

A Convenient Synthesis of Novel Isoxazolidine and Isoxazole Isoquinolinones Fused Hybrids


Abstract
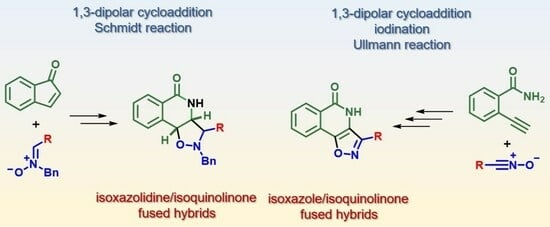
:
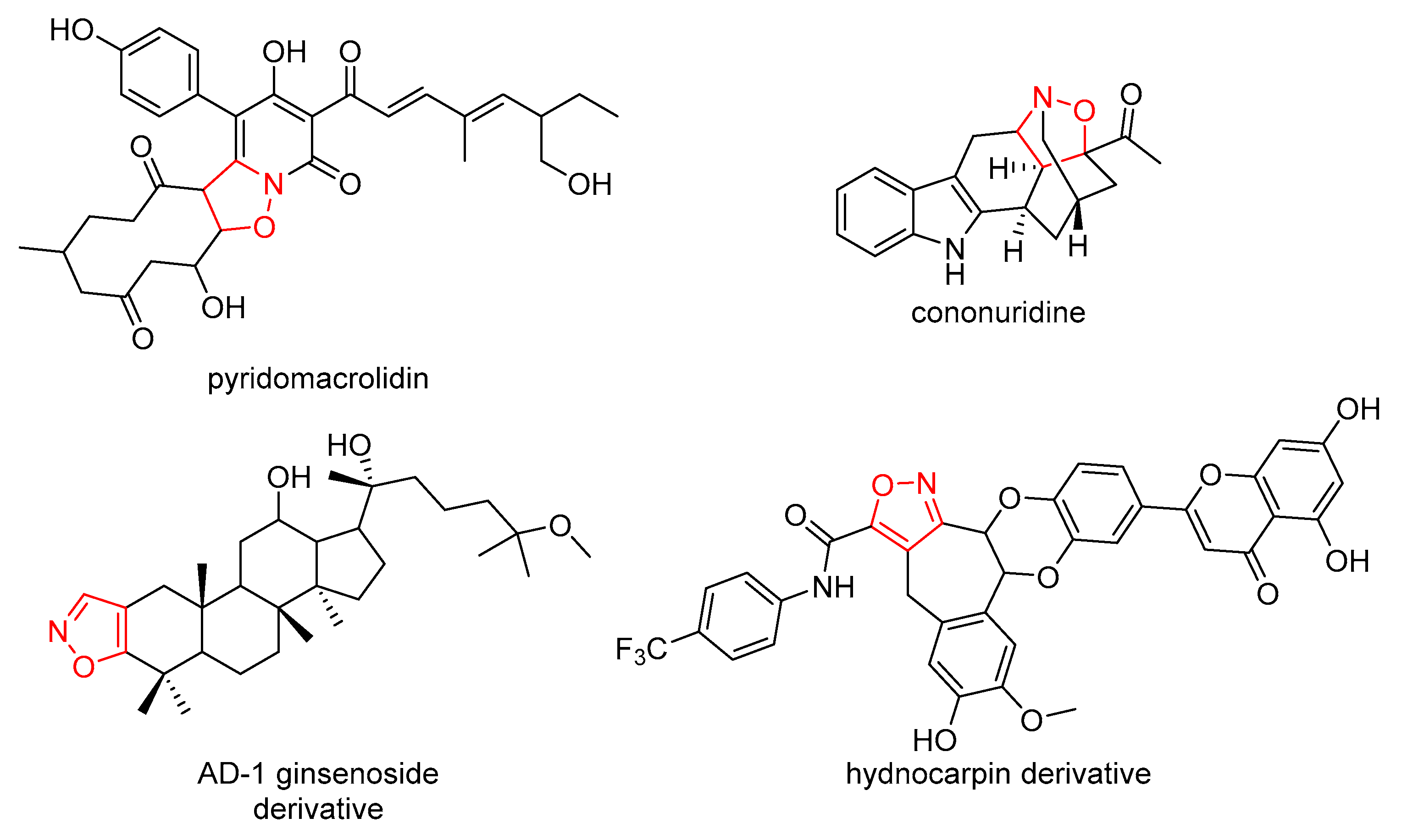
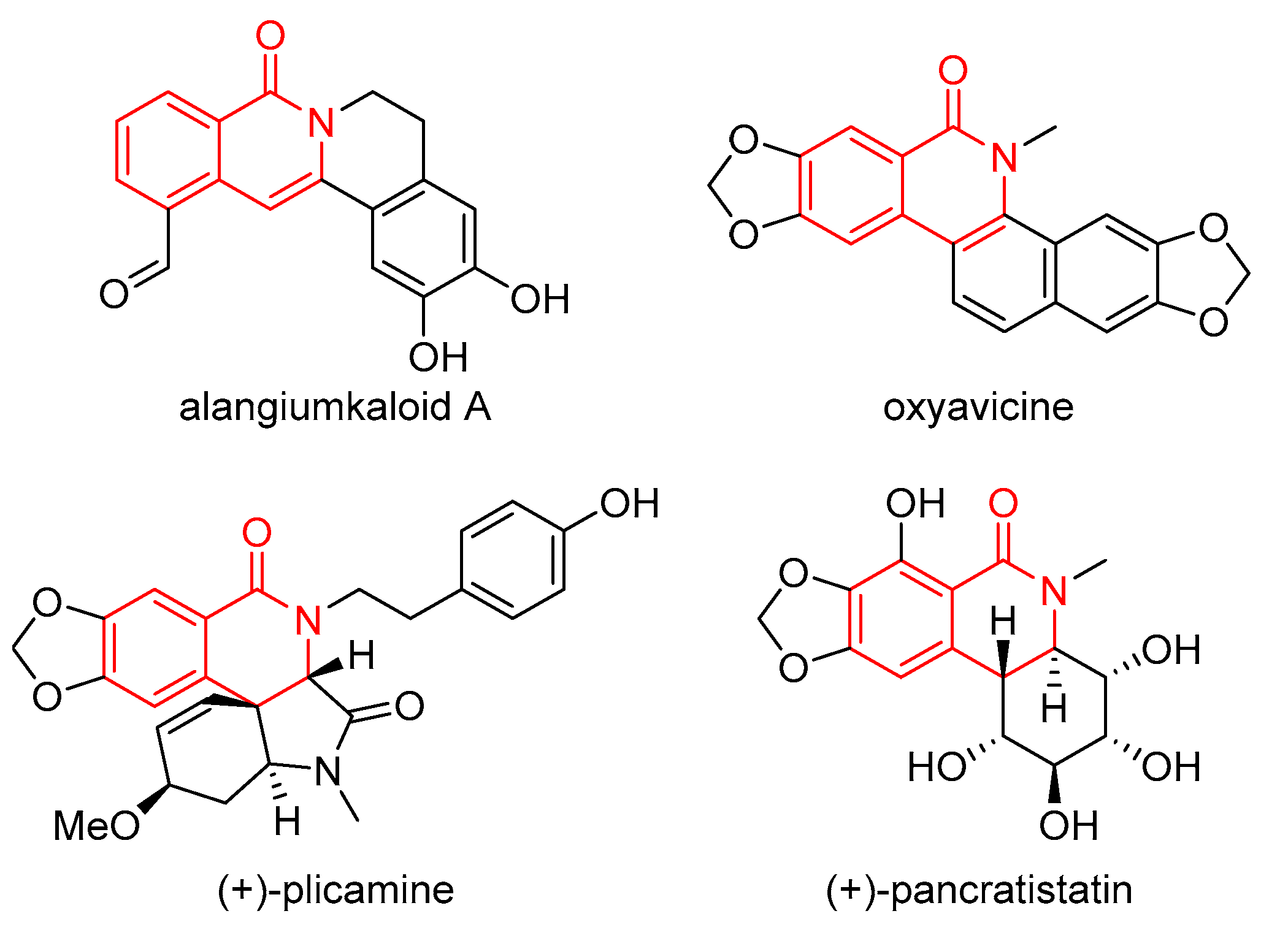
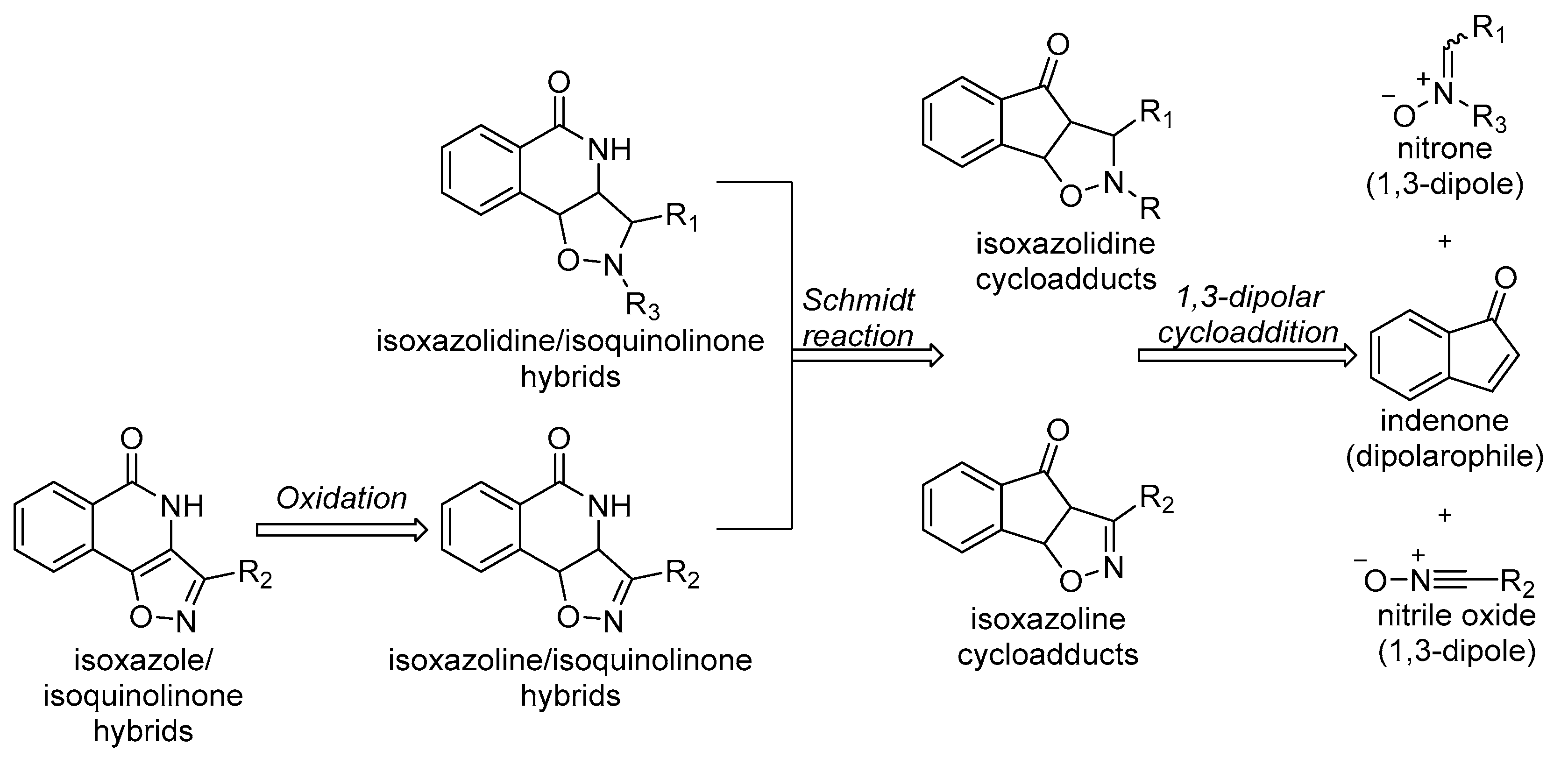
1. Introduction
2. Results and Discussion
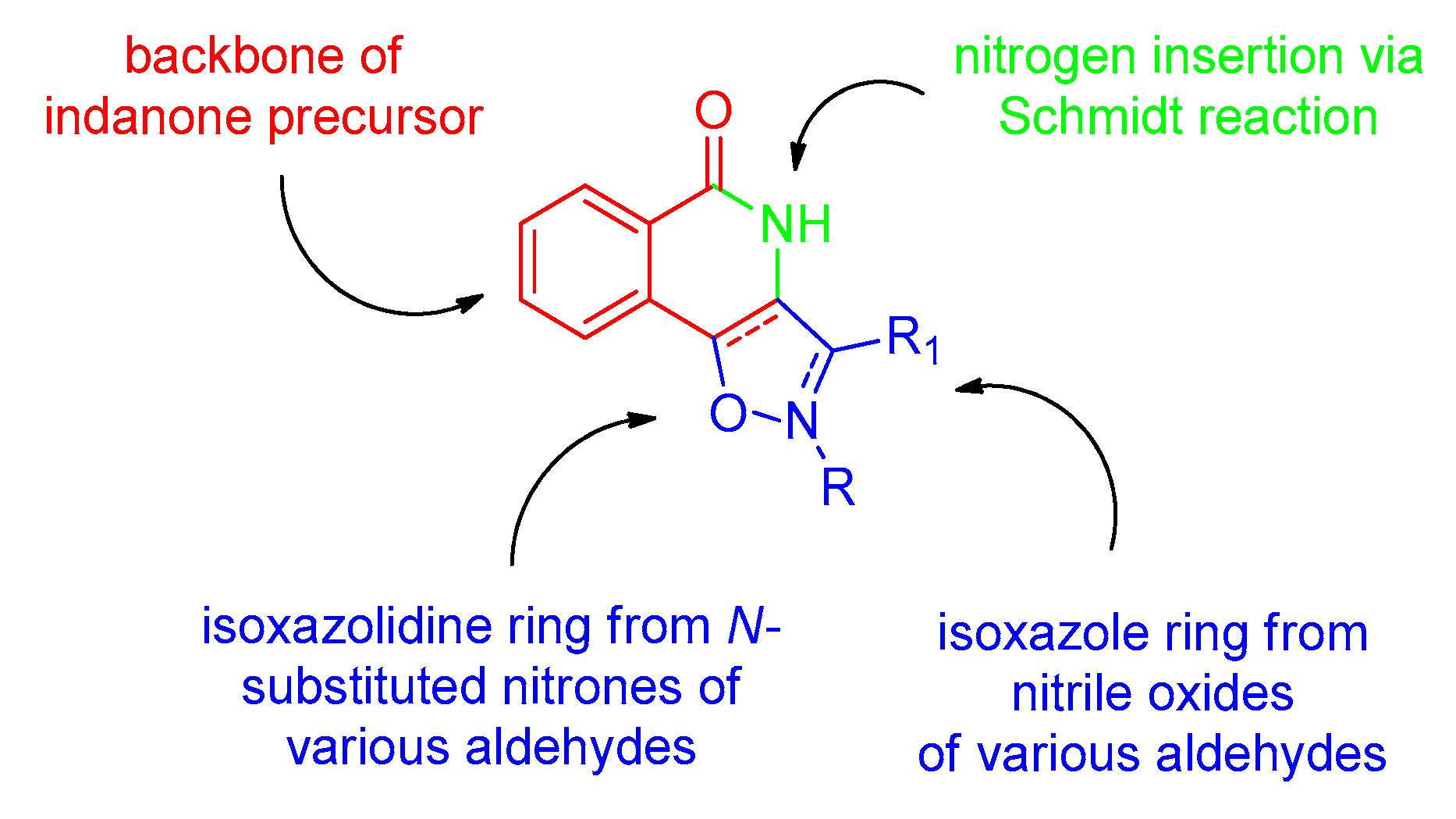
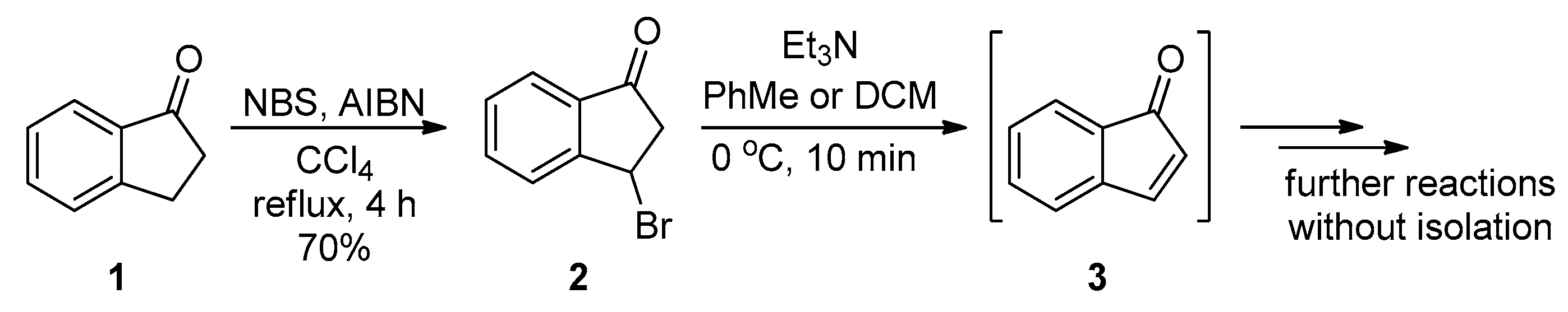
2.1. Synthesis of the Starting Materials
2.2. Synthesis of Isoxazolidine/Isoquinolinone Hybrids
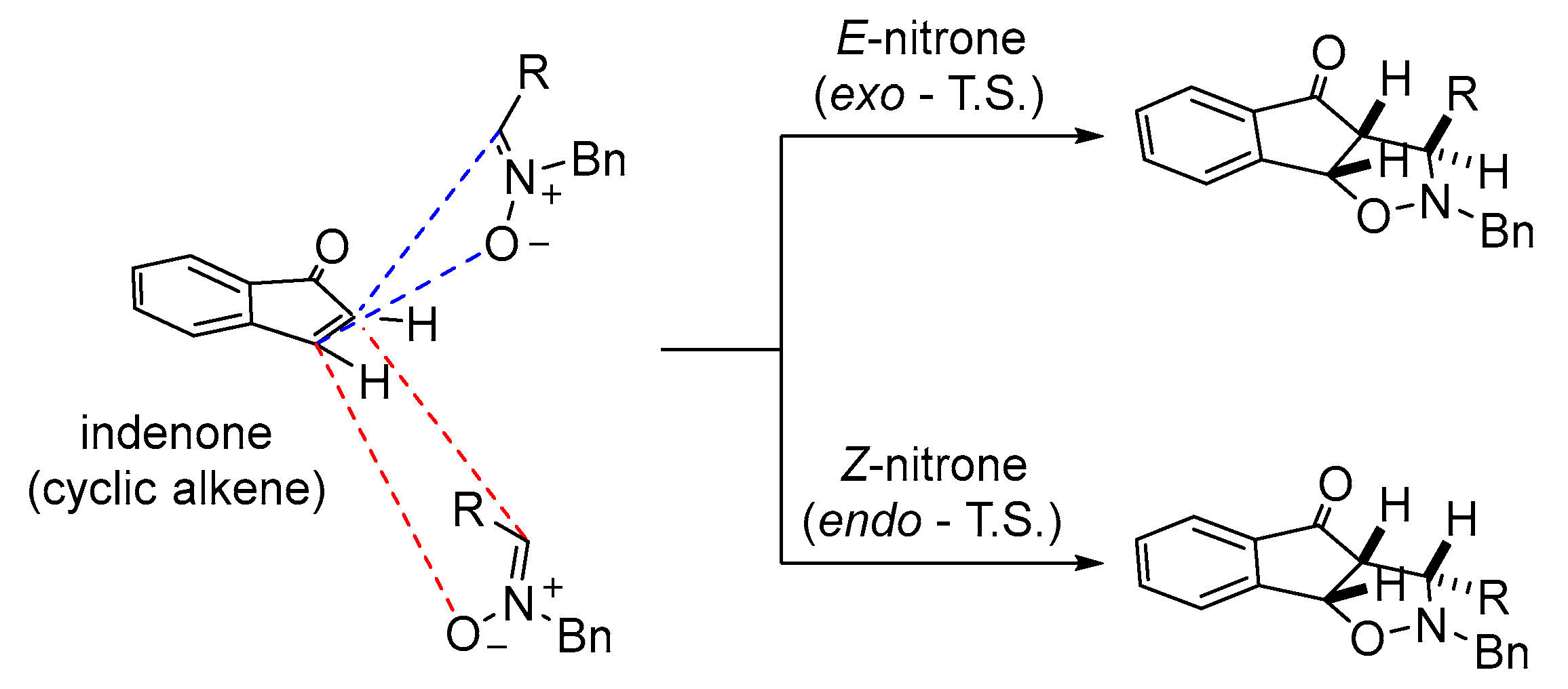
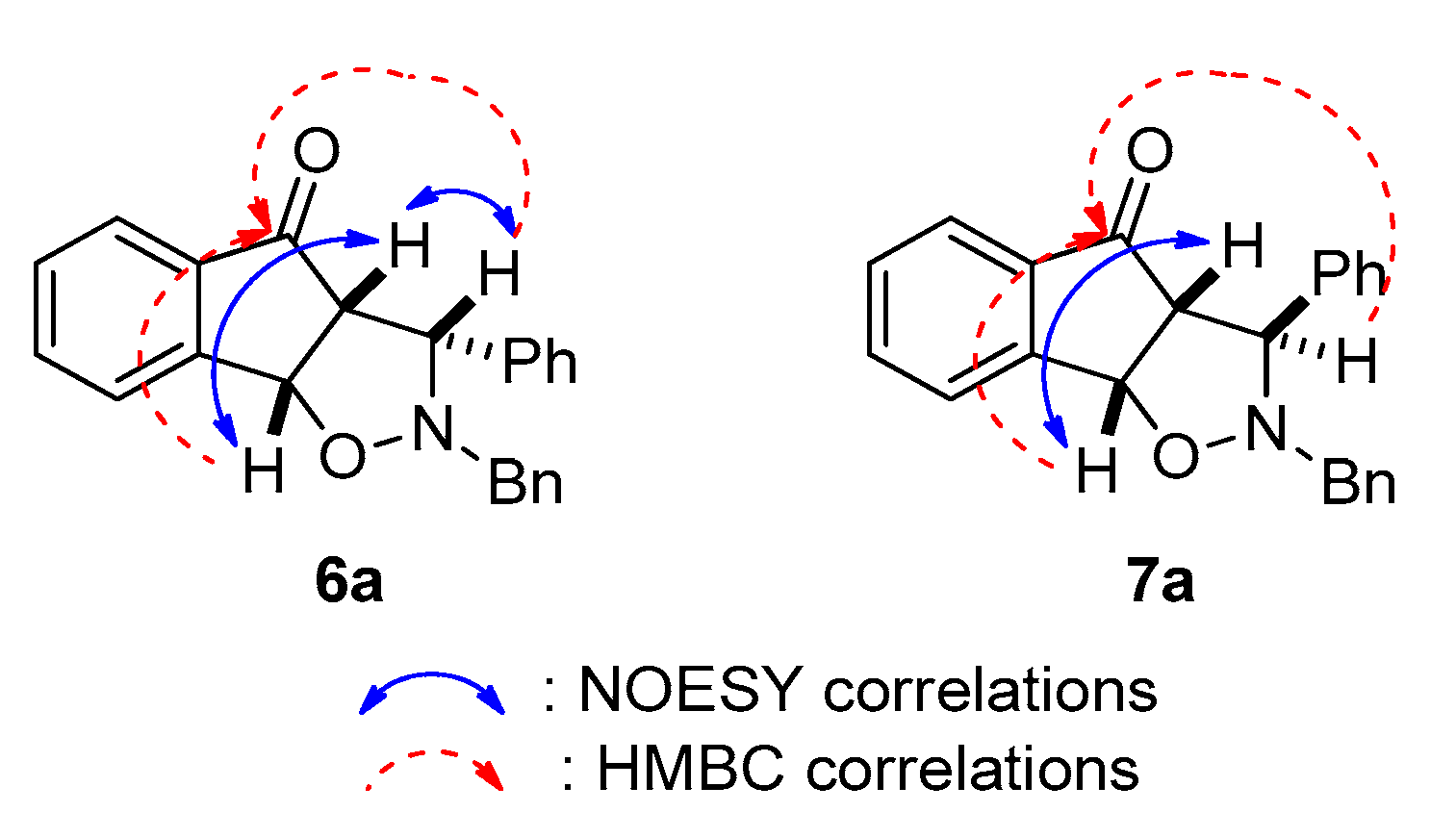
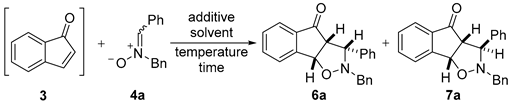
2.2.1. Optimisation Studies for the 1,3-dipolar Cycloaddition Reaction between Indenone and Nitrones
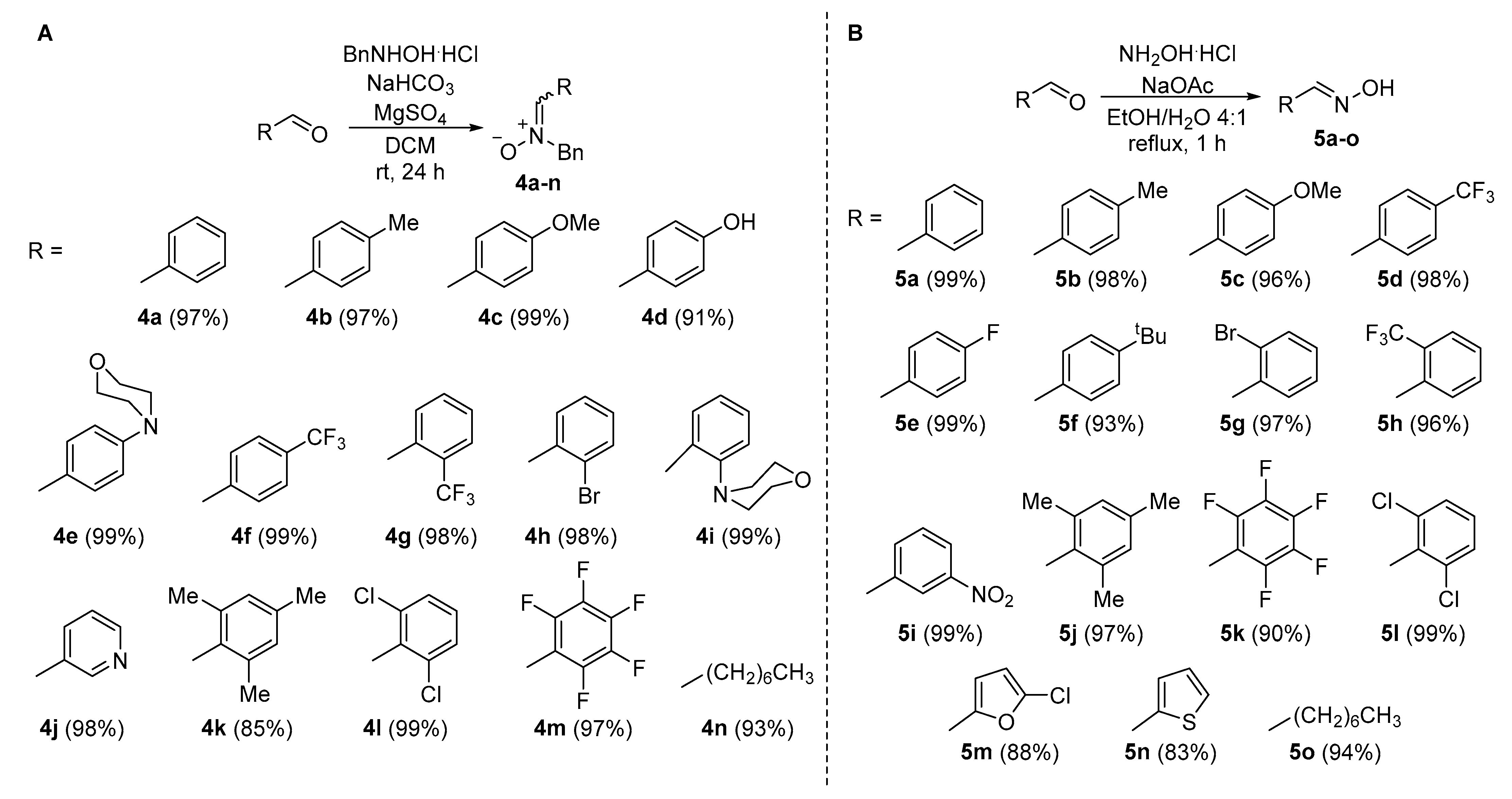
2.2.2. Synthesis of Diverse Isoxazolidines
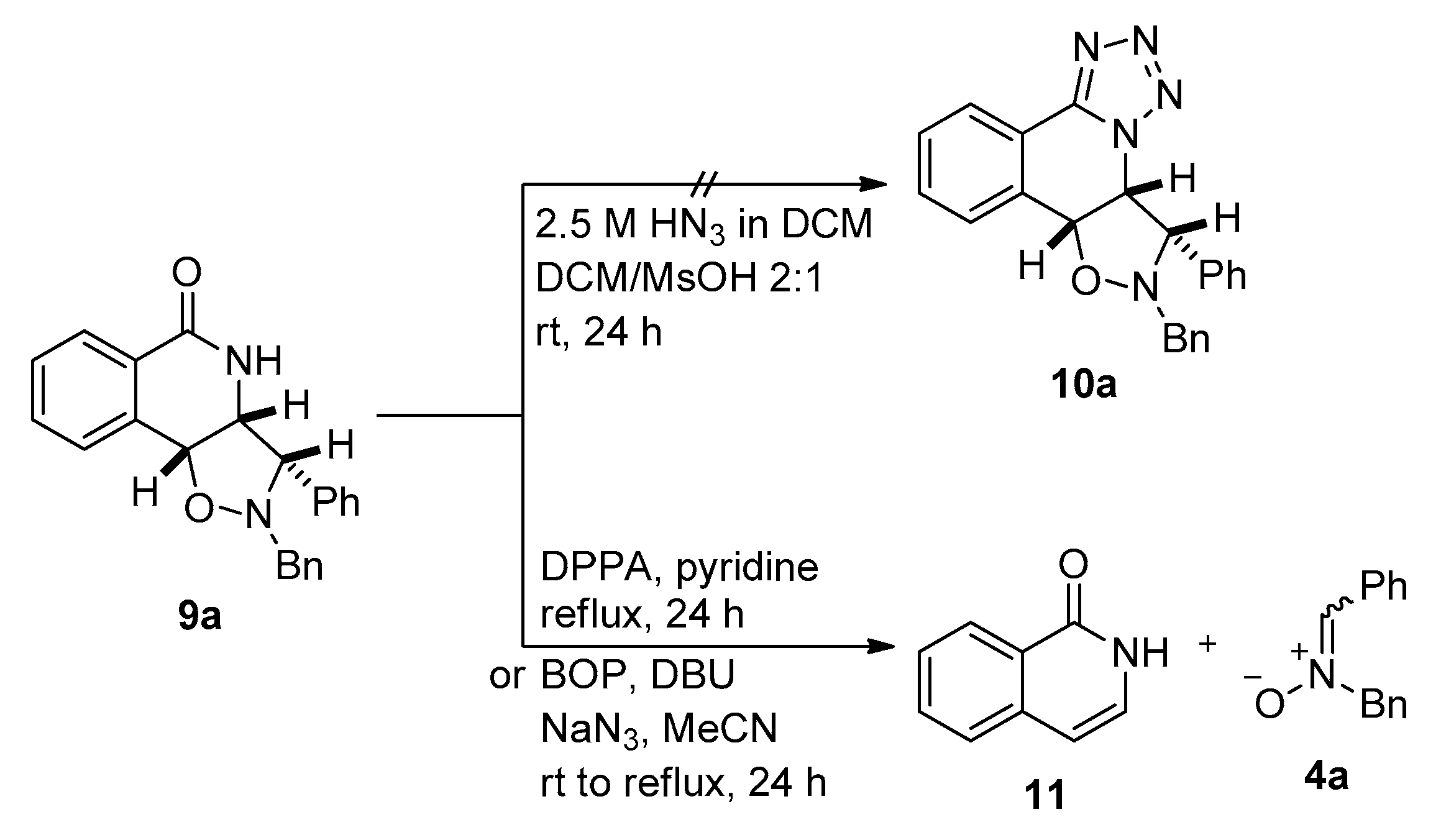
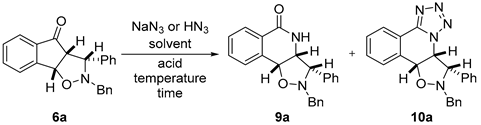
2.2.3. Optimisation of the Schmidt Reaction of Isoxazolidine Adducts
2.2.4. Synthesis of the Targeted Isoxazolidine/Tetrahydroisoquinolinone Hybrids
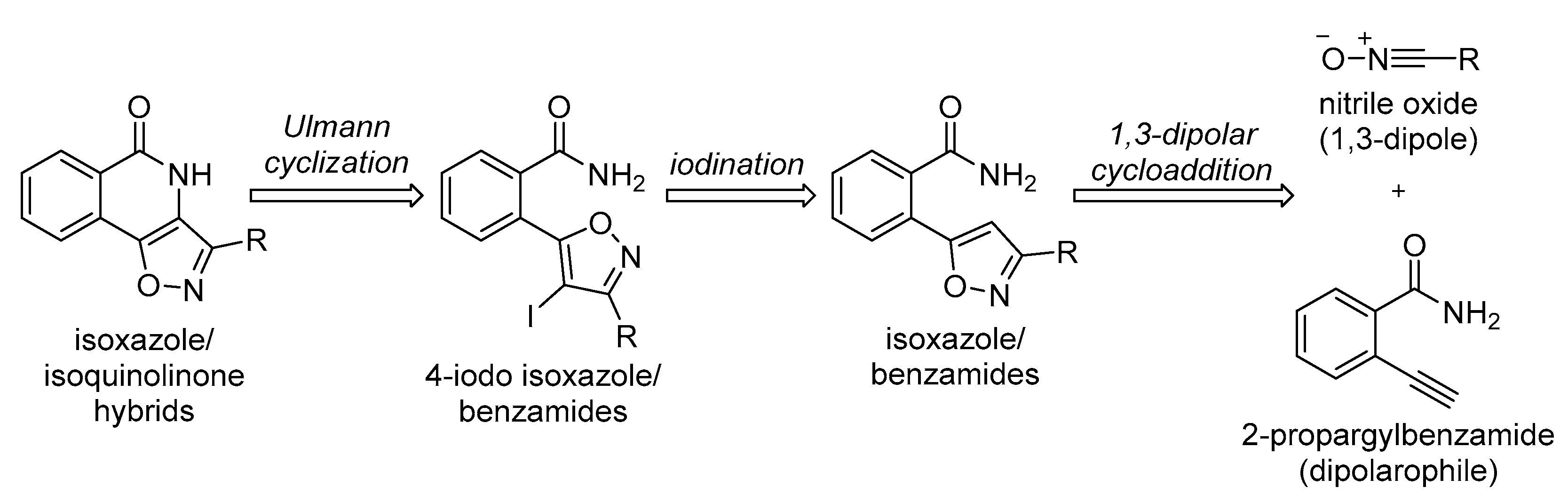
2.3. Synthesis of Isoxazole/Isoquinolinone Hybrids
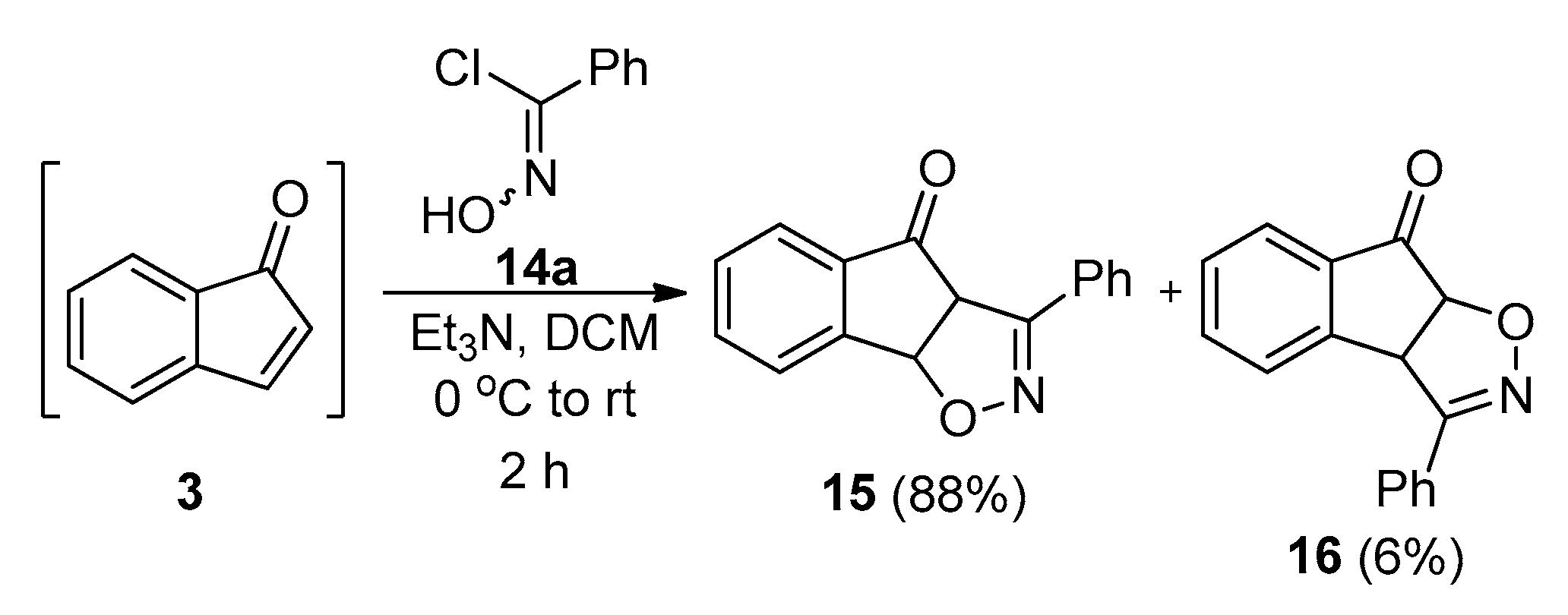
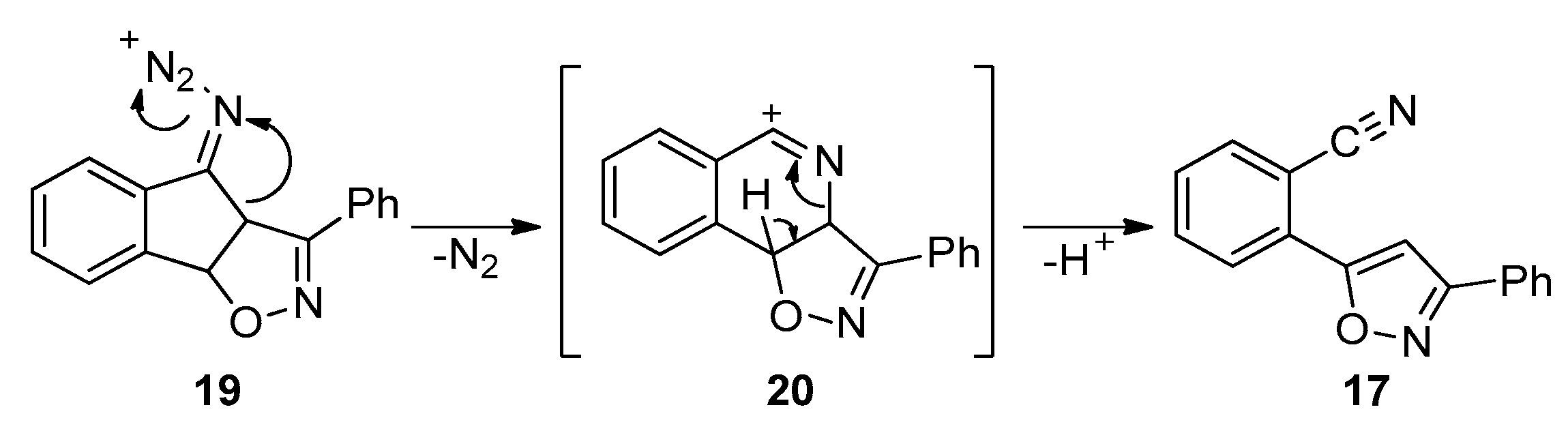
2.3.1. Model 1,3-dipolar Cycloaddition Reaction between Indenone and Benzonitrile Oxide and the Following Schmidt Reaction
2.3.2. Revision of the Synthetic Plan
2.3.3. Isoxazole/Benzamides via an 1,3-dipolar Cycloaddition Reaction
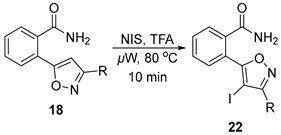
2.3.4. Iodination of the Isoxazole Ring
2.3.5. Optimisation Studies for the Ullmann Reaction
2.3.6. Building a Library of Isoxazole/Isoquinolinone Hybrids via the Ullmann-Type Cyclisation
3. Experimental Section
3.1. General Information
3.2. Synthesis of 3-Bromo-2,3-dihydro-1H-inden-1-one (2)
3.3. General Procedure for the Preparation of Nitrones 4
3.4. General Procedure for the Preparation of Oximes 5
3.5. General Procedure for the Preparation of Isoxazolidine Cycloadducts 6 and 7
3.6. General Procedure for the Preparation of Hybrids 9 and 12
3.7. Synthesis of Isoxazoline Cycloadducts 15 and 16
3.8. Synthesis of Nitrile 17 and Benzamide 18a from 15
3.9. Synthesis of 2-Propargylbenzamide (21)
3.10. General Procedure for the Preparation of Hydroxamoyl Chlorides 14
3.11. General Procedure for the Preparation of Isoxazole/Benzamides 18
3.12. General Procedure for the Preparation of 4-Iodo Isoxazoles 22
3.13. General Procedure for the Preparation of Isoxazole/Isoquinolinones 23
3.14. Synthesis of Isoquinolinone 24 via Reductive Deiodination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breugst, M.; Reissig, H.-U. The Huisgen Reaction: Milestones of the 1,3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [PubMed]
- Berthet, M.; Cheviet, T.; Dujardin, G.; Parrot, I.; Martinez, J. Isoxazolidine: A Privileged Scaffold for Organic and Medicinal Chemistry. Chem. Rev. 2016, 116, 15235–15283. [Google Scholar] [CrossRef] [PubMed]
