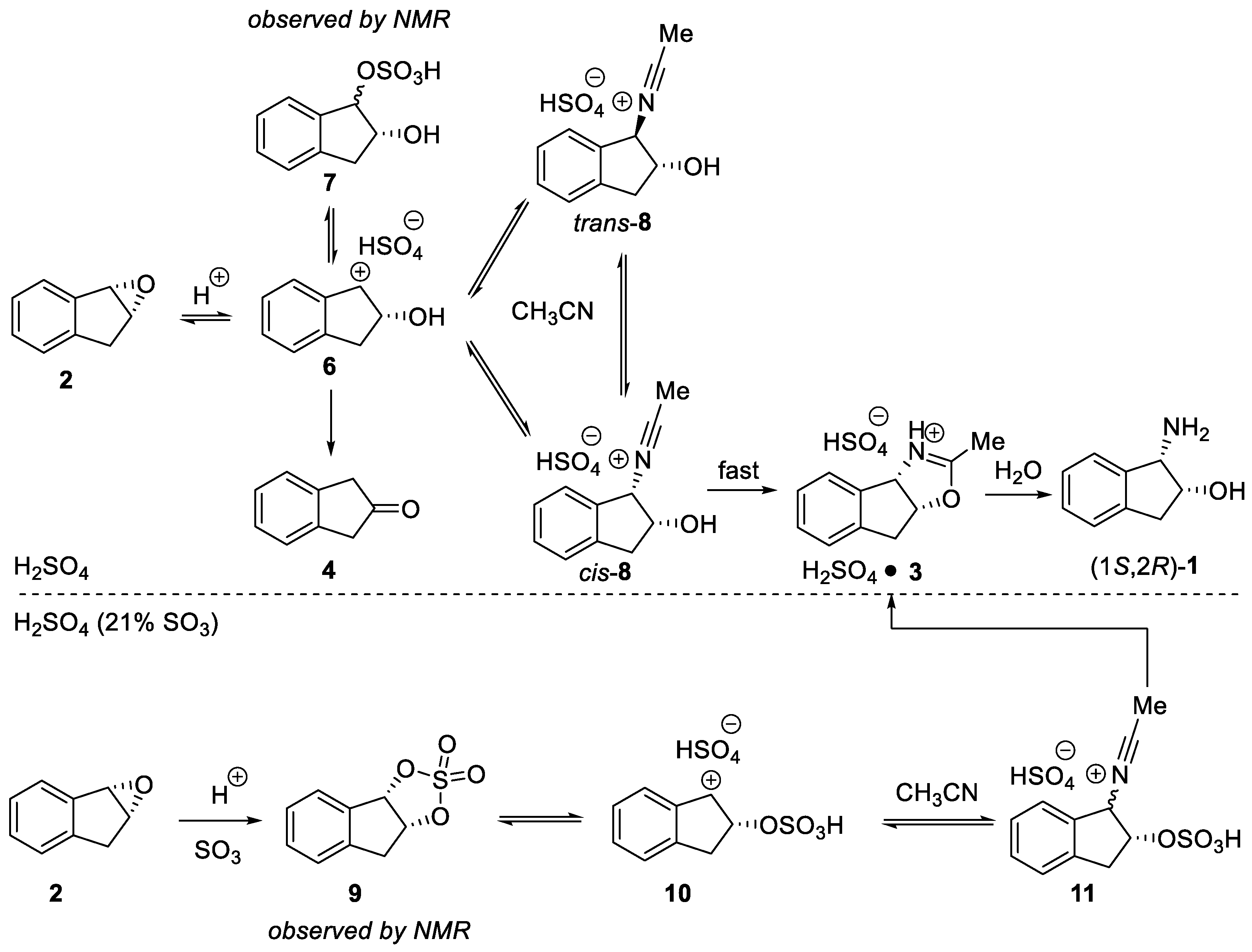
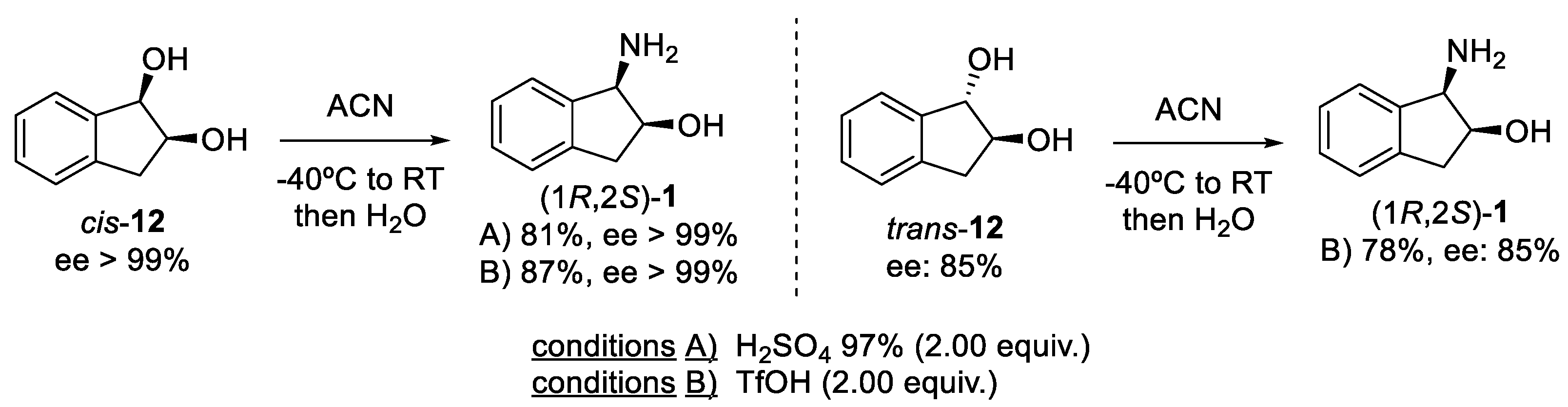
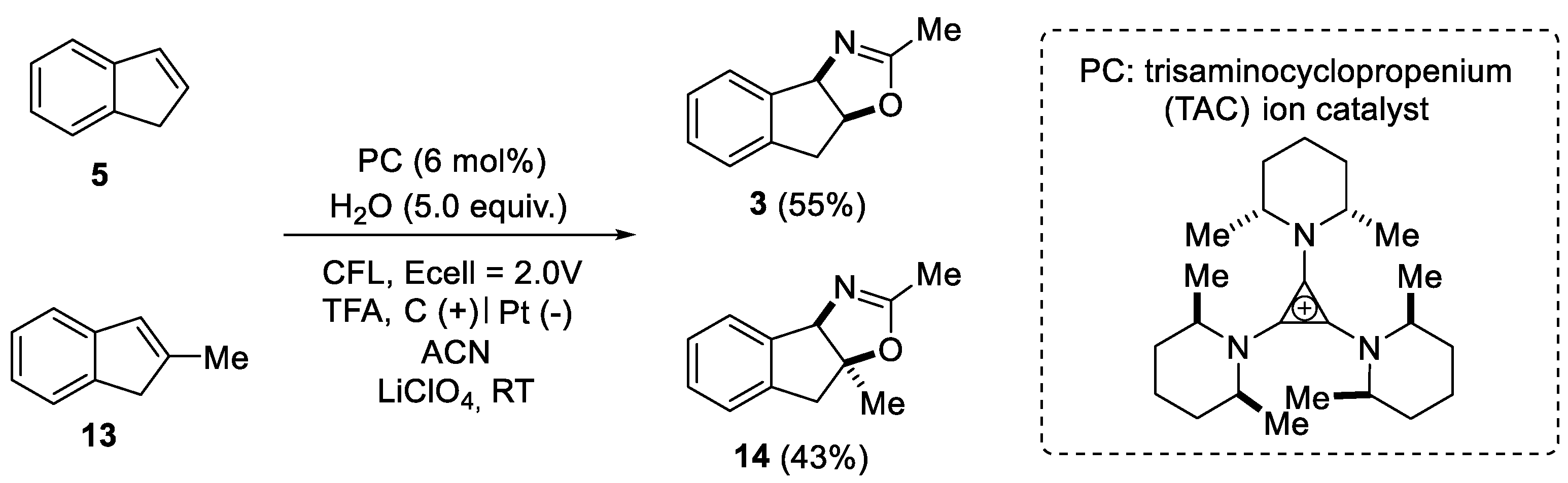
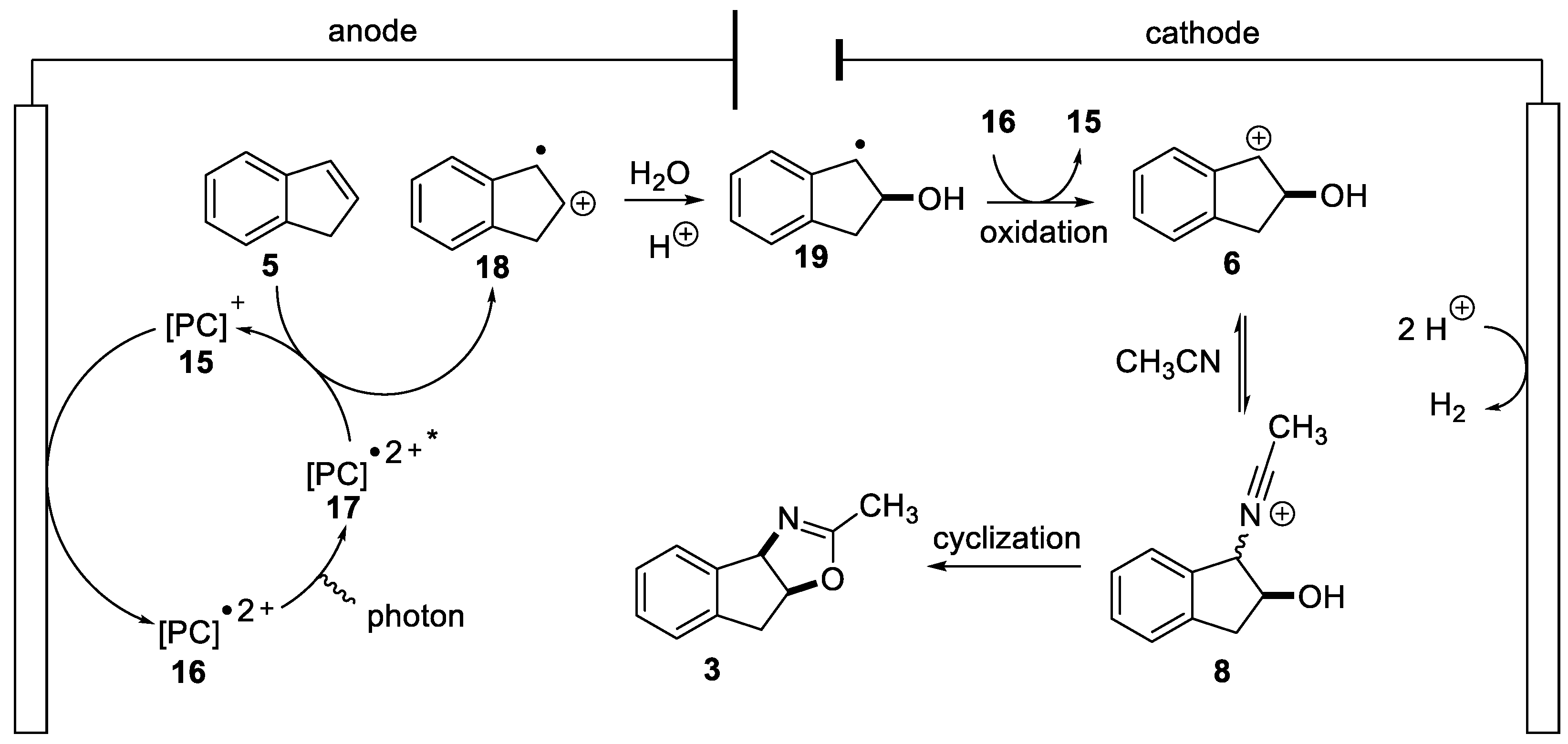
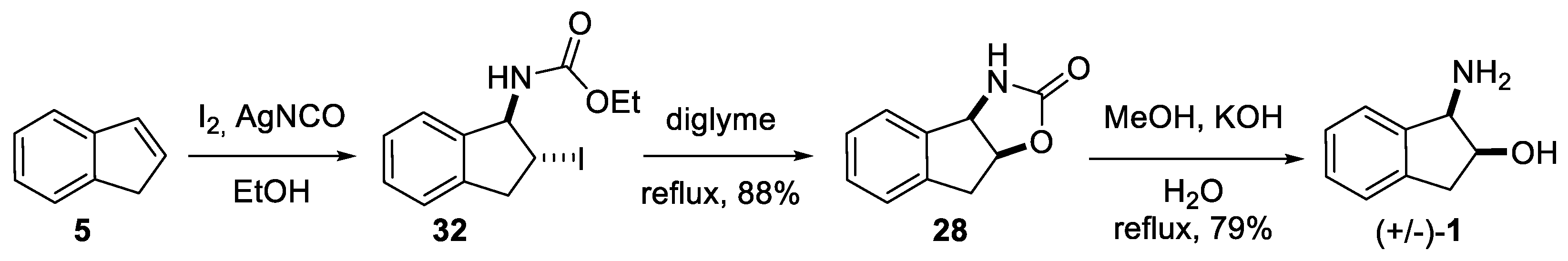
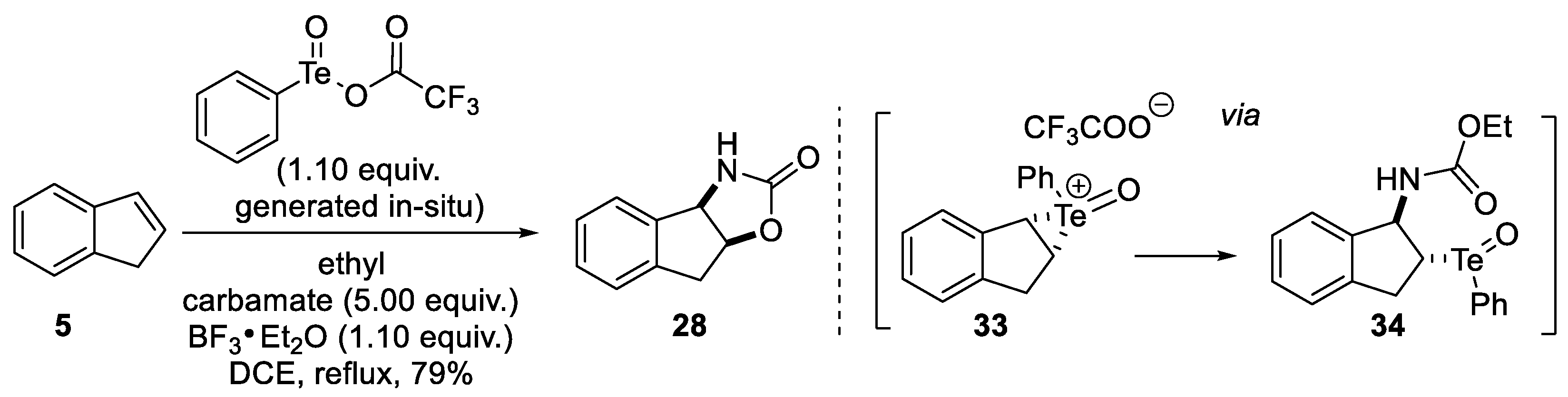
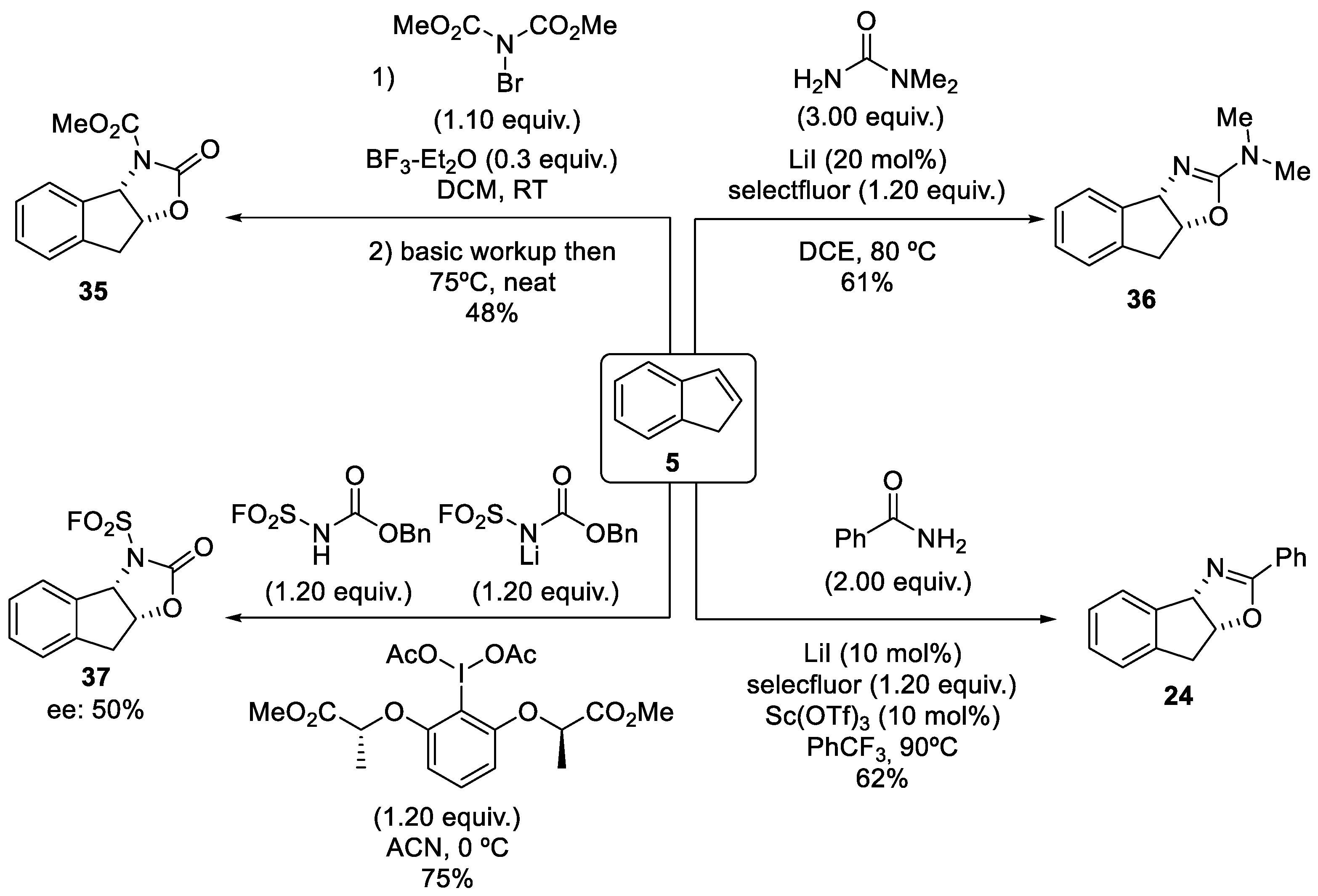
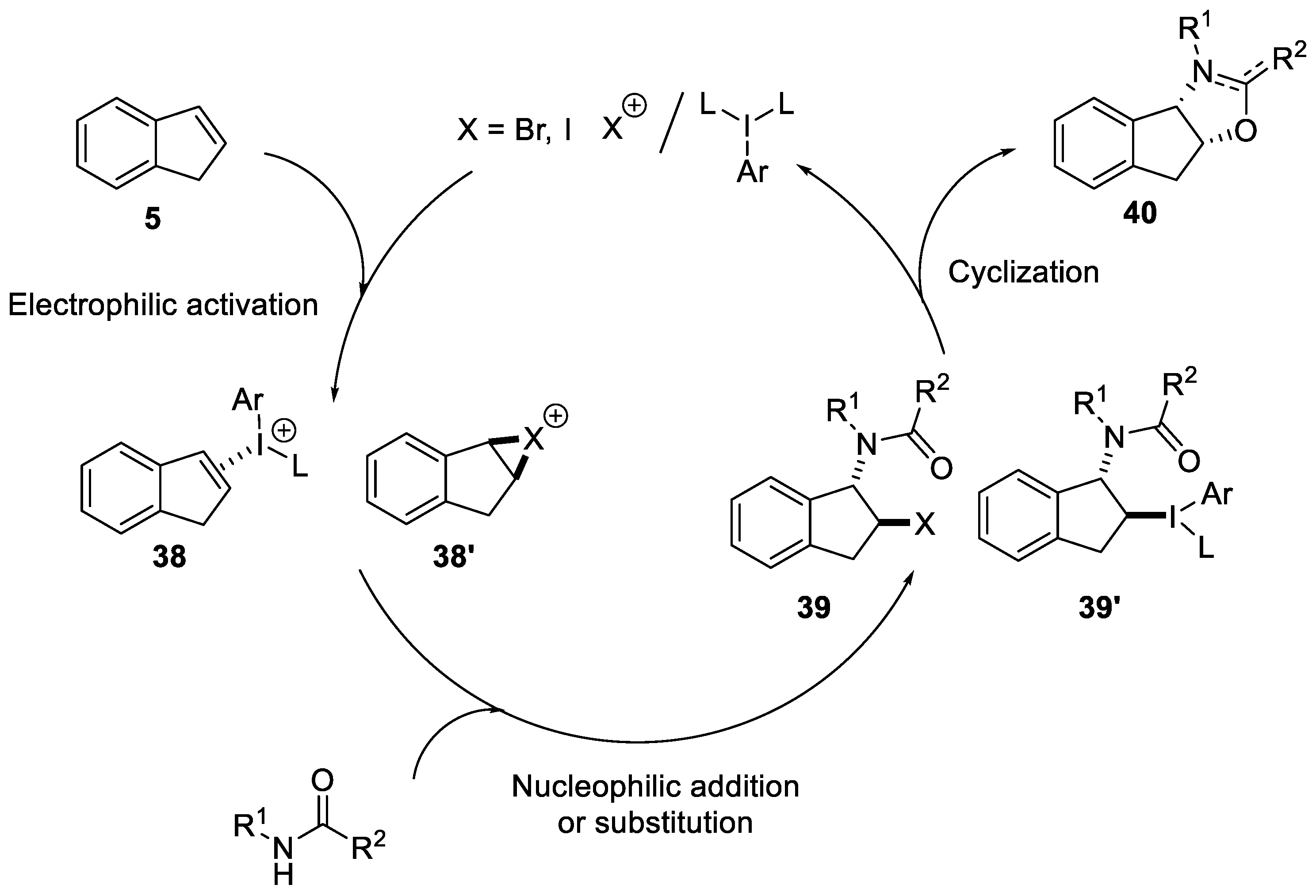
2.1.1. Nitrilium Trapping—Ritter Type Reaction
2.1.2. Intramolecular Amide Cyclization
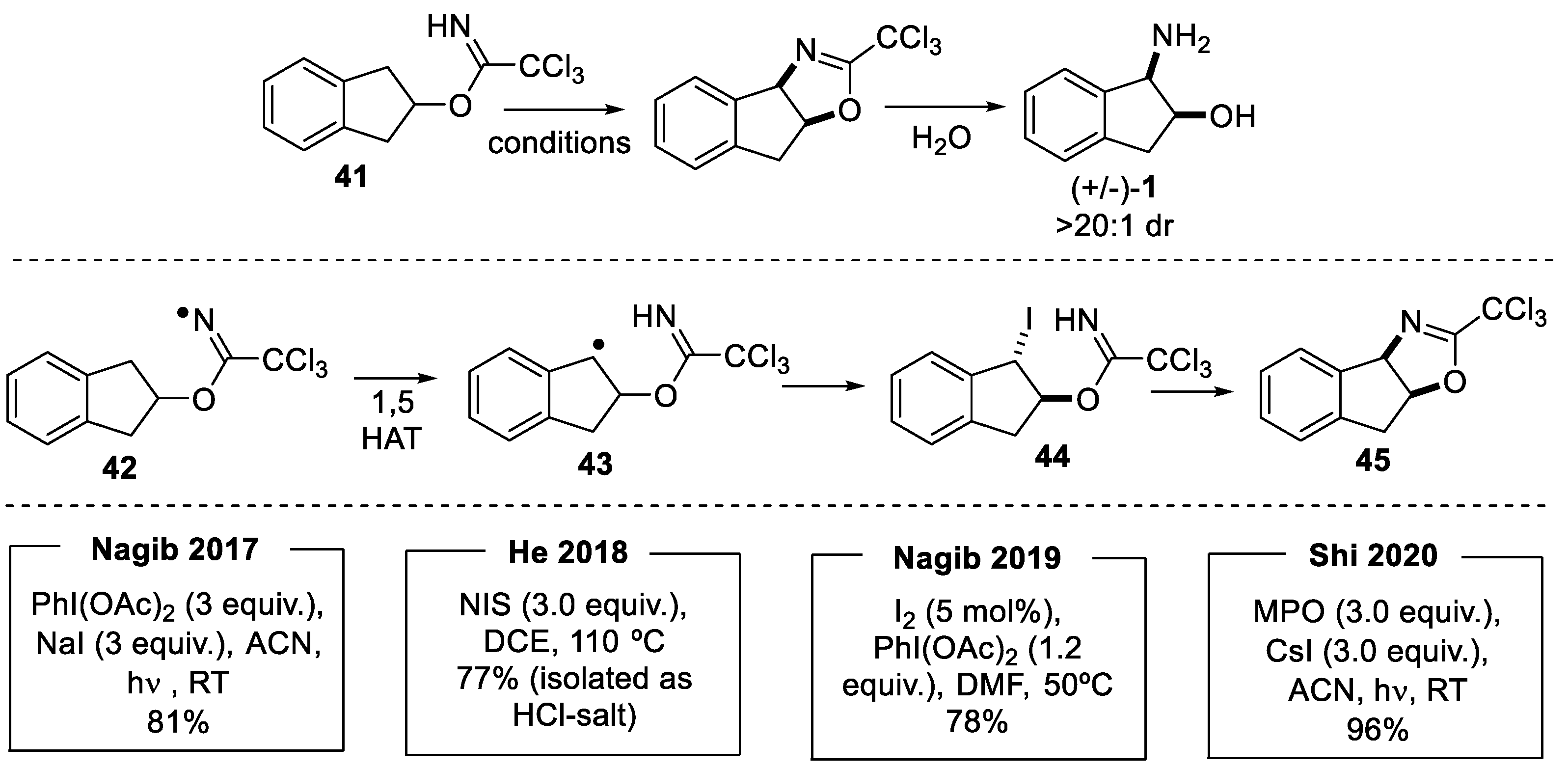
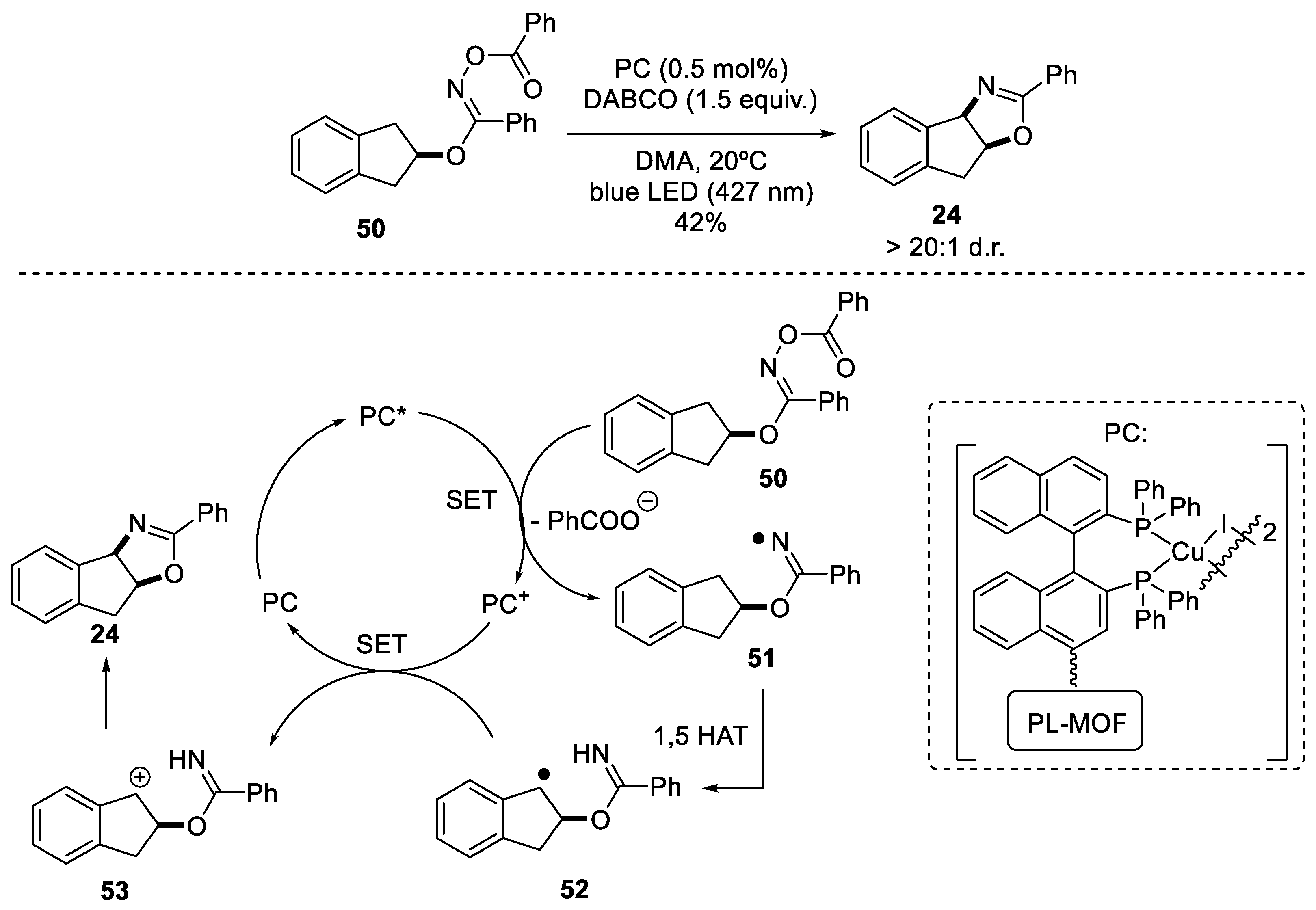
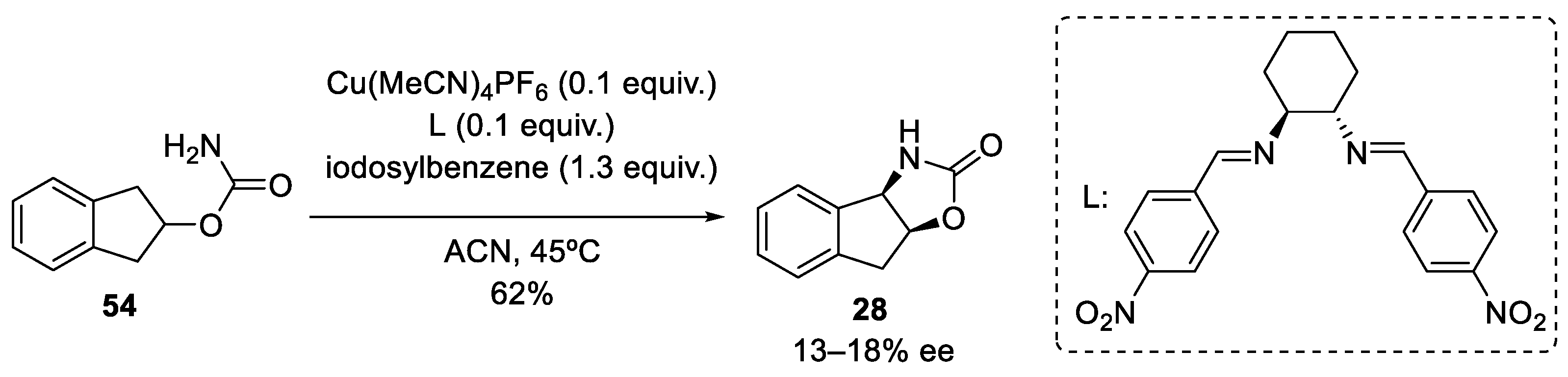
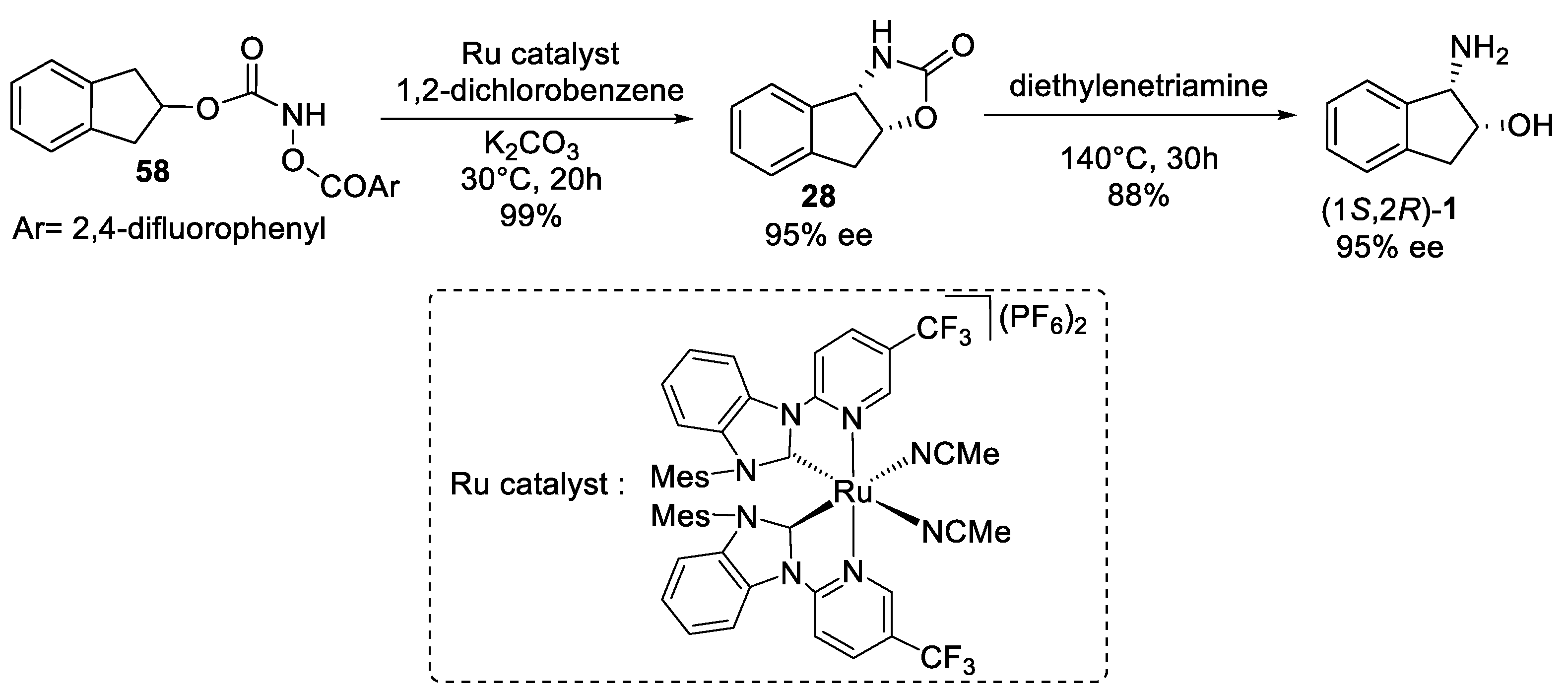
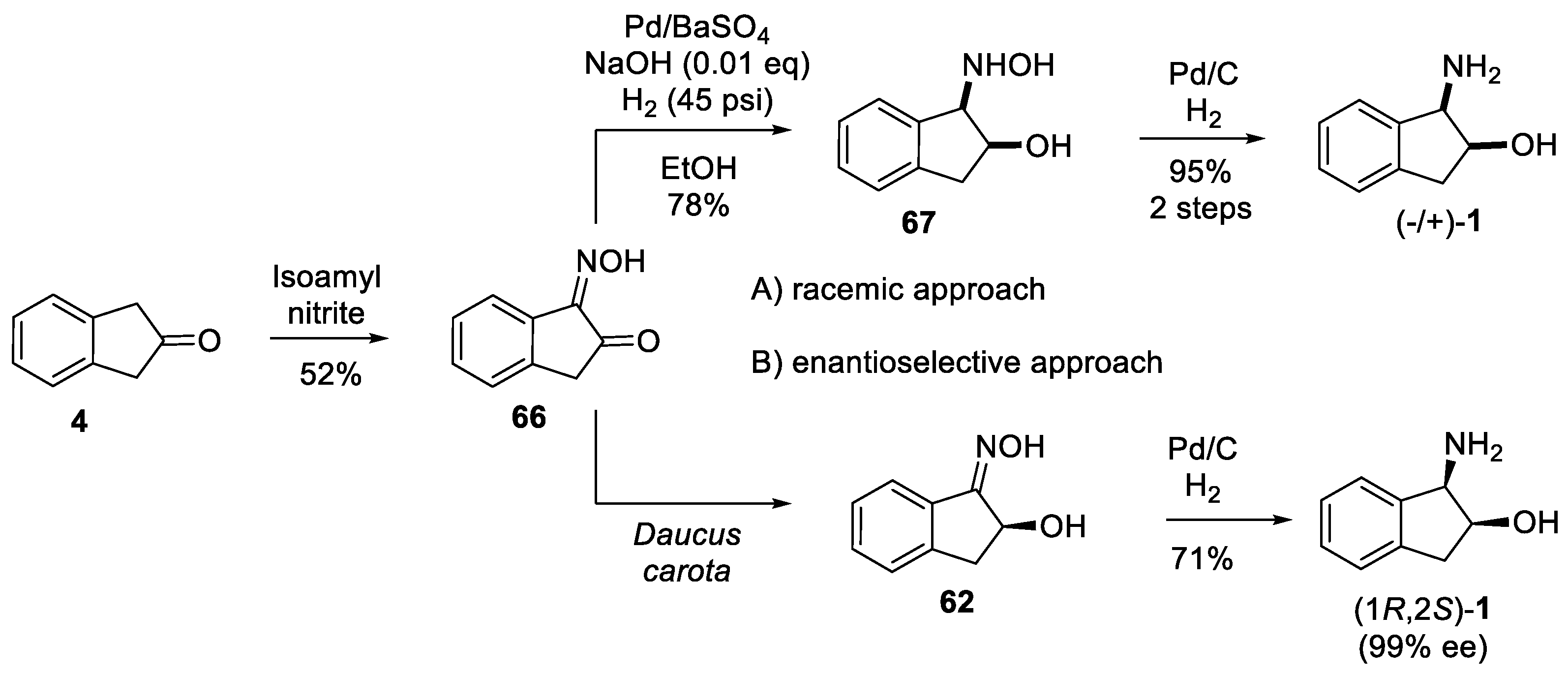
2.1.3. Benzylic Csp3-H Amination
- Benzylic Csp3-H amination via radical pathway
- Benzylic Csp3-H amination via nitrene chemistry
2.2. cis Stereochemistry Controlled by Epimerization
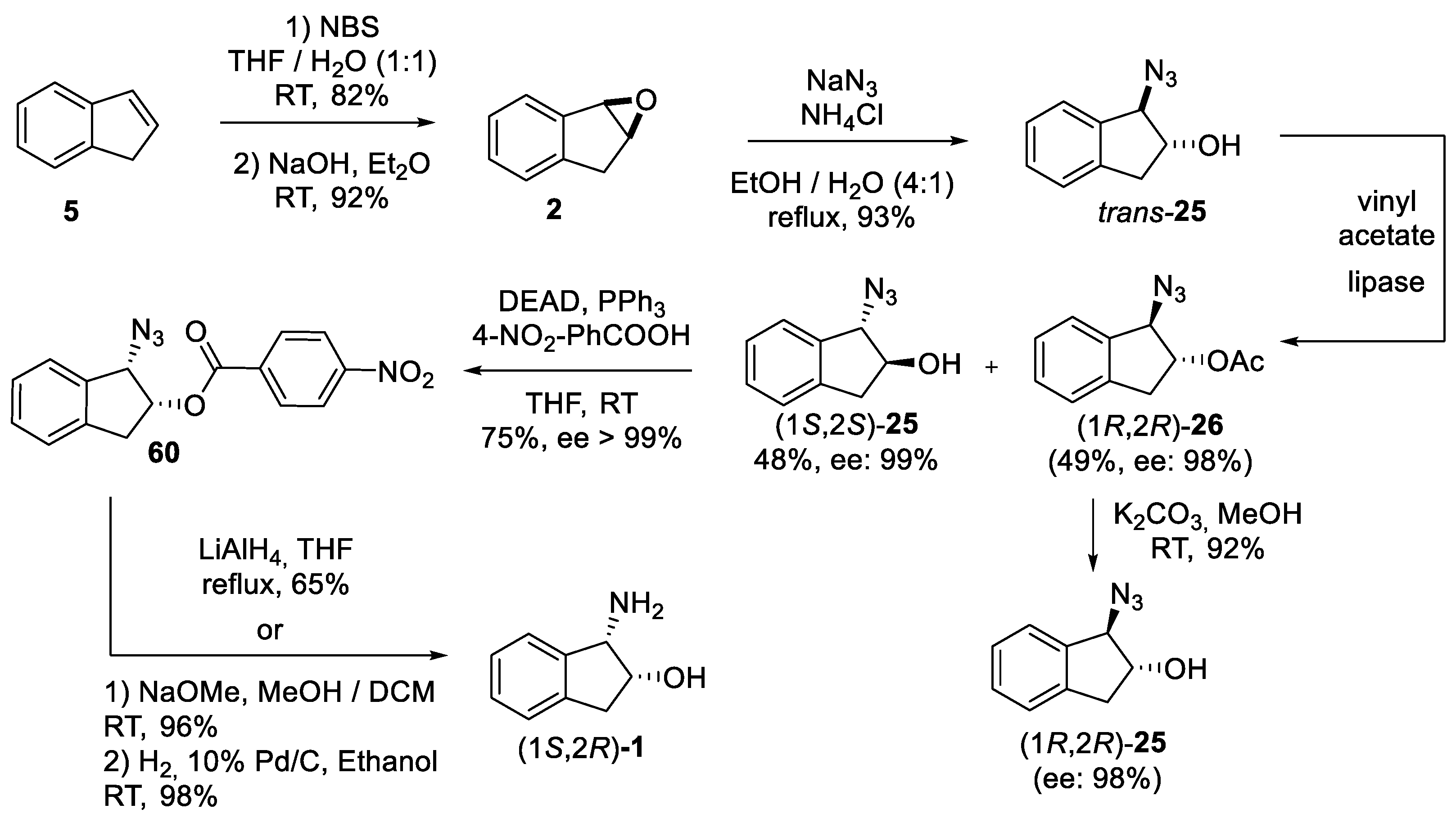
2.2.1. Epimerization via SN2 at C1 Position
2.2.2. Epimerization via Mitsunobu Reaction at C2 Position
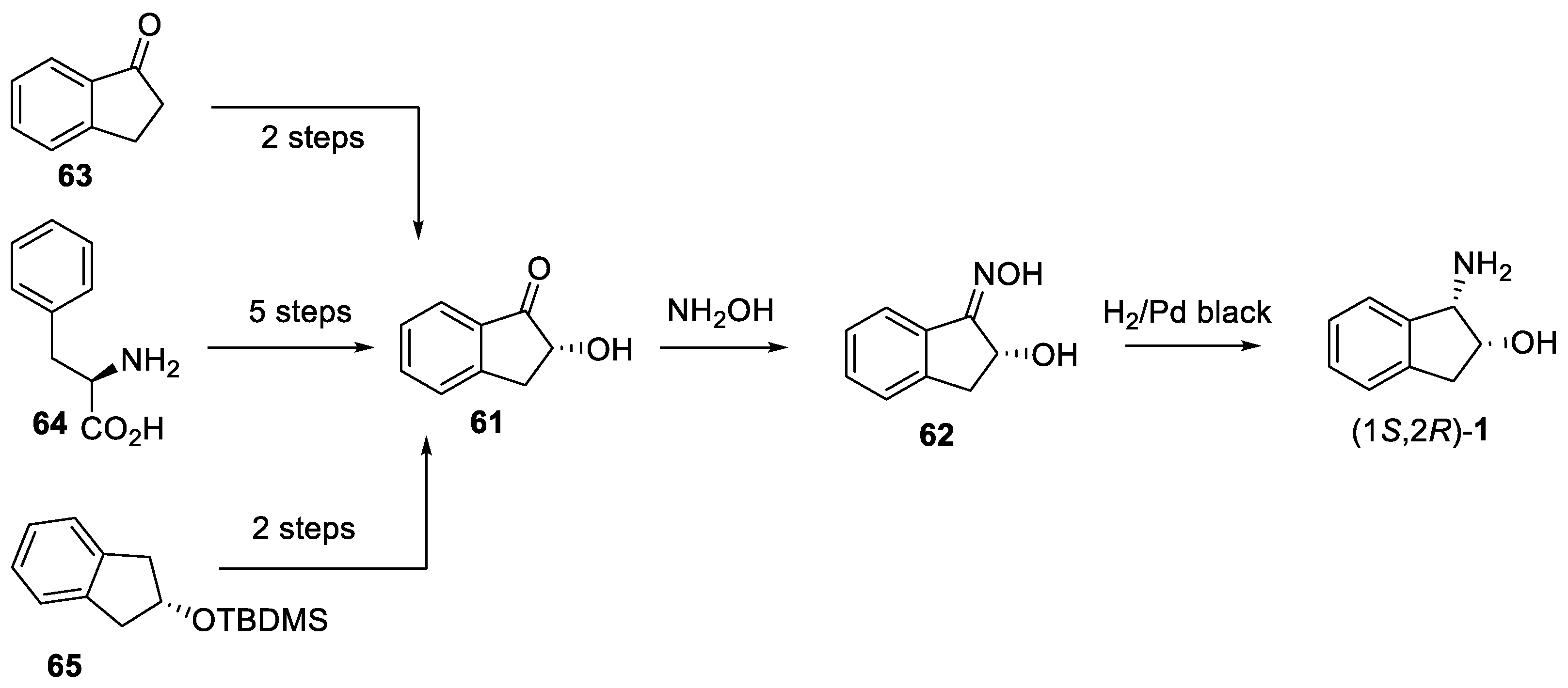
2.3. cis Stereochemistry Controlled by Diastereoselective Imino Alcohol Reduction
3. Syntheses from Non-Indane Skeleton
3.1. Synthesis from (E)-Cinnamate Ethyl Ester
3.2. Synthesis from 7,3-Xylofuranose Derivative
3.3. Synthesis from D-Phenylalanine
4. Resolution
4.1. Chemical Resolution
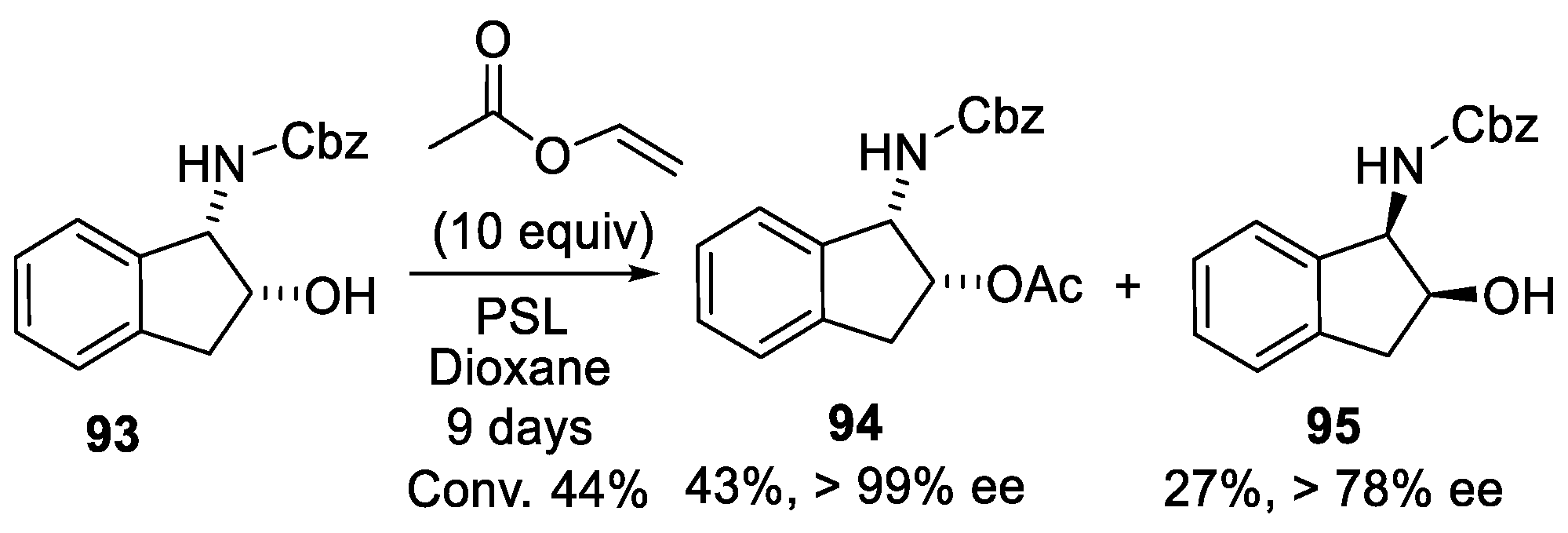
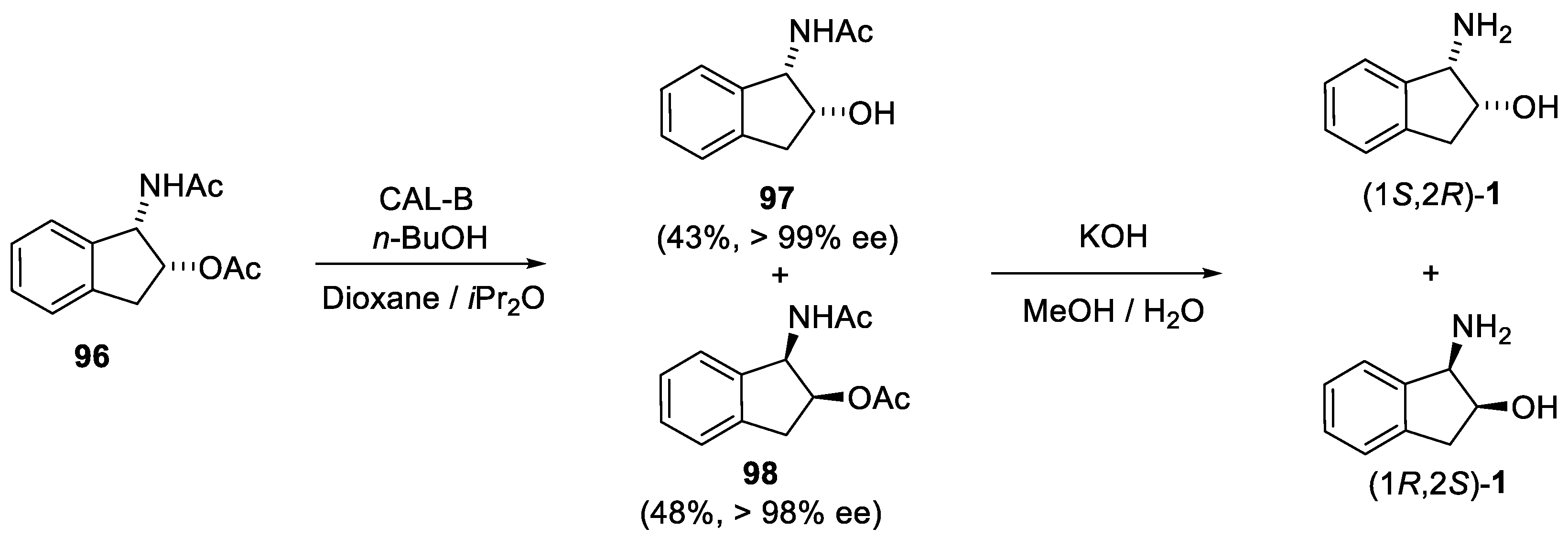
4.2. Enzymatic Resolution
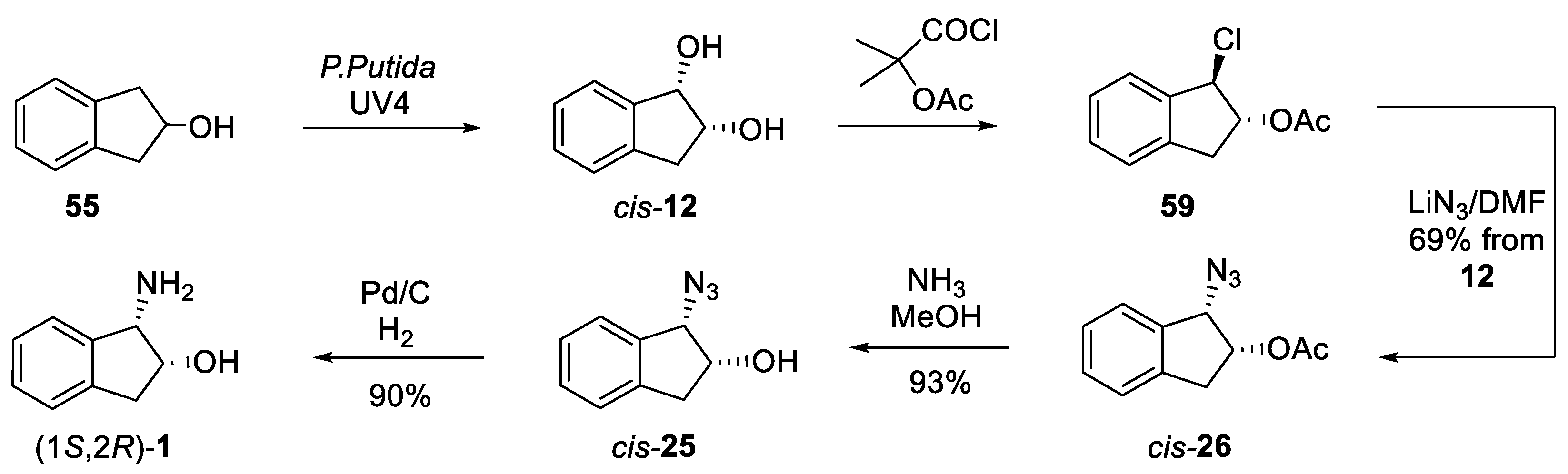
5. Substituted cis-1-Amino-2-Indanol
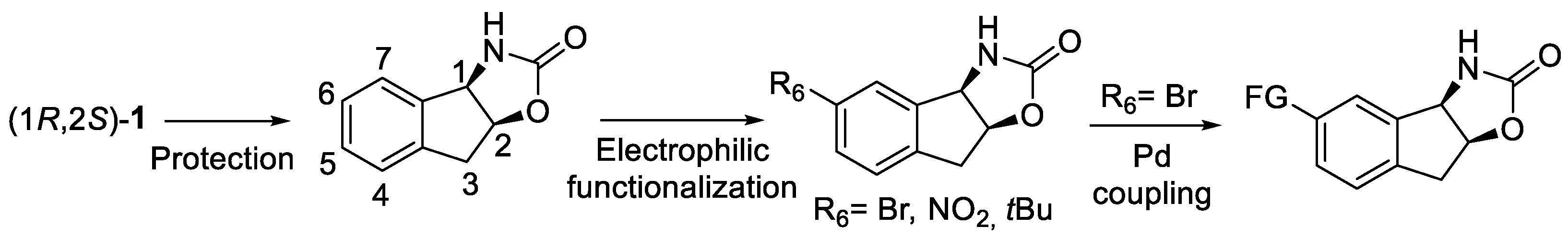
5.1. Post-Functionalization
5.2. Pre-Functionalization
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saddique, F.A.; Zahoor, A.F.; Faiz, S.; Naqvi, A.A.R.; Usman, M.; Ahmad, M. Recent trends in ring opening of epoxides by amines as nucleophiles. Synth. Commun. 2016, 46, 831–868. [Google Scholar] [CrossRef]
- Weng, C.; Zhang, H.; Xiong, X.; Lu, X.; Zhou, Y. Evolution of Epoxides to Synthesize β-amino Alcohols. Asian J. Org. Chem. 2014, 26, 3761–3768. [Google Scholar] [CrossRef]
- Karjalainen, O.K.; Koskinen, A.M.P. Diastereoselective synthesis of vicinal amino alcohols. Org. Biomol. Chem. 2012, 10, 4311–4326. [Google Scholar] [CrossRef]
- Bergmeier, S.C. The Synthesis of Vicinal Amino Alcohols. Tetrahedron 2000, 56, 2561–2576. [Google Scholar] [CrossRef]
- Desimoni, G.; Faita, G.; Jørgensen, K.A. C2-Symmetric Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2006, 106, 3561–3651. [Google Scholar] [CrossRef]
- Desimoni, G.; Faita, G.; Quadrelli, P. Pyridine-2,6-bis(oxazolines), Helpful Ligands for Asymmetric Catalysts. Chem. Rev. 2003, 103, 3119–3154. [Google Scholar] [CrossRef]
- Babu, S.A.; Krishnan, K.K.; Ujwaldev, S.M.; Anilkumar, G. Applications of Pybox Complexes in Asymmetric Catalysis. Asian J. Org. Chem. 2018, 7, 1033–1053. [Google Scholar] [CrossRef]
- Higashijima, S.; Itoh, H.; Senda, Y.; Nakano, S. cis-1-Amino-1,2,3,4-tetrahydro-2-naphthalenol: Resolution and application to the catalytic enantioselective reduction of ketones. Tetrahedron Asymmetry 1997, 8, 3107–3110. [Google Scholar] [CrossRef]
- Gallou, I.; Senanayake, C.H. cis-1-Amino-2-indanol in Drug Design and Applications to Asymmetric Processes. Chem. Rev. 2006, 106, 2843–2874. [Google Scholar] [CrossRef]
- Dorsey, B.D.; Levin, R.B.; McDaniel, S.L.; Vacca, J.P.; Guare, J.P.; Darke, P.L.; Zugay, J.A.; Emini, E.A.; Schleif, W.A.; Quintero, J.C.; et al. RL-735,524: The Design of a Potent and Orally Bioavailable HIV Protease Inhibitor. J. Med. Chem. 1994, 37, 3443–3451. [Google Scholar] [CrossRef]
- Reider, P.J. Advances in AIDS Chemotherapy: The Asymmetric Synthesis of CRIXIVAN®. Chimia 1997, 51, 306–308. [Google Scholar] [CrossRef]
- Abdel-Rahman, H.; Kimura, T.; Hidaka, K.; Kiso, A.; Nezami, A.; Freire, E.; Hayashi, Y.; Kiso, Y. Design of inhibitors against HIV, HTLV-I, and Plasmodium falciparum aspartic proteases. Biol. Chem. 2004, 385, 1035–1039. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Fidanze, S.; Senanayakeb, C.H. cis-1-Aminoindan-2-ol in Asymmetric Syntheses. Synthesis 1998, 1998, 937–961. [Google Scholar] [CrossRef]
