
Reaction of Picolinamides with Ketones Producing a New Type of Heterocyclic Salts with an Imidazolidin-4-One Ring

 , ,
, , 

Abstract
:1. Introduction
2. Results
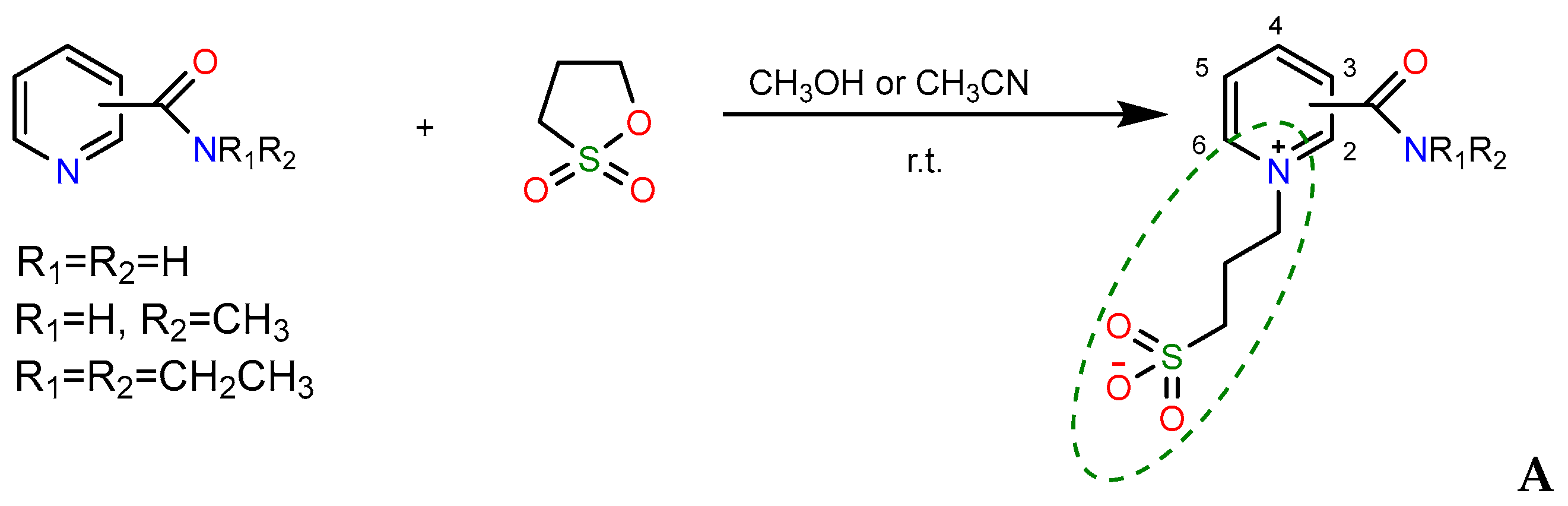
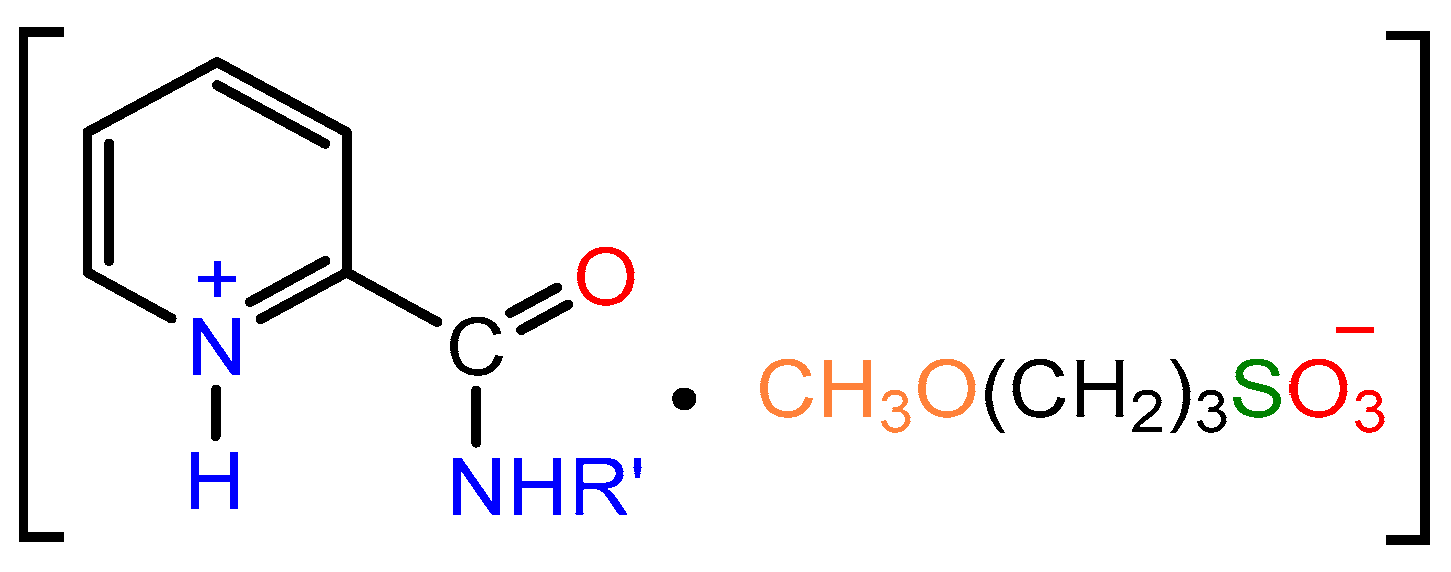