- Sysak, A.; Obmińska-Mrukowicz, O. Isoxazole Ring as A Useful Scaffold in A Search for New Therapeutic Agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Q.; Tang, H.; Pan, X. Isoxazole/Isoxazoline Skeleton in the Structural Modification of Natural Products: A Review. Pharmaceuticals 2023, 16, 228. [Google Scholar] [CrossRef] [PubMed]
- Chiacchioa, M.A.; Giofrèb, S.V.; Romeob, R.; Romeob, G.; Chiacchio, U. Isoxazolidines as Biologically Active Compounds. Curr. Org. Synth. 2016, 13, 726–749. [Google Scholar] [CrossRef]
- Perrone, M.G.; Vitale, P.; Panella, A.; Ferorelli, S.; Contino, M.; Lavecchia, A.; Scilimati, A. Isoxazole-Based-Scaffold Inhibitors Targeting Cyclooxygenases (COXs). ChemMedChem 2016, 11, 1172–1187. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Kakinuma, N.; Uchida, K.; Hashimoto, R.; Yanagisawa, T.; Nakagawa, A. Pyridovericin and Pyridomacrolidin: Novel Metabolites from Entomopathogenic Fungi, Beauveria Bassiana. J. Antibiot. 1998, 51, 596–598. [Google Scholar] [CrossRef]
- Nge, C.-E.; Sim, K.-S.; Lim, S.-H.; Thomas, N.F.; Low, Y.-Y.; Kam, T.-S. A Hexacyclic, Iboga-Derived Monoterpenoid Indole with a Contracted Tetrahydroazepine C-Ring and Incorporation of an Isoxazolidine Moiety, a Seco-Corynanthean, an Aspidosperma- Aspidosperma Bisindole with Anticancer Properties, and the Absolute Configuration of the Pyridopyrimidine Indole Alkaloid, Vernavosine. J. Nat. Prod. 2016, 79, 2709–2717. [Google Scholar]
- Ma, L.; Miao, D.; Lee, J.J.; Li, T.; Chen, Y.; Su, G.; Zhao, Y. Synthesis and Biological Evaluation of Heterocyclic Ring-Fused Dammarane-Type Ginsenoside Derivatives as Potential Anti-Tumor Agents. Bioorg. Chem. 2021, 116, 105365. [Google Scholar] [CrossRef]
- Arya, J.S.; Joseph, M.M.; Sherin, D.R.; Nair, J.B.; Manojkumar, T.K.; Maiti, K.K. Exploring Mitochondria-Mediated Intrinsic Apoptosis by New Phytochemical Entities: An Explicit Observation of Cytochrome c Dynamics on Lung and Melanoma Cancer Cells. J. Med. Chem. 2019, 62, 8311–8329. [Google Scholar] [CrossRef]
- Rao, L.B.; Sreenivasulu, C.; Kishore, D.R.; Satyanarayana, G. Trending Strategies for the Synthesis of Quinolinones and Isoquinolinones. Tetrahedron 2022, 127, 133093. [Google Scholar] [CrossRef]
- Humphries, P.S.; Benbow, J.W.; Bonin, P.D.; Boyer, D.; Doran, S.D.; Frisbie, R.K.; Piotrowski, D.W.; Balan, G.; Bechle, B.M.; Conn, E.L.; et al. Synthesis and SAR of 1,2,3,4-Tetrahydroisoquinolin-1-ones as Novel G-Protein-Coupled Receptor 40 (GPR40) Antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 2400–2403. [Google Scholar] [CrossRef] [PubMed]
- Crosby, I.T.; Shin, J.K.; Capuano, B. The Application of the Schmidt Reaction and Beckmann Rearrangement to the Synthesis of Bicyclic Lactams: Some Mechanistic Considerations. Aust. J. Chem. 2010, 63, 211–226. [Google Scholar] [CrossRef]
- López, L.; Selent, J.; Ortega, R.; Masaguer, C.F.; Domínguez, E.; Areias, F.; Brea, J.; Loza, M.I.; Sanz, F.; Pastor, M. Synthesis, 3D-QSAR, and Structural Modeling of Benzolactam Derivatives with Binding Affinity for the D2 and D3 Receptors. ChemMedChem 2010, 5, 1300–1317. [Google Scholar] [CrossRef] [PubMed]