- Senanayake, C.; Roberts, F.E.; DiMichele, L.M.; Ryan, K.M.; Liu, J.; Fredenburgh, L.E.; Foster, B.S.; Douglas, A.W.; Larsen, R.D.; Verhoeven, T.R.; et al. The Behavior of Indene Oxide in the Ritter Reaction: A Simple Route to cis-Aminoindanol. Tetrahedron Lett. 1995, 36, 3993–3996. [Google Scholar] [CrossRef]
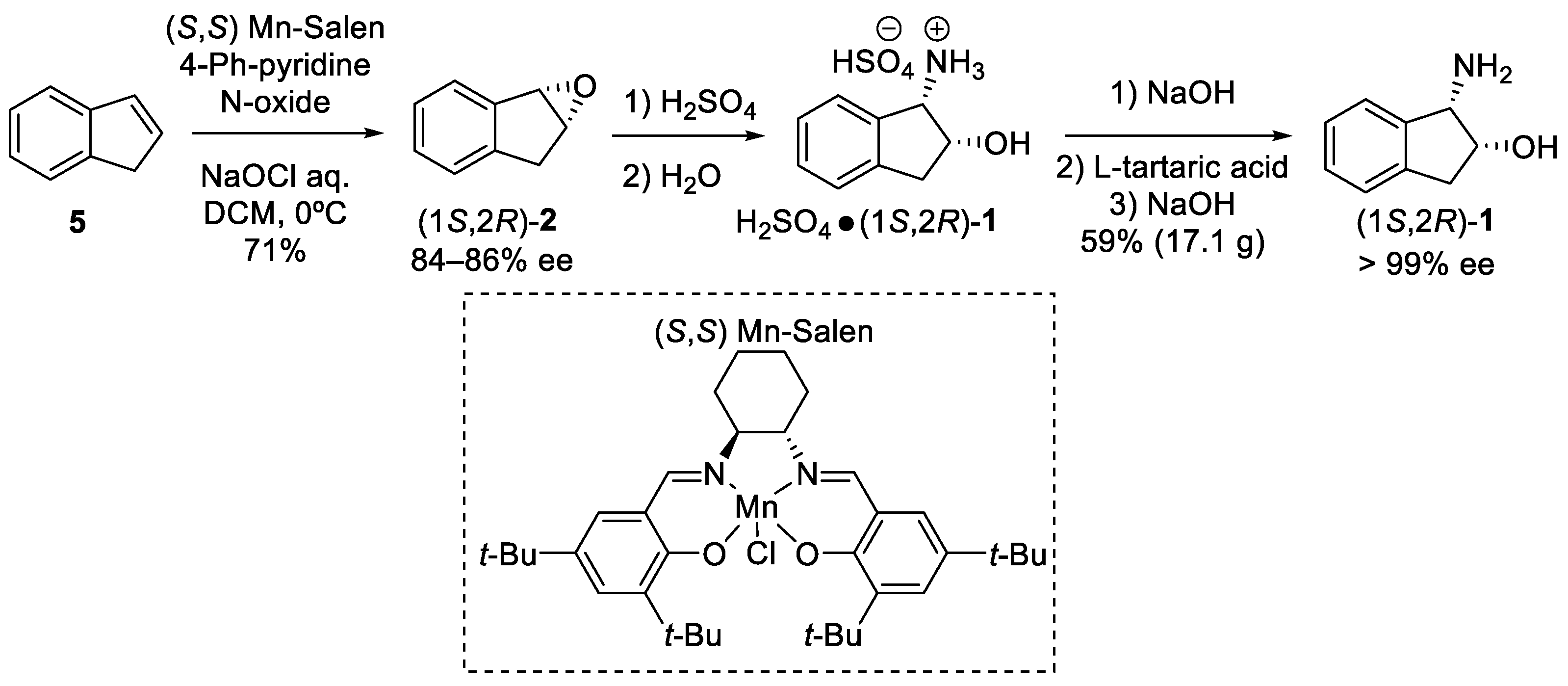
- Larrow, J.F.; Roberts, E.; Verhoeven, T.R.; Ryan, K.M.; Senanayake, C.H.; Reider, P.J.; Jacobsen, E.N. (1S,2R)-1-AMINOINDAN-2-OL. Org. Synth. 1998, 76, 46. [Google Scholar]
- Senanayake, C.H.; DiMichele, L.M.; Liu, J.; Fredenburgh, L.E.; Ryan, K.M.; Roberts, F.E.; Larsen, R.D.; Verhoeven, R.R.; Reider, P.J. Regio- and Stereocontrolled Syntheses of Cyclic Chiral cis-Amino Alcohols from 1,2-Diols or Epoxides. Tetrahedron Lett. 1995, 36, 7615–7618. [Google Scholar] [CrossRef]
- Alexander, G.; Medvedev, A.G.; Grishanov, D.A.; Mikhaylov, A.A.; Churakov, A.V.; Tripol’skaya, T.A.; Ottenbacher, R.V.; Bryliakov, K.P.; Shames, A.I.; Lev, O.; et al. V Triphenyllead Hydroperoxide: A 1D Coordination Peroxo Polymer, Single-Crystal-to-Single-Crystal Disproportionation to a Superoxo/Hydroxo Complex, and Application in Catalysis. Inorg. Chem. 2022, 61, 8193–8205. [Google Scholar]
- Huang, J.; Liu, S.; Ma, Y.; Cai, J. Chiral salen Mn (III) immobilized on ZnPS-PVPA through alkoxyl-triazole for superior performance catalyst in asymmetric epoxidation of unfunctionalized olefins. J. Organomet. Chem. 2019, 886, 27–33. [Google Scholar] [CrossRef]
- Kratky, J.; Eggerichs, D.; Heine, T.; Hofmann, S.; Sowa, P.; Weiße, R.H.; Tischler, D.; Sträter, N. Structural and Mechanistic Studies on Substrate and Stereoselectivity of the Indole Monooxygenase VpIndA1: New Avenues for Biocatalytic Epoxidations and Sulfoxidations. Angew. Chem. Int. Ed. 2023, 62, e202300657. [Google Scholar] [CrossRef] [PubMed]
- Toda, H.; Imae, R.; Itoh, N. Efficient biocatalysis for the production of enantiopure (S)-epoxides using a styrene monooxygenase (SMO) and Leifsonia alcohol dehydrogenase (LSADH) system. Tetrahedron Asymmetry 2012, 23, 1542–1549. [Google Scholar] [CrossRef]
- Martínez-Montero, L.; Tischler, D.; Süss, P.; Schallmey, A.; Franssen, M.C.R.; Hollmann, F.; Paul, C.E. Asymmetric azidohydroxylation of styrene derivatives mediated by a biomimetic styrene monooxygenase enzymatic cascade. Catal. Sci. Technol. 2021, 11, 5077–5085. [Google Scholar] [CrossRef]
- He, G.-H.; Liu, Y.-L.; Liu, H.; Lu, X.-B. Enantioselective Resolution Copolymerization of Racemic cis-Epoxides and Cyclic Anhydrides Mediated by Multichiral Bimetallic Chromium Complexes. Macromolecules 2022, 55, 3869–3876. [Google Scholar] [CrossRef]
- Zhang, J.; Reddy, J.; Roberge, C.; Senanayake, C.; Greashman, R.; Chartrain, M. Chiral Bio-Resolution of Racemic Indene Oxide by Fungal Epoxide Hydrolases. J. Ferment. Bioeng. 1995, 80, 244–246. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Lo Meo, P.; Riela, S.; D’Anna, F.; Noto, R. Lipase-catalyzed resolution of β-hydroxy selenides. Tetrahedron Asymmetry 2006, 17, 2713–2721. [Google Scholar] [CrossRef]
- Igarashi, Y.; Otsutomo, S.; Harada, M.; Nakano, S.; Watanabe, S. Lipase-Mediated Resolution of Indene Bromohydrin. Synthesis 1997, 5, 549–552. [Google Scholar] [CrossRef]
- Hao, B.; Gunaratna, M.J.; Zhang, M.; Weerasekara, S.; Seiwald, S.N.; Nguyen, V.T.; Meier, A.; Hua, D.H. Chiral-Substituted Poly-N-vinylpyrrolidinones and Bimetallic Nanoclusters in Catalytic Asymmetric Oxidation Reactions. J. Am. Chem. Soc. 2016, 138, 16839–16848. [Google Scholar] [CrossRef]