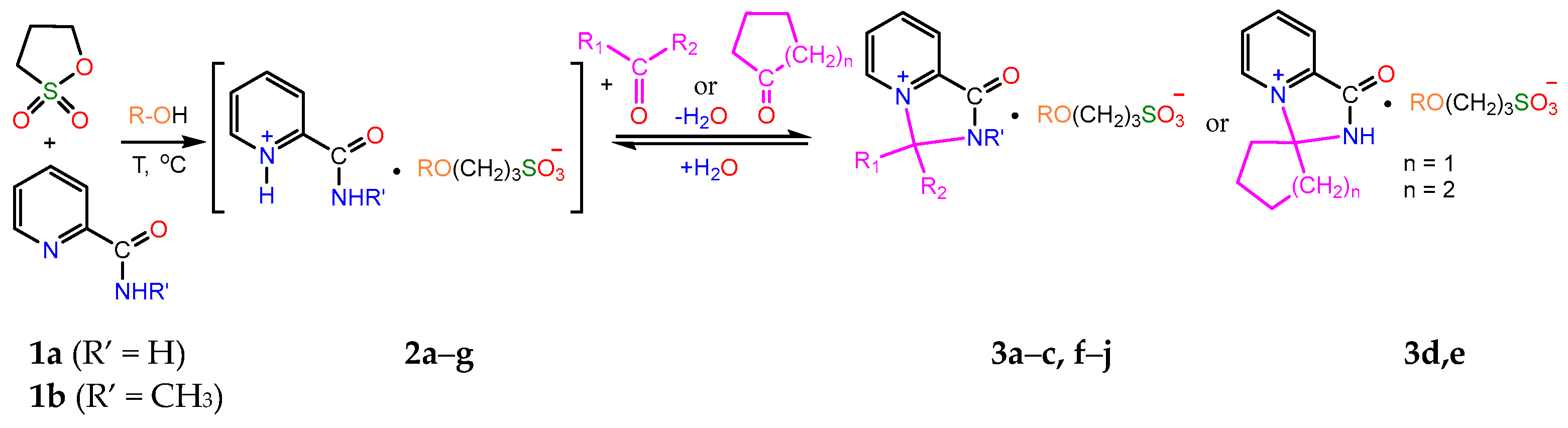
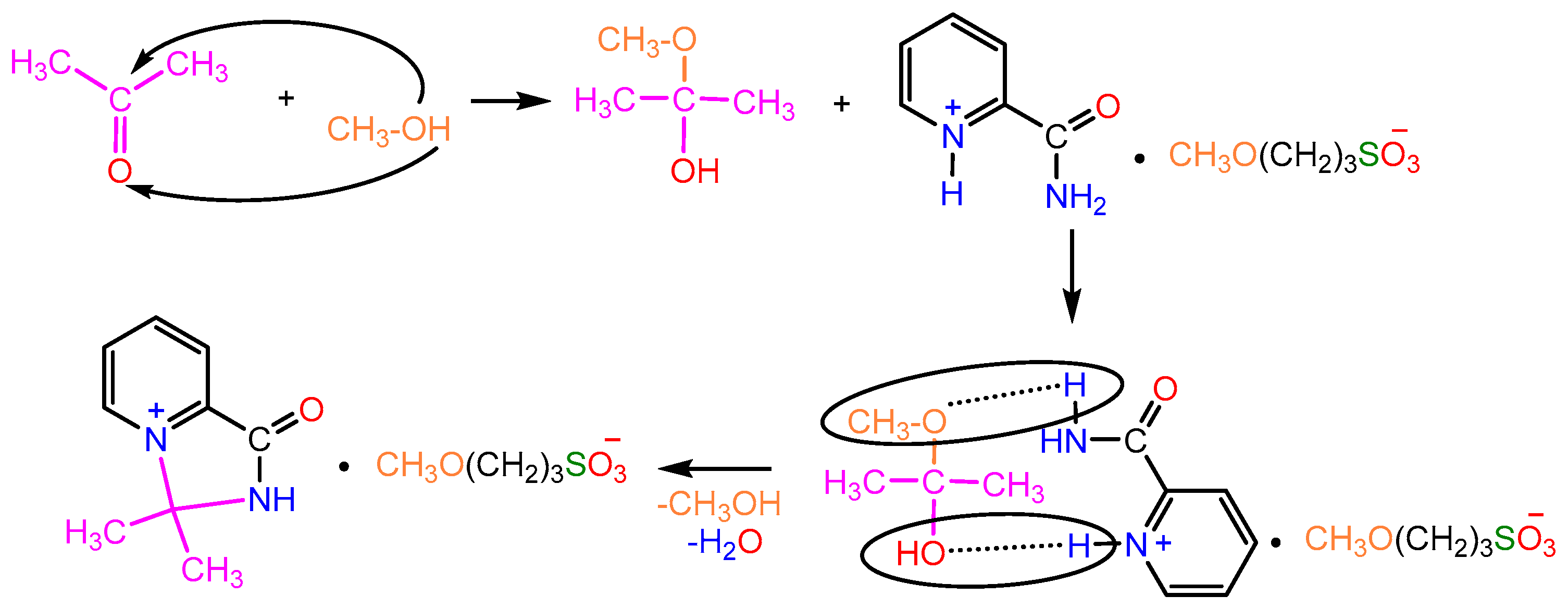
2.1. Synthesis
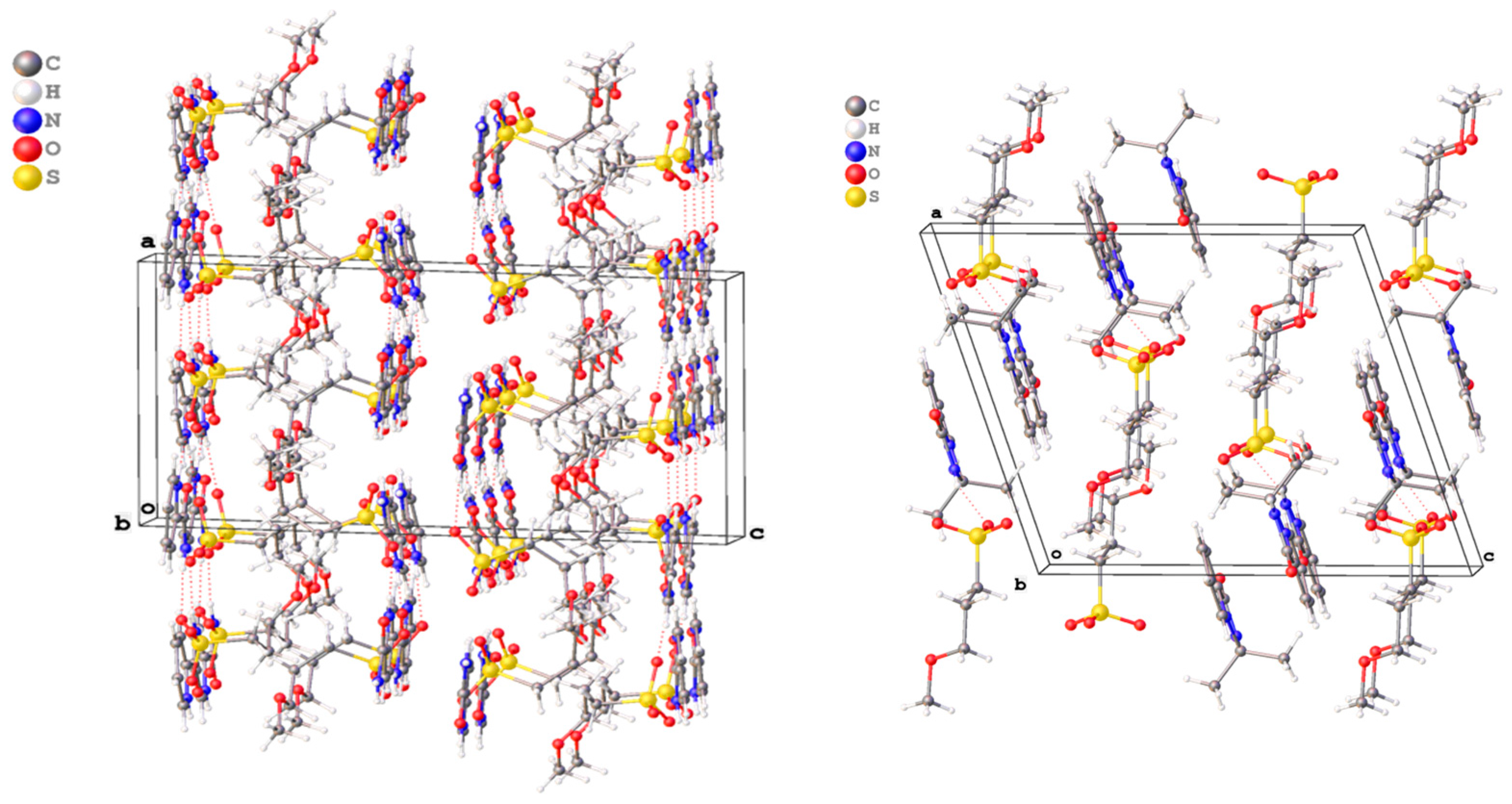