- Pailee, P.; Prachyawarakorn, V.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. Protoberberine Alkaloids and Cancer Chemopreventive Properties of Compounds from Alangium salviifolium. Eur. J. Org. Chem. 2011, 2011, 3809–3814. [Google Scholar] [CrossRef]
- Arthur, H.R.; Hui, W.H.; Ng, Y.L. An Examination of the Rutaceae of Hong Kong. Part III. The Alkaloid, Avicine, from Zanthoxylum Avicennae. J. Chem. Soc. 1959, 803, 4007–4009. [Google Scholar] [CrossRef]
- Zhan, G.; Zhou, J.; Liu, R.; Liu, T.; Guo, G.; Wang, J.; Xiang, M.; Xue, Y.; Luo, Z.; Zhang, Y.; et al. Galanthamine, Plicamine, and Secoplicamine Alkaloids from Zephyranthes candida and Their Anti-acetylcholinesterase and Anti-inflammatory Activities. J. Nat. Prod. 2016, 79, 760–766. [Google Scholar] [CrossRef]
- Manpadi, M.; Kornienko, A. Total Syntheses of Pancratistatin. A Review. Org. Prep. Proced. Int. 2008, 40, 107–161. [Google Scholar] [CrossRef]
- Hauser, F.M.; Zhou, M.; Sun, Y. Facile Synthesis of Indenones from Indanones: A New Procedure. Synth. Commun. 2001, 31, 77–80. [Google Scholar] [CrossRef]
- Lu, X.; Schneider, U. Aza-Morita-Baylis-Hillman Reactions Catalyzed by a Cyclopropenylidene. Chem. Commun. 2016, 52, 12980–12983. [Google Scholar] [CrossRef]
- Chandrasekhar, B.; Ahn, S.; Ryu, J.-S. Synthesis of 4-Isoxazolines through Gold(I)-Catalyzed Cyclization of Propargylic N-Hydroxylamines. J. Org. Chem. 2016, 81, 6740–6749. [Google Scholar] [CrossRef] [PubMed]
- He, C.-T.; Han, X.-L.; Zhang, Y.-X.; Du, Z.-T.; Si, C.-M.; Wei, B.-G. Sc(OTf)3-Catalyzed [3 + 2]-Cycloaddition of Nitrones with Ynones. Org. Biomol. Chem. 2021, 19, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, o.; Mulani, I.; De Niro, A.; Maiuolo, L.; Nardi, M.; Russo, B.; Avnet, S. Efficient Synthesis of Isoxazolidine-Substituted Bisphosphonates By 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 2011, 67, 5635–5641. [Google Scholar] [CrossRef]
- Poulsen, P.H.; Vergura, S.; Monleón, A.; Jørgensen, D.K.B.; Jørgensen, K.A. Controlling Asymmetric Remote and Cascade 1,3-Dipolar Cycloaddition Reactions by Organocatalysis. J. Am. Chem. Soc. 2016, 138, 6412–6415. [Google Scholar] [CrossRef] [PubMed]
- Delso, I.; Terejo, T. 1H–15N HMBC as a Valuable Tool for the Identification and Characterization of Nitrones. Tetrahedron Lett. 2007, 48, 4101–4104. [Google Scholar] [CrossRef]
- Chakraborty, B.; Chettri, E. Synthesis of Some Novel Class of Regioselective Spiro Isoxazolidine Derivatives via 1,3-Dipolar Cycloaddition Reaction of N-Benzyl-C-fluorosubstituted Phenyl Nitrones in Ionic Liquid. J. Heterocycl. Chem. 2018, 55, 1157–1165. [Google Scholar] [CrossRef]
- Katahara, S.; Kobayashi, S.; Fujita, K.; Matsumoto, T.; Sato, T.; Chida, N. An Iridium-Catalyzed Reductive Approach to Nitrones from N-Hydroxyamides. J. Am. Chem. Soc. 2016, 138, 5246–5249. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Batra, S. Direct Transformation of Arylamines to Aryl Halides via Sodium Nitrite and N-Halosuccinimide. Chem. Eur. J. 2018, 24, 14622–14626. [Google Scholar] [CrossRef]