- Köhler, V.; Mao, J.; Heinisch, T.; Pordea, A.; Sardo, A.; Wilson, Y.M.; Knörr, L.; Creus, M.; Prost, J.-C.; Schirmer, T.; et al. OsO4·Streptavidin: A Tunable Hybrid Catalyst for the Enantioselective cis-Dihydroxylation of Olefins. Angew. Chem. Int. Ed. 2011, 50, 10863–10866. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Meng, Y.; Haeffner, F.; Leon, R.M.; Crockett, M.P.; Morken, J.P. Carbohydrate/DBU Cocatalyzed Alkene Diboration: Mechanistic Insight Provides Enhanced Catalytic Efficiency and Substrate Scope. J. Am. Chem. Soc. 2018, 140, 3663–3673. [Google Scholar] [CrossRef]
- Zhu, J.; Sun, X.-T.; Wang, X.-D.; Wu, L. Enantioselective Dihydroxylation of Alkenes Catalyzed by 1,4-Bis(9-O-dihydroquinidinyl) phthalazine-Modified Binaphthyl-Osmium Nanoparticles. ChemCatChem 2018, 10, 1788–1792. [Google Scholar] [CrossRef]
- Tetour, D.; Paška, T.; Máková, V.; Holubová, B.N.; Karpíšková, J.; Řezanka, M.; Brus, J.; Hodačová, J. Cinchonine-based organosilica materials as heterogeneous catalysts of enantioselective alkene dihydroxylation. J. Catal. 2021, 404, 493–500. [Google Scholar] [CrossRef]
- Nakano, S.; Igarashi, Y.; Nohira, H. Lipase-mediated kinetic resolution of cis-1,2-indandiol and the Ritter reaction of its mono-acetate. Tetrahedron Asymmetry 2001, 12, 59–62. [Google Scholar] [CrossRef]
- Huang, H.; Lambert, T.H. Regiodivergent Electrophotocatalytic Aminooxygenation of Aryl Olefins. J. Am. Chem. Soc. 2022, 144, 18803–18809. [Google Scholar] [CrossRef] [PubMed]
- Lutz, R.E.; Wayland, R.L. Further Studies on the Stability of β-Hydroxyethylamines toward the Oppenauer Oxidation. cis- and trans-1-Amino-2-indanols. J. Am. Chem. Soc. 1951, 73, 1639–1641. [Google Scholar]
- Gao, Y.; Hong, Y.; Nie, X.; Bakale, R.P.; Feinberg, R.R.; Zepp, C.M. Optically Pure 1-Amino 2-Indanols. U.S. Patent 5,616,808, 1997. [Google Scholar]
- Ghosh, A.K.; Kincaid, J.F.; Haske, M.G. A Convenient Enzymatic Route to Optically Active l-Aminoindan-2-ol: Versatile Ligands for HIV-1 Protease Inhibitors and Asymmetric Syntheses. Synthesis 1997, 5, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Didier, E.; Loubinoux, B.; Ramos Tombo, G.M.; Rihs, G. Chemo-Enzymatic Synthesis of 1,2- and 1,3- Amino-Alcohols and their Use in the Enantioselective Reduction of Acetophenone and Anti-Acetophenone Oxine Methyl Ether with Borane. Tetrahedron 1991, 47, 4941–4958. [Google Scholar] [CrossRef]
- Hassner, A.; Lorber, M.E.; Heathcock, C. Addition of Iodine Isocyanate to Olefins. Scope and Synthetic Utility. J. Org. Chem. 1967, 32, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.X.; Aso, Y.; Otsubo, T.; Ogura, F. Aminotellurinylation of Olefins and Its Utilization for Synthesis of 2-Oxazolidinones. J. Org. Chem. 1989, 54, 4398–4404. [Google Scholar] [CrossRef]
- Hu, N.X.; Aso, Y.; Ogura, O.J. Amidotellurinylation of Olefins and a Novel One-Pot Synthesis of 4,5-Dihydro-oxazoles from Olefins. J. Chem. Soc. Perkin Trans. 1 1989, 1775–1780. [Google Scholar] [CrossRef]
- Kuszpit, M.R.; Giletto, M.B.; Jones, C.L.; Bethel, T.K.; Tepe, J.J. Hydroxyamination of Olefins Using Br-N-(CO2Me)2. J. Org. Chem. 2015, 80, 1440–1445. [Google Scholar] [CrossRef]
- Wu, F.; Alom, N.-E.; Ariyarathna, J.P.; Naß, J.; Li, W. Regioselective Formal [3+2] Cycloadditions of Urea Substrates with Activated and Unactivated Olefins for Intermolecular Olefin Aminooxygenation. Angew. Chem. Int. Ed. 2019, 58, 11676–11680. [Google Scholar] [CrossRef]
- Wu, F.; Kaur, N.; Alom, N.-E.; Li, W. Chiral Hypervalent Iodine Catalysis Enables an Unusual Regiodivergent Intermolecular Olefin Aminooxygenation. JACS Au 2021, 1, 734–741. [Google Scholar] [CrossRef]