2.2. X-ray Study
2.3. Reactions of Hydrolysis of Compounds 3a, 3d and 3e
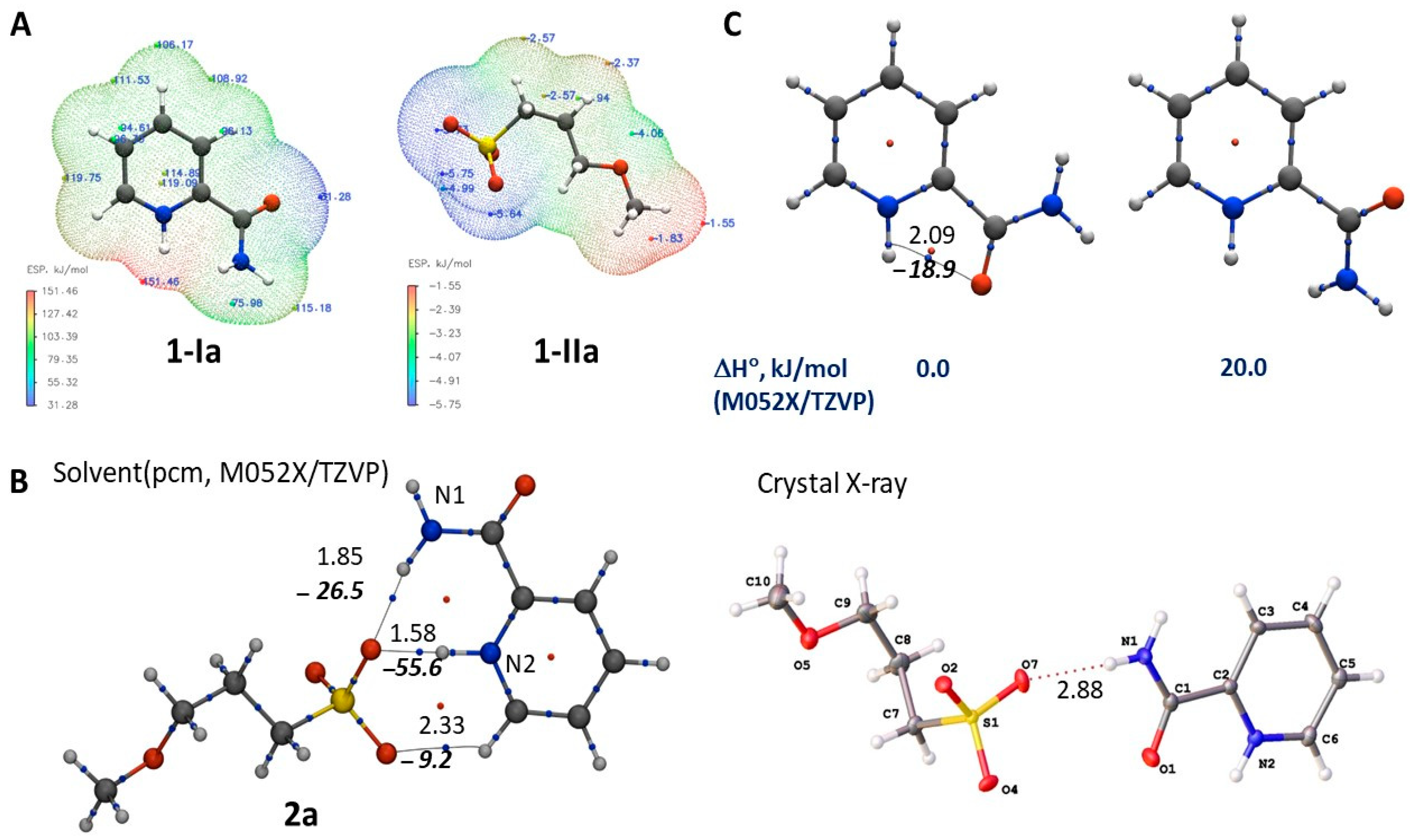
2.4. Theoretical Study