- Schierle, S.; Neumann, S.; Heitel, P.; Willems, S.; Kaiser, A.; Pollinger, J.; Merk, D. Design and Structural Optimization of Dual FXR/PPARδ Activators. J. Med. Chem. 2020, 63, 8369–8379. [Google Scholar] [CrossRef]
- Di Nunno, L.; Vitale, P.; Scilimati, A.; Simonea, L.; Capitelli, F. Stereoselective Dimerization of 3-Arylisoxazoles to Cage-Shaped Bis-b-lactams syn 2,6-Diaryl-3,7-diazatricyclo[4.2.0.02,5]-octan-4,8-diones Induced by Hindered Lithium Amides. Tetrahedron 2007, 63, 12388–12395. [Google Scholar] [CrossRef]
- Tambara, K.; Dan Pantos, G. Conversion of Aldoximes into Nitriles and Amides Under Mild Conditions. Org. Biomol. Chem. 2013, 11, 2466–2472. [Google Scholar] [CrossRef] [PubMed]
- Steiger, S.A.; Li, C.; Backos, D.S.; Reigan, P.; Natale, N.R. Dimeric Isoxazolyl-1,4-Dihydropyridines Have Enhanced Binding at the Multi-Drug Resistance Transporter. Bioorg. Med. Chem. 2017, 25, 3223–3234. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lu, M. Metal-Free: A Novel and Efficient Aerobic Oxidation of Primary Amines to Oximes Using N,N′,N″-Trihydroxyisocyanuric Acid and Acetaldoxime as Catalysts in Water. Synlett 2014, 25, 1873–1878. [Google Scholar] [CrossRef]
- McIntosh, M.L.; Naffziger, M.R.; Ashburn, B.O.; Zakharov, L.N.; Carter, R.G. Highly Regioselective Nitrile Oxide Dipolar Cycloadditions with ortho-Nitrophenyl Alkynes. Org. Biomol. Chem. 2012, 10, 9204–9213. [Google Scholar] [CrossRef] [PubMed]
- Jawalekar, A.M.; Reubsaet, E.; Rutjes, F.P.J.T.; van Delft, F.L. Synthesis of Isoxazoles by Hypervalent Iodine-Induced Cycloaddition of Nitrile Oxides to Alkynes. Chem. Commun. 2011, 47, 3198–3200. [Google Scholar] [CrossRef] [PubMed]
- Kanemasa, S.; Matsuda, H.; Kamimurac, A.; Kakinami, T. Synthesis of Hydroximoyl Chlorides from Aldoximes and Benzyltrimethylammonium Tetrachloroiodate (BTMA ICl4). Tetrahedron 2000, 56, 1057–1064. [Google Scholar] [CrossRef]
- Suzuki, K.; Watanabe, T.; Murahashi, S.-I. Oxidation of Primary Amines to Oximes with Molecular Oxygen using 1,1-Diphenyl-2-picrylhydrazyl and WO3/Al2O3 as Catalysts. J. Org. Chem. 2013, 78, 2301–2310. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Arceo, E.; García, J.M.; López, R.; González, A.; Linden, A. Lewis Acid Catalyzed Asymmetric Cycloadditions of Nitrones: A’-Hydroxy Enones as Efficient Reaction Partners. Angew. Chem. Int. Ed. 2005, 44, 6187–6190. [Google Scholar] [CrossRef]
- Cao, G.; Zhou, S.; Teng, D. Synthesis of Spiroisoxazolidinyl-Benzoisothiazolines by 1,3-Dipolar Cycloaddition of Benzoisothiazole-2,2-dioxide-3-ylidenes with Nitrones. Tetrahedron 2017, 73, 2329–2333. [Google Scholar] [CrossRef]
- Śnieżek, M.; Stecko, S.; Panfil, I.; Furman, B.; Urbańczyk-Lipkowska, Z.; Chmielewski, M. Thermal and Sc(OTf)3 Catalyzed 1,3-Dipolar Cycloaddition of Open-Chain Nitrones to α,β-Unsaturated Lactones: Combined Experimental and Computational Studies. Tetrahedron Asymmetry 2013, 24, 89–103. [Google Scholar] [CrossRef]