- Wata, C.; Hashimoto, T. Organoiodine-Catalyzed Enantioselective Intermolecular Oxyamination of Alkenes. J. Am. Chem. Soc. 2021, 143, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Wappes, E.A.; Nakafuku, K.M.; Nagib, D.A. Directed β C−H Amination of Alcohols via Radical Relay Chaperones. J. Am. Chem. Soc. 2017, 139, 10204–10207. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.-Q.; Chen, X.-Y.; Chen, G.; He, G. Radical-mediated intramolecular β–C(sp3)–H amidation of alkylimidates: Facile synthesis of 1,2-amino alcohols. Chem. Commun. 2018, 54, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Stateman, L.M.; Wappes, E.A.; Nakafuku, K.M.; Edwards, K.M.; Nagib, D.A. Catalytic β C–H amination via an imidate radical Relay. Chem. Sci. 2019, 10, 2693–2699. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Fu, K.; Fang, Y.; Zjou, J.; Shi, L. Site-Specific C(sp3)–H Aminations of Imidates and Amidines Enabled by Covalently Tethered Distonic Radical Anions. Angew. Chem. Int. Ed. 2020, 59, 20682–20690. [Google Scholar] [CrossRef]
- Nakafuku, K.M.; Zhang, Z.; Wappes, E.A.; Stateman, L.M.; Chen, A.D.; Nagib, D.A. Enantioselective radical C–H amination for the synthesis of β-amino alcohols. Nat. Chem. 2020, 12, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Xia, Q.; Wand, D.; Jin, J.-K.; Li, Z.; Liand, Q.-J.; Sun, M.-Y.; Liu, D.; Liu, L.-J.; Shu, H.-X.; et al. Metal-Organic Framework Supported Copper Photoredox Catalysts for Iminyl Radical-Mediated Reactions. Angew. Chem. Int. Ed. 2023, 62, e202300233. [Google Scholar] [CrossRef] [PubMed]
- Barman, D.N.; Nicholas, K.M. Copper-Catalyzed Intramolecular C–H Amination. Eur. J. Org. Chem. 2011, 2011, 908–911. [Google Scholar] [CrossRef]
- Barman, D.N.; Liu, P.; Houk, K.N.; Nicholas, K.M. On the Mechanism of Ligand-Assisted, Copper-Catalyzed Benzylic Amination by Chloramine-T. Organometallics 2010, 29, 3404–3412. [Google Scholar] [CrossRef]
- Noshita, M.; Shimizu, Y.; Morimoto, H.; Oshsima, T. Diethylenetriamine-Mediated Direct Cleavage of Unactivated Carbamates and Ureas. Org. Lett. 2016, 18, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Espino, C.G.; Du Bois, J. A Rh-Catalyzed C-H Insertion Reaction for the Oxidative Conversion of Carbamates to Oxazolidinones. Angew. Chem. Int. Ed. 2001, 40, 598–600. [Google Scholar] [CrossRef]
- Cui, Y.; He, C. A Silver-Catalyzed Intramolecular Amidation of Saturated C-H Bonds. Angew. Chem. Int. Ed. 2004, 43, 4210–4212. [Google Scholar] [CrossRef] [PubMed]
- Espino, C.G.; Wehn, P.M.; Chow, J.; Du Bois, J. Synthesis of 1,3-Difunctionalized Amine Derivatives through Selective C-H Bond Oxidation. J. Am. Chem. Soc. 2001, 123, 6935–6936. [Google Scholar] [CrossRef]
- Liang, J.-L.; Yuan, S.-X.; Huang, J.-S.; Yu, W.-Y.; Che, C.-M. Highly Diastereo- and Enantioselective Intramolecular Amidation of Saturated C-H Bonds Catalyzed by Ruthenium Porphyrins. Angew. Chem. Int. Ed. 2002, 41, 3465–3468. [Google Scholar] [CrossRef]
- Liu, P.; Wong, E.L.-M.; Yuen, A.W.-H.; Che, C.-M. Highly Efficient Alkene Epoxidation and Aziridination Catalyzed by Iron(II) Salt + 4,4′,4″-Trichloro-2,2′:6′,2″-terpyridine/ 4,4″-Dichloro-4′-O-PEG-OCH3-2,2′:6′,2″-terpyridine. Org. Lett. 2008, 10, 3275–3278. [Google Scholar] [CrossRef]
- Liu, Y.; Guan, X.; Wong, E.L.-M.; Liu, P.; Huang, J.-S.; Che, C.-M. Nonheme Iron-Mediated Amination of C(sp3)−H Bonds. Quinquepyridine-Supported Iron-Imide/Nitrene Intermediates by Experimental Studies and DFT Calculations. J. Am. Chem. Soc. 2013, 135, 7194–7204. [Google Scholar] [CrossRef]
- Liang, J.-L.; Yuan, S.-X.; Huang, J.-S.; Che, C.-M. Intramolecular C-N Bond Formation Reactions Catalyzed by Ruthenium Porphyrins: Amidation of Sulfamate Esters and Aziridination of Unsaturated Sulfonamides. J. Org. Chem. 2004, 69, 3610–3619. [Google Scholar] [CrossRef]
- Zhang, J.; Chan, P.W.H.; Che, C.-M. Enantioselective intramolecular amidation of sulfamate esters catalyzed by chiral manganese(III) Schiff-base complexes. Tetrahedron Lett. 2005, 46, 5403–5408. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Jung, H.; Kim, D.; Park, J.; Chang, S. Versatile Cp*Co(III)(LX) Catalyst System for Selective Intramolecular C−H Amidation Reactions. J. Am. Chem. Soc. 2020, 142, 12324–12332. [Google Scholar] [CrossRef]