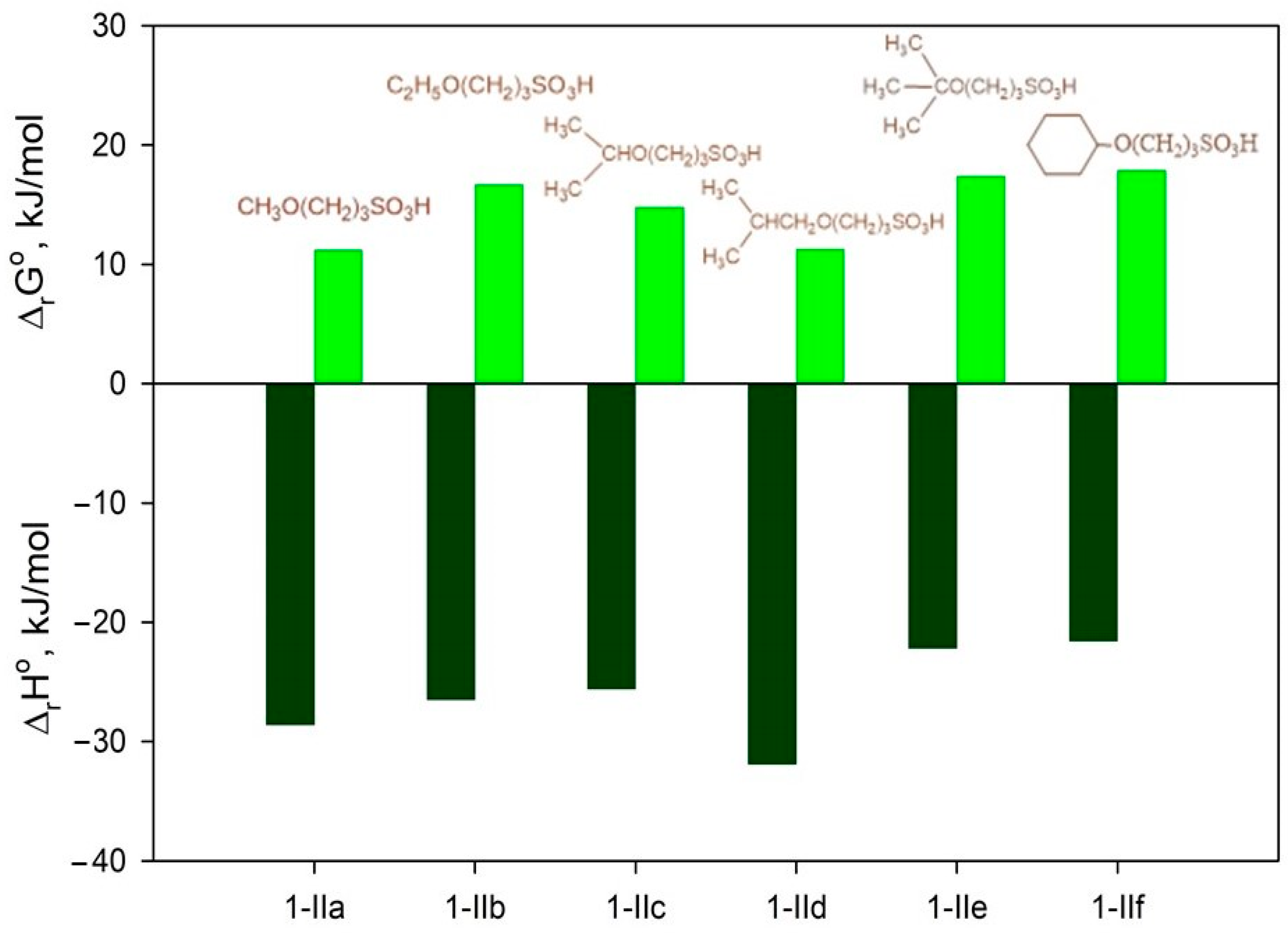
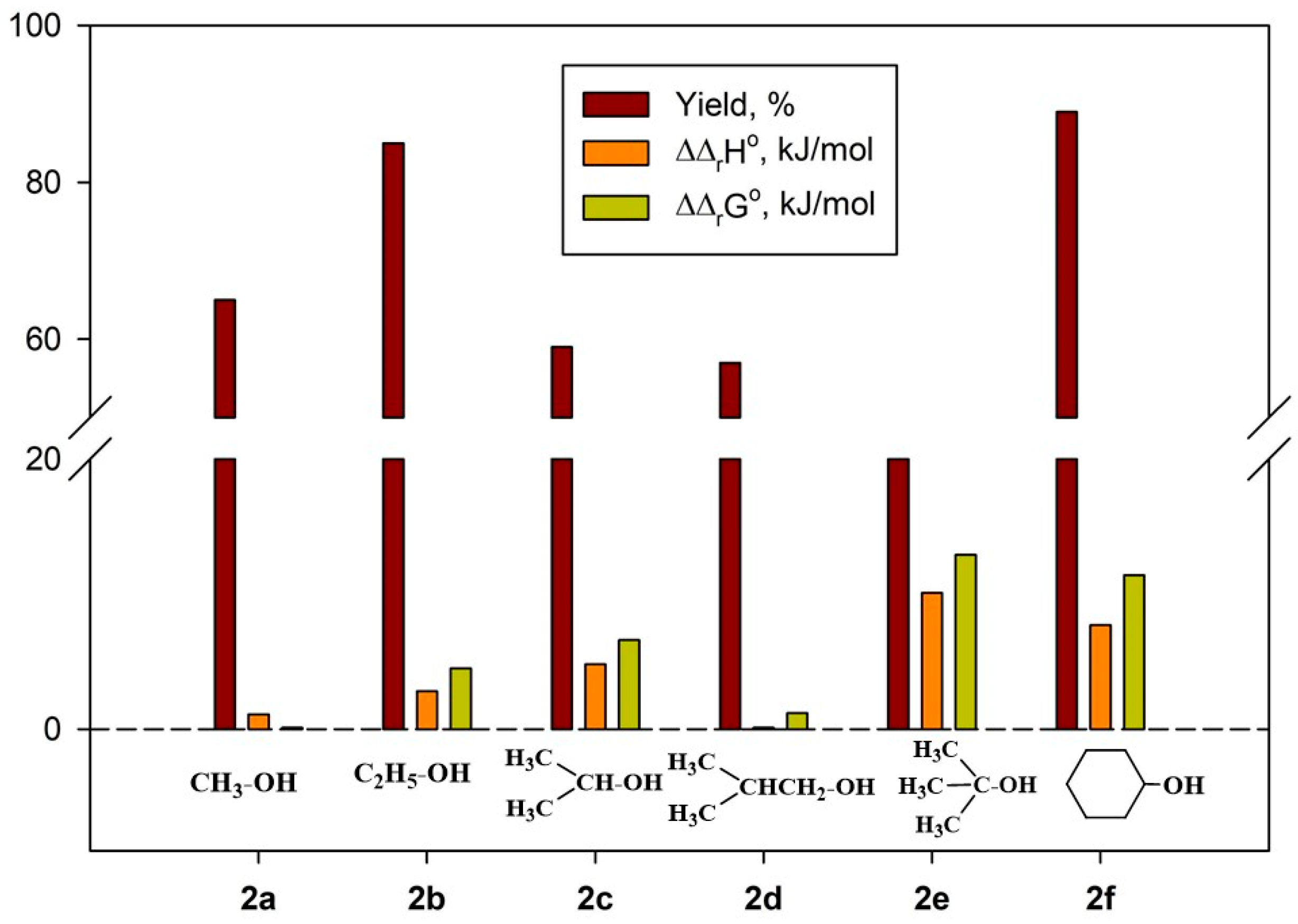
2.4.1. Thermodynamic Parameters of Formation for Compounds 2a–f
2.4.2. Structures of the Cation–Anion Complexes
2.4.3. Thermodynamic Parameters of 2a–g Formation