- Minami, S.; Tomita, M.; Takamatsu, H.; Uyeo, S. The Schmidt Reaction with Some Tetralone and Indanone Derivatives. Chem. Pharm. Bull. 1965, 13, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.; Lockhart, I.M. The Schmidt Reaction with Aromatic Ketones. J. Chem. Soc. 1965, 4806–4812. [Google Scholar] [CrossRef]
- Conley, R.T. Schmidt Reactions in Polyphosphoric Acid. I. Rearrangement of Ketones. J. Org. Chem. 1958, 23, 1330–1333. [Google Scholar] [CrossRef]
- Škorić, D.Đ.; Klisurić, O.R.; Jakimov, D.S.; Sakač, M.N.; Csanádi, J.J. Synthesis of New Bile Acid-Fused Tetrazoles Using the Schmidt Reaction. Beilstein J. Org. Chem. 2021, 17, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Koldobskii, G.I.; Ostrovskii, V.A. Application of the Schmidt Reaction for the Preparation of Tetrazoles. Chem. Heterocycl. Compd. 1975, 11, 626–635. [Google Scholar] [CrossRef]
- Ishihara, K.; Shioiri, T.; Matsugi, M. An Expeditious Approach to Tetrazoles from Amides Utilizing Phosphorazidates. Org. Lett. 2020, 22, 6244–6247. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.-K.; Wacharasindhu, S.; Levins, G.C.; Lin, M.; Tabei, K.; Mansour, T.S. The Scope and Mechanism of Phosphonium-Mediated SNAr Reactions in Heterocyclic Amides and Ureas. J. Org. Chem. 2007, 72, 10194–10210. [Google Scholar] [CrossRef]
- Stotani, S.; Gatta, V.; Medda, F.; Padmanaban, M.; Karawajczyk, A.; Tammela, P.; Giordanetto, F.; Tzalis, D.; Collina, S. A Versatile Strategy for the Synthesis of 4,5-Dihydroxy-2,3-Pentanedione (DPD) and Related Compounds as Potential Modulators of Bacterial Quorum Sensing. Molecules 2018, 23, 2545. [Google Scholar] [CrossRef]
- VerHaeghe, D.G.; Weber, G.S.; Pappalardo, P.A. Beckmann Fragmentation Versus Beckmann Rearrangement in Dehydronorcamphor Derivatives. Tetrahedron Lett. 1989, 30, 4041–4044. [Google Scholar] [CrossRef]
- Brahmchari, D.; Verma, A.K.; Mehta, S. Regio- and Stereoselective Synthesis of Isoindolin-1-ones through BuLi-Mediated Iodoaminocyclization of 2-(1-Alkynyl)benzamides. J. Org. Chem. 2018, 83, 3339–3347. [Google Scholar] [CrossRef]
- Morita, T.; Fuse, S.; Nakamura, H. Generation of an 4-Isoxazolyl Anion Species: Facile Access to Multifunctionalized Isoxazoles. Angew. Chem. Int. Ed. 2016, 55, 1–6. [Google Scholar] [CrossRef]
- Quana, Z.-J.; Xiaa, H.-D.; Zhanga, Z.; Daa, Y.-X. Ligand-free CuTC-catalyzed N-arylation of Amides, Anilines and 4-aminoantipyrine: Synthesis of N-arylacrylamides, 4- amido-N-phenylbenzamides and 4-amino (N-phenyl)antipyrenes. Appl. Organometal. Chem. 2014, 28, 81–85. [Google Scholar] [CrossRef]
- Cantillo, D.; de Frutos, O.; Rincon, J.A.; Mateos, C.; Kappe, C.O. A Scalable Procedure for Light-Induced Benzylic Brominations in Continuous Flow. J. Org. Chem. 2014, 79, 223–229. [Google Scholar] [CrossRef]