- Lebel, H.; Huard, K.; Lectard, S. N-Tosyloxycarbamates as a Source of Metal Nitrenes: Rhodium-Catalyzed C-H Insertion and Aziridination Reactions. J. Am. Chem. Soc. 2005, 127, 14198–14199. [Google Scholar] [CrossRef] [PubMed]
- Huard, K.; Lebel, H. N-Tosyloxycarbamates as Reagents in Rhodium-Catalyzed C-H Amination Reactions. Chem. Eur. J. 2008, 14, 6222–6230. [Google Scholar] [CrossRef]
- Lebel, H.; Mamani Laparra, L.; Khalifa, M.; Trudel, C.; Audubert, C.; Szponarski, M.; Dicaire Leduc, C.; Azek, E.; Ernzerhof, M. Synthesis of oxazolidinones: Rhodium-catalyzed C–H amination of N-mesyloxycarbamates. Org. Biomol. Chem. 2017, 15, 4144–4158. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Jung, H.; Kim, D.; Chang, S. Multidimensional Screening Accelerates the Discovery of Rhodium Catalyst Systems for Selective Intra- and Intermolecular C−H Amidations. ACS Catal. 2022, 12, 8127–8138. [Google Scholar] [CrossRef]
- Grelier, G.; Rey-Rodriguez, R.; Darses, B.; Retailleau, P.; Dauban, P. Catalytic Intramolecular C(sp3)–H Amination of Carbamimidates. Eur. J. Org. Chem. 2017, 2017, 1880–1883. [Google Scholar] [CrossRef]
- Milczek, E.; Boudet, N.; Blakey, S. Enantioselective C-H Amination Using Cationic Ruthenium(II)–pybox Catalysts. Angew. Chem. Int. Ed. 2008, 47, 6825–6828. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, M.; Kitagaki, S.; Anada, M.; Hashimoto, S. Enantioselective Intramolecular C–H Amidation of Sulfamate Esters Catalyzed by Chiral Dirhodium(II) Carboxylates. Heterocycles 2006, 69, 527–537. [Google Scholar] [CrossRef]
- Reddy, R.P.; Davies, H.M.L. Dirhodium Tetracarboxylates Derived from Adamantylglycine as Chiral Catalysts for Enantioselective C-H Aminations. Org. Lett. 2006, 8, 5013–5016. [Google Scholar] [CrossRef]
- Zhou, Z.; Tan, Y.; Shen, X.; Ivlev, S.; Meggers, E. Catalytic enantioselective synthesis of β-amino alcohols by nitrene insertion. Sci. China Chem. 2021, 64, 452–458. [Google Scholar] [CrossRef]
- Hu, Q.; Guo, C. Platinum/Copper Dual-Catalyzed Asymmetric Vinylogous Addition Reactions for the Synthesis of Functionalized Indoles. Angew. Chem. Int. Ed. 2023, 62, e202305638. [Google Scholar] [CrossRef]
- Buchsteiner, M.; Martinez-Rodriguez, L.; Jerabek, P.; Pozo, I.; Patzer, M.; Nöthling, N.; Lehmann, C.W.; Fürstner, A. Catalytic Asymmetric Fluorination of Copper Carbene Complexes: Preparative Advances and a Mechanistic Rationale. Chem. Eur. J. 2020, 26, 2509–2515. [Google Scholar] [CrossRef]
- Liu, C.; Yi, J.-C.; Zheng, Z.-B.; Tang, Y.; Dai, L.-X.; You, S.-L. Enantioselective Synthesis of 3a-Amino-Pyrroloindolines by Copper-Catalyzed Direct Asymmetric Dearomative Amination of Tryptamines. Angew. Chem. Int. Ed. 2016, 55, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Sun, G.; Yao, W.; Zhu, Y.; Wang, R.; Cai, L.; Liu, K.; Zhang, Q.; Liu, X.-W.; Wan, Q. 3-Aminodeoxypyranoses in Glycosylation: Diversity-Oriented Synthesis and Assembly in Oligosaccharides. Angew. Chem. Int. Ed. 2017, 56, 5227–5231. [Google Scholar] [CrossRef]
- Allison, B.D.; Mani, N.S. Gram-Scale Synthesis of a β-Secretase 1 (BACE 1) Inhibitor. ACS Omega 2017, 2, 397–408. [Google Scholar] [CrossRef]
- Zeng, J.; Wang, R.; Yao, W.; Zhang, S.; Sun, G.; Liao, Z.; Menga, L.; Wan, Q. Diversified synthesis and α-selective glycosylation of 3-amino-2,3,6-trideoxy sugars. Org. Chem. Front. 2018, 5, 3391–3395. [Google Scholar] [CrossRef]
- Strambeanu, I.I.; White, M.C. Catalyst-Controlled C−O versus C−N Allylic Functionalization of Terminal Olefins. J. Am. Chem. Soc. 2013, 135, 12032–12037. [Google Scholar] [CrossRef] [PubMed]
- Cochran, B.M.; Michael, F.E. Metal-Free Oxidative Cyclization of Urea-Tethered Alkenes with Hypervalent Iodine. Org. Lett. 2008, 10, 5039–5042. [Google Scholar] [CrossRef] [PubMed]
- Lakshman, M.K.; Chaturvedi, S.; Zajc, B.; Gibson, D.T.; Resnick, S.M. A General Chemoenzymatic Synthesis of Enantiopure cis β-Amino Alcohols from Microbially Derived cis-Glycols. Synthesis 1998, 1998, 1352–1356. [Google Scholar] [CrossRef]
- Boyd, D.R.; Sharma, N.D.; Bowers, N.L.; Goodrich, P.A.; Groocock, M.R.; Blacker, A.J.; Clarke, D.A.; Howard, T.; Dalton, H. Stereoselective Dioxygenase-Catalysed Benzylic Hydroxylation at Prochiral Methylene Groups in the Chemoenzymatic Synthesis of Enantiopure Vicinal Aminoindanols. Tetrahedron Asymmetry 1996, 7, 1559–1562. [Google Scholar] [CrossRef]
- Ogasawara, K.; Takahashi, M. Lipase-Mediated Resolution of trans-1-Azidoindan-2-ol: A New Route to Optically Pure cis-1-Aminoindan-2-ol. Synthesis 1996, 1996, 954–958. [Google Scholar]
- Kajiro, H.; Mitamura, S.; Mori, A.; Hiyama, T.A. Practical Synthesis of (1S,2R)-1-Amino-2-indanol, a Key Component of an HIV Protease Inhibitor, Indinavir. Bull. Chem. Soc. Jpn. 1999, 72, 1093–1100. [Google Scholar] [CrossRef]
- Ayhan, S.; Demir, A.S.; Hamamci, H.; Doganel, F.; Ozgul, E. Chemoenzymatic synthesis of (1S,2 R)-1-amino-2-indanol, a key intermediate of HIV protease inhibitor, indinavir. J. Mol. Catal. B Enzym. 2000, 9, 157–161. [Google Scholar]
- Komiya, N.; Noji, S.; Murahashi, S.-I. Manganese Catalyzed Asymmetric Oxidation of Alkanes to Optically Active Ketones Bearing Asymmetric Center at the α-Position. Tetrahedron Lett. 1998, 39, 7921–7924. [Google Scholar] [CrossRef]
- Nguyen, T.H.; Ma, E. Efficient diastereoselective synthesis of cis-2-amino-1-indanol derivatives and cis- and trans-1-amino-2-indanol via Pd-catalyzed hydrogenation. Synth. Commun. 2021, 51, 3717–3728. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, G.S.K.K.; Sabitha, G.; Krishna, A.D.; Prasad, A.R.; Rahaman, H.U.R.; Rao, K.V.; Rao, A.B. Daucus carota and baker’s yeast mediated bio-reduction of prochiral ketones. Tetrahedron Asymmetry 2007, 18, 717–723. [Google Scholar] [CrossRef]
- Kim, E.J.; An, K.M.; Ko, S.Y. Enantioselective Synthesis of Cyclic Amino Alcohols: Cis-1-Amino-2-indanol. Bull. Korean Chem. Soc. 2006, 27, 2019–2022. [Google Scholar] [CrossRef]
- Ko, S.Y. Unusual Regioselection in the Mitsunobu Reactions of syn-2,3-Dihydroxy Esters: Synthesis of Statine and Its Diastereomer. J. Org. Chem. 2002, 67, 2689–2691. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Barrales, A.; Pérez-Bautista, J.A.; Cruz-Gregorio, S.; Meza-Léon, R.L.; Quintero, L.; Cortezano-Arellano, O.; Sartillo-Piscil, F. Chiron approach and the [4 + 2] Diels-Alder cycloaddition of 2-pyrones for the synthesis of cis-(-)-aminoindan-2-ol. Results Chem. 2021, 3, 100174–100177. [Google Scholar] [CrossRef]
- Ramirez, E.; Sánchez, M.; Meza-Léon, R.L.; Quintero, L.; Sartillo-Piscil, F. Selective Wittig olefination in aqueous media for the rapid preparation of unsaturated 7,3-lactone-α-d-xylofuranose derivatives. Tetrahedron Lett. 2010, 51, 2178–2180. [Google Scholar] [CrossRef]
- Kajiro, J.; Mitamura, S.-I.; Mori, A.; Hiyama, T. A Practical Synthesis of (1S, 2R)-1-Amino-2-indanol, a Key Component of HIV Protease Inhibitor, Indinavir. Synlett 1998, 1, 51–52. [Google Scholar] [CrossRef]
- Urban, F.J.; Moore, B.S.J. Synthesis of optically active 2-benzyldihydrobenzopyrans for the hypoglycemic agent englitazone. Heterocyclic. Chem. 1992, 29, 431–438. [Google Scholar] [CrossRef]
- Thompson, W.J.; Fitzgerald, P.M.D.; Holloway, M.K.; Emini, E.A.; Darke, P.L.; McKeever, B.M.; Schleif, W.A.; Quintero, J.C.; Zugay, J.A.; Tucker, T.J.; et al. Synthesis and Antiviral Activity of a Series of HIV-1 Protease Inhibitors with Functionality Tethered to the Pt or P/ Phenyl Substituents: X-ray Crystal Structure Assisted Design. Med. Chem. 1992, 35, 1685–1701. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T.; Yamamoto, K.; Momose, Y.; Yoshida, H.; Nagaoka, Y.; Fuji, K. Kinetic resolution of amino alcohol derivatives with a chiral nucleophilic catalyst: Access to enantiopure cyclic cis-amino alcohols. Chem. Commun. 2001, 2700–2701. [Google Scholar] [CrossRef]