- Wang, A.; Lv, P.; Liu, Y. 4,5-Dihydro-1,2,4-oxadiazole as a Single Nitrogen Transfer Reagent: Synthesis of Functionalized Isoxazoles Assisted by Sc(OTf)3 or Au(I)/ Sc(OTf)3 Synergistic Catalysis. Org. Lett. 2023, 25, 4377–4382. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 3 (equiv.) | 4a 1 (equiv.) | Solvent 2 | Temp. (°C) | Additive (0.25 equiv.) | Time (h) | 6a 5 | 7a 5 | Yield 5 (comb.) |
| 1 | 1 | 1 | PhMe | 110 | - | 24 | 40% | 30% | 70% |
| 2 | 1 | 1 | DCM | 25 | - | 72 | 18% | 7% | 25% |
| 3 | 1 | 1 | none | 80 | - | 2 | 32.5% | 15.5% | 48% |
| 4 | 1 | 1 | DCM | 25 | Cu(OTf)2 | 24 | 10% | <1% | 10% |
| 5 | 1 | 1 | PhMe | 40 | Sc(OTf)3 | 72 | 43% | 20% | 63% |
| 6 | 1 | 1 | PhMe | 40 | Mg(OTf)2 | 72 | 39.5% | 25.5% | 65% |
| 7 | 1.2 | 1 | PhMe | 60 | Zn(OTf)2 | 24 | 50% | 28% | 78.5% |
| 8 | 1.2 | 1 | PhMe | 60 | Zn(OTf)2 | 48 | 54.5% | 25.5% | 80% |
| 9 | 1.2 | 1 | PhMe | 60 | In(OTf)3 | 48 | 35% | 20.5% | 55.5% |
| 10 | 1.2 | 1 | PhMe | 60 | AgOTf | 72 | 45.5% | 36% | 81.5% |
| 11 3 | 1.2 | 1 | PhMe | 80 | Zn(OTf)2 | 24 | 50% | 34% | 84% |
| 12 | 1.2 | 1 | PhMe | 80 | AgOTf | 24 | 50% | 40% | 90% |
| 13 | 1.2 | 1 | PhMe | 80 | Cu(OTf)2 | 24 | 31% | 19% | 50% |
| 14 4 | 1.2 | 1 | PhMe | 80 | Zn(OTf)2 | 3 | 34.5% | 18.5% | 53% |
| 15 4 | 1.2 | 1 | PhMe | 100 | AgOTf | 4 | 46% | 34% | 80% |
| 16 3 | 1 | 1 | PhMe | 25 | SnCl4 | 24 | - | - | - |
| 17 3 | 1 | 1 | PhMe | 25 | TiCl4 | 24 | - | - | - |
| 18 | 1.2 | 1 | PhMe | 80 | Ag2CO3 | 24 | 45% | 35.5% | 80.5% |
| 19 | 1.2 | 1 | PhMe | 80 | Ag2O | 24 | 45% | 35% | 80% |
 | |||||
|---|---|---|---|---|---|
| Entry | R = | endo Cycloadduct | exo Cycloadduct | Regio Cycloadduct | Yield 1 (comb.) |
| 1 | 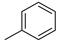 | 6a (50%) | 7a (40%) | - | 90% |
| 2 |  | 6b (40%) | 7b (37%) | - | 77% |
| 3 |  | 6c (47%) | 7c (24%) | - | 71% |
| 4 |  | 6d (45%) | 7d (26%) | - | 71% |
| 5 | 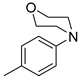 | 6e (43%) | 7e (26%) | - | 69% |
| 6 | 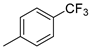 | 6f (54%) | 7f (24%) | - | 78% |
| 7 | 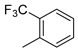 | 6g (31%) | 7g (24%) | 8g (16%) | 71% |
| 8 | 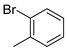 | 6h (28%) | 7h (20%) | 8h (23%) | 71% |
| 9 | 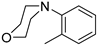 | 6i (42%) | 7i (34%) | - | 76% |
| 10 | 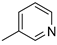 | 6j (55%) | 7j (27%) | - | 82% |
| 11 | 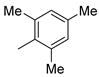 | 6k (32%) | 7k (20%) | - | 52% |
| 12 |  | 6l (40%) | - | - | 40% |
| 13 | 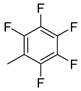 | 6m (18%) | 7m (37%) | - | 55% |
| 14 |  | 6n (21%) | 7n (55%) | - | 76% |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Acid | Reagent | Solvent 4 | Temp. (°C) | Time (h) | 9a 5 | 10a 5 | Yield 5 (comb.) |
| 1 1 | aq. HCl (37%) | NaN3 | PhMe | 25 | 24 | - | - | rsm |
| 2 1 | aq. HCl (37%) | NaN3 | PhMe | 50 | 24 | - | - | rsm |
| 3 2 | aq. HCl (37%) | NaN3 | - | 80 | 24 | - | - | rsm |
| 4 2 | TFA | NaN3 | - | 25 | 24 | - | - | rsm |
| 5 2 | TFA | NaN3 | - | 72 | 24 | - | - | rsm |
| 6 1 | TfOH | NaN3 | PhMe | 25 | 24 | - | - | rsm |
| 7 2 | TfOH | NaN3 | - | 25 | 24 | - | - | rsm |
| 8 1 | H2SO4 | NaN3 | PhMe | 80 | 24 | 15% | - | 15% |
| 9 1 | MsOH | NaN3 | PhMe | 25 | 24 | <10% | - | ~10% + rsm |
| 10 3 | MsOH | HN3 | PhMe | 25 | 48 | 76% | 13% | 89% |
| 11 3 | MsOH | HN3 | DCM | 25 | 48 | 78% | 14% | 92% |
 | ||||
|---|---|---|---|---|
| Entry | R = | Lactam 1 | Tetrazole 1 | Combined Yield 1 |
| 1 | 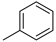 | 9a (78%) | 10a (14%) | 89% |
| 2 | 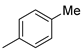 | 9b (59%) | 10b (19%) | 78% |
| 3 |  | 9c (62%) | - | 62% |
| 4 |  | 9d (54%) | - | 54% |
| 5 | 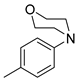 | 9e (81%) | - | 81% |
| 6 | 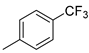 | 9f (70%) | - | 70% |
| 7 | 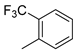 | 9g (65%) | 10g (12%) | 77% |
| 8 |  | 9h (65%) | 10h (17%) | 82% |
| 9 |  | 9i (51%) | - | 51% |
| 10 |  | 9j (53%) | - | 53% |
| 11 | 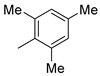 | 9k (90%) | - | 90% |
| 12 | 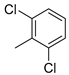 | 9l (72%) | 10l (13%) | 85% |
| 13 | 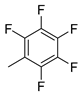 | 9m (65%) | - | 65% |
| 14 | 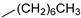 | 9n (69%) | 10n (11%) | 80% |
 | ||||
|---|---|---|---|---|
| Entry | R = | Lactam 1 | Tetrazole 1 | Combined Yield 1 |
| 1 | 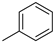 | 12a (82%) | 13a (14%) | 96% |
| 2 |  | 12b (72%) | 13b (14%) | 86% |
| 3 | 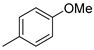 | 12c (83%) | 13c (14%) | 97% |
| 4 | 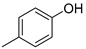 | 12d (75%) | - | 75% |
| 5 | 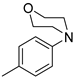 | 12e (84%) | 13e (14%) | 98% |
| 6 |  | 12f (85%) | 13f (14%) | 99% |
| 7 | 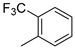 | 12g (82%) | 13g (14%) | 96% |
| 8 | 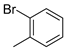 | 12h (87%) | 13h (12%) | 99% |
| 9 | 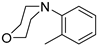 | 12i (84%) | 13i (8%) | 92% |
| 10 |  | 12j (64%) | - | 64% |
| 11 | 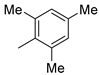 | - | - | decomposed |
| 13 | 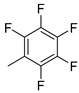 | 12m (81%) | 13m (12%) | 93% |
| 14 | 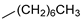 | 12n (90%) | 13n (9%) | 99% |
 | |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 | 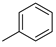 | 18a (84%) | 9 | 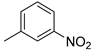 | 18i (83%) |
| 2 | 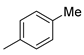 | 18b (89%) | 10 2 |  | 18j (90%) |
| 3 | 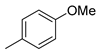 | 18c (87%) | 11 3 |  | 18k (84%) |
| 4 |  | 18d (86%) | 12 2 | 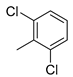 | 18l (93%) |
| 5 |  | 18e (88%) | 13 |  | 18m (80%) |
| 6 | 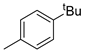 | 18f (91%) | 14 |  | 18n (79%) |
| 7 | 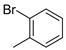 | 18g (92%) | 15 | 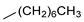 | 18o (92%) |
| 8 |  | 18h (89%) | |||
 | |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 |  | 22a (97%) | 9 3 | 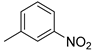 | 22i (99%) |