- Pelotier, B.; Priem, G.; Macdonald, S.J.F.; Anson, M.S.; Uptonc, R.J.; Campbell, I.B. Convenient method for the kinetic resolution of β-aminoalcohols. Tetrahedron Lett. 2005, 46, 9005–9007. [Google Scholar] [CrossRef]
- Sakuraia, R.; Sakaib, K. Resolution of racemic cis-1-amino-2-indanol by diastereomeric salt formation with (S)-2-phenylpropionic acid. Tetrahedron Asymmetry 2003, 14, 411–413. [Google Scholar] [CrossRef]
- Askin, D.; Cianciosi, S.J.; Hoerrner, R.S. Reductive Amination Process. U.S. Patent 5,508,404, 6 April 1996. [Google Scholar]
- Luna, A.; Maestro, A.; Astorga, C.; Gotor, V. Enzymatic resolution of (±)-cis- and (±)-trans-1-aminoindan-2-ol and (±)-cis- and (±)-trans-2-aminoindan-1-ol. Tetrahedron Asymmetry 1999, 10, 1969–1977. [Google Scholar] [CrossRef]
- Anilkumar, A.T.; Goto, K.; Takahashi, T.; Ishizaki, K.; Kaga, H. Lipase-mediated resolution of cis-1-amino-2-indanol, the key component of the HIV protease inhibitor indinavir. Tetrahedron Asymmetry 1999, 10, 2501–2503. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Choi, Y.-S.; Yang, S.; Yang, W.R.; Jeong, J.-H. Continuous-Flow Kinetic Resolution of (±)-cis-1-Amino-2-indanol by Lipase-Catalyzed N-Acetylation. Synlett 2015, 26, 1981–1984. [Google Scholar]
- Chen, S.; Wu, L.; Shao, Q.; Yang, G.; Zhang, W. Pd(II)-Catalyzed Asymmetric 1,6-Conjugate Addition of Arylboronic Acids to Meldrum’s Acid-Derived Dienes. Chem. Commun. 2018, 54, 2522–2525. [Google Scholar] [CrossRef]
- Cao, S.; Hong, W.; Ye, Z.; Gong, L. Photocatalytic three-component asymmetric sulfonylation via direct C(sp3)–H functionalization. Nat. Commun. 2021, 12, 2377–2387. [Google Scholar] [CrossRef]
- Singh, R.; Bhamidipati, S. Tricyclic Carbamate JAK Inhibitors. WO 2010/039518 A2, 8 April 2010. [Google Scholar]
- Xu, H.; Wolf, C. Asymmetric Synthesis of Chiral 1,3-Diaminopropanols: Bisoxazolidine-Catalyzed C–C Bond Formation with α-Keto Amides. Angew. Chem. Int. Ed. 2011, 50, 12249–12252. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kobayashi, T.; Mori, H.; Katsumura, S. Development of Highly Stereoselective Asymmetric 6π-Azaelectrocyclization of Conformationally Flexible Linear 1-Azatrienes. From Determination of Multifunctional Chiral Amines, 7-Alkyl cis-1-Amino-2-indanols, to Application as a New Synthetic Strategy: Formal Synthesis of 20-Epiuleine. J. Org. Chem. 2004, 69, 5906–5925. [Google Scholar] [PubMed]
- Kobayashi, T.; Tanaka, K.; Miwa, J.; Katsumura, S. Synthesis of new chiral auxiliaries for 6p-azaelectrocyclization: 4- and 7-alkyl substituted cis-1-amino-2-indanols. Tetrahedron Asymmetry 2004, 15, 185–188. [Google Scholar] [CrossRef]
- Liu, S.Y.; Katsumura, S. An improved synthesis of (-)-7-isopropyl-cis-1-amino-2-indanolthe chiral auxiliary for 6π-azaelectrocyclization. Chin. Chem. Lett. 2009, 20, 1204–1206. [Google Scholar] [CrossRef]
- Liang, F.; Gibney, M.; Yeh, V.; Li, X.; Molteni, V.; Shaw, D.; Berman, A.M.; Lewis, S. Compounds and Compositions as PDGFR Kinase Inhibitors. WO 2013/033620 A1, 7 March 2013. [Google Scholar]
- Alley, K.A.; Clarkson, A.J.; Uehara, A.; Johnson, J.S. A Site-Specific Synthetic Route to Substituted Inda(box) Ligands. Org. Lett. 2023, 25, 9108–9113. [Google Scholar] [CrossRef]
- Marino, S.T.; Stachurska-Buczek, D.; Huggins, D.A.; Krywult, B.M.; Sheehan, C.S.; Nguyen, T.; Choi, N.; Parsons, J.G.; Griffiths, P.G.; James, I.W.; et al. Synthesis of Chiral Building Blocks for Use in Drug Discovery. Molecules 2004, 9, 405–426. [Google Scholar] [CrossRef]
- Marcin, L.R.; Good, A.C.; Wu, Y.-J.; Zuev, D.S.; Olson, R.E.; Wang, N. Diaminopropane Derived Macrocycles as Inhibitors of Beta Amyloid Production. U.S. Patent 2008/0194535 A1, 10 August 2008. [Google Scholar]