| 2 | 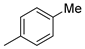 | 22b (99%) | 10 | 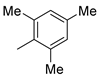 | - |
| 3 2 | 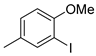 | 22c (88%) | 11 |  | 22k (99%) |
| 4 | 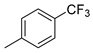 | 22d (99%) | 12 | 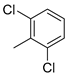 | 22l (98%) |
| 5 |  | 22e (97%) | 13 | 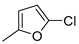 | - |
| 6 |  | 22f (97%) | 14 |  | - |
| 7 |  | 22g (99%) | 15 | 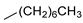 | 22o (98%) |
| 8 | 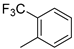 | 22h (99%) | |||
 | |||||
|---|---|---|---|---|---|
| Entry 1 | Copper Source (equiv.) | Base (2 equiv.) | Solvent 2 | Temp. (°C) | Yield 5 |
| 1 | CuI (0.15) | tBuONa | tBuOH | 100 | 16% |
| 2 | CuI (0.15) | tBuONa | 1,4-dioxane | 100 | 20% |
| 3 3 | CuI (0.15) | Cs2CO3 | DMF | 100 | 15% |
| 4 | CuI (1) | Cs2CO3 | DMF | 100 | 56% |
| 5 | CuTC (0.15) | K3PO4 | DMF | 100 | 50% |
| 6 | CuTC (0.25) | K3PO4 | DMF | 80 | 61% |
| 7 | CuTC (0.15) | tBuONa | DMF | 100 | 55% |
| 8 | CuTC (0.15) | Cs2CO3 | DMF | 100 | 56% |
| 9 | CuTC (0.25) | Cs2CO3 | DMF | 80 | 73% |
| 10 | CuTC (0.25) | K2CO3 | DMF | 80 | 78% |
| 11 | CuTC (0.25) | K2CO3 | DMF | 120 | - |
| 12 | CuTC (0.25) | K2CO3 | DMSO | 80 | 31% |
| 13 4 | CuTC (0.25) | K2CO3 | DMF | 80 | 48% |
 | |||||
|---|---|---|---|---|---|
| Entry | R = | Yield 1 | Entry | R = | Yield 1 |
| 1 | 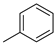 | 23a (78%) | 7 |  | 23g (58%) |
| 2 |  | 23b (67%) | 8 | 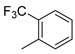 | 23h (61%) |
| 3 | 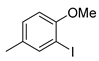 | 23c (93%) | 9 | 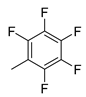 | 23k (29%) |
| 4 | 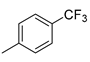 | 23d (81%) | 10 | 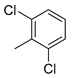 | 23l (46%) |
| 5 | 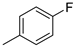 | 23e (89%) | 11 | 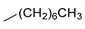 | 23o (80%) |
| 6 | 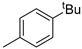 | 23f (78%) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouzounthanasis, K.A.; Rizos, S.R.; Koumbis, A.E. A Convenient Synthesis of Novel Isoxazolidine and Isoxazole Isoquinolinones Fused Hybrids. Molecules 2024, 29, 91. https://doi.org/10.3390/molecules29010091
Ouzounthanasis KA, Rizos SR, Koumbis AE. A Convenient Synthesis of Novel Isoxazolidine and Isoxazole Isoquinolinones Fused Hybrids. Molecules. 2024; 29(1):91. https://doi.org/10.3390/molecules29010091
Chicago/Turabian StyleOuzounthanasis, Konstantinos A., Stergios R. Rizos, and Alexandros E. Koumbis. 2024. "A Convenient Synthesis of Novel Isoxazolidine and Isoxazole Isoquinolinones Fused Hybrids" Molecules 29, no. 1: 91. https://doi.org/10.3390/molecules29010091
APA StyleOuzounthanasis, K. A., Rizos, S. R., & Koumbis, A. E. (2024). A Convenient Synthesis of Novel Isoxazolidine and Isoxazole Isoquinolinones Fused Hybrids. Molecules, 29(1), 91. https://doi.org/10.3390/molecules29010091