- Gross, M.F.; Beaudoin, S.; McNaughton-Smith, G.; Amato, G.S.; Castle, N.A.; Huang, C.; Zou, A.; Yu, W. Aryl sulfonamido indane inhibitors of the Kv1.5 ion channel. Bioorg. Med. Chem. Lett. 2007, 17, 2849–2853. [Google Scholar] [CrossRef]
- McCarron, A.S.; Sells, T.B.; Stirling, M.; Stroud, S.G. Inhibitors of NEDD8-Activating Enzyme. WO 2013/028832 A2, 19 August 2013. [Google Scholar]
- Graham, S.L.; Hungate, R.W.; Huff, J.L.; Lyle, T.A.; Young, S.D.; Britcher, S.F.; Scholz, T.H.; Payne, L.S.; Dorsey, B.D. HIV Protease Inhibitors Having Polyether Substituents. U.S. Patent 747156, 27 May 1992. [Google Scholar]
- Kobayashi, Y.; Kinbara, K.; Sato, M.; Saigo, K. Synthesis, Absolute Configuration, and Application of Enantiopure trans-1-Aminobenz[f]indan-2-ol. Chirality. 2005, 17, 108–112. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kurasawa, T.; Kinbara, K.; Saigo, K. Rational Design of CH/π Interaction Sites in a Basic Resolving Agent. J. Org. Chem. 2004, 69, 7436–7441. [Google Scholar] [CrossRef]
- Jin, Y.X.; Tan, G.; Choi, H.J.; Hyun, M.H. A Facile Synthesis of Optically Active Four Stereoisomers of 1-Aminobenz[f]indan-2-ol. Bull. Korean Chem. Soc. 2006, 27, 755–758. [Google Scholar] [CrossRef]
- Rendina, V.L.; Goetz, S.A.; Neitzel, A.E.; Kaplan, H.Z.; Kingsbury, J.S. Scalable synthesis of a new enantiomerically pure π-extended rigid amino indanol. Tetrahedron Lett. 2012, 53, 15–18. [Google Scholar] [CrossRef]
- Powers, T.; Steeneck, C.; Biesinger, R.; Bluhm, H.; Deng, H.; Dodd, R.; Gallagher JR, B.M.; Gege, C.; Hochgurtel, M.; Kiely, A.; et al. Multicyclic Bis-Amide MMP Inhibitors. U.S. Patent 2006/0173183 A, 3 August 2006. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R4 | R5 | R6 | R7 | Strategy |
|---|---|---|---|---|---|
| 1 | H | H | Br | H | Bromination [71,72,101,102] |
| 2 | H | H | tBu | H | Friedel–Crafts [71] |
| 3 | H | H | Cy | H | Bromination/Coupling [71,72,101] |
| 4 | H | H | NO2 | H | Nitration [103] |
| 5 | H | H | Ar and derivatives | H | Bromination/Coupling [72,101,102] |
| 6 | H | H | iPr | H | Bromination/Coupling [72,73] |
| 7 | H | Ad | Ad | H | Bromination/Coupling [72] |
| Entry | R4 | R5 | R6 | R7 | Strategy |
|---|---|---|---|---|---|
| 1 | H | H | H | Me | Strategy A—Ritter [104,105,106] Strategy C—Reduction [86] |
| 2 | H | H | H | iPr | Strategy A—Ritter [105,106,107] Strategy C—Reduction [86] |
| 3 | H | H | F | H | Strategy A—Ritter [108] |
| 4 | H | H | Br | H | Strategy A—Ritter [109] |
| 5 | H | H | OMe | H | Strategy A—Ritter [110] Strategy B—Mitsunobu [111] |
| 6 | H | H | NO2 | H | Strategy A—Ritter [112] |
| 7 | H | Cl | H | H | Strategy A—Ritter [113] |
| 8 | H | F | H | H | Strategy A—Ritter [108] |
| 9 | H | Br | H | H | Strategy C—Reduction [86,119] |
| 10 | Me | H | H | H | Strategy A—Ritter [106] Strategy C—Reduction [86] |
| 11 | F | H | H | H | Strategy C—Reduction [114] |
| 12 | H | Br | Br | H | Strategy C—Reduction [86] |
| 13 | H | Π-extended (biaryl) | H | Strategy A—Ritter [115,116] Strategy B—Mitsunobu [117] | |
| 14 | H | H | Π-extended (biaryl) | Strategy A—Ritter [118] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendas, I.; Gastaldi, S.; Suppo, J.-S. Strategies for Accessing cis-1-Amino-2-Indanol. Molecules 2024, 29, 2442. https://doi.org/10.3390/molecules29112442
Mendas I, Gastaldi S, Suppo J-S. Strategies for Accessing cis-1-Amino-2-Indanol. Molecules. 2024; 29(11):2442. https://doi.org/10.3390/molecules29112442
Chicago/Turabian StyleMendas, Inès, Stéphane Gastaldi, and Jean-Simon Suppo. 2024. "Strategies for Accessing cis-1-Amino-2-Indanol" Molecules 29, no. 11: 2442. https://doi.org/10.3390/molecules29112442
APA StyleMendas, I., Gastaldi, S., & Suppo, J.-S. (2024). Strategies for Accessing cis-1-Amino-2-Indanol. Molecules, 29(11), 2442. https://doi.org/10.3390/molecules29112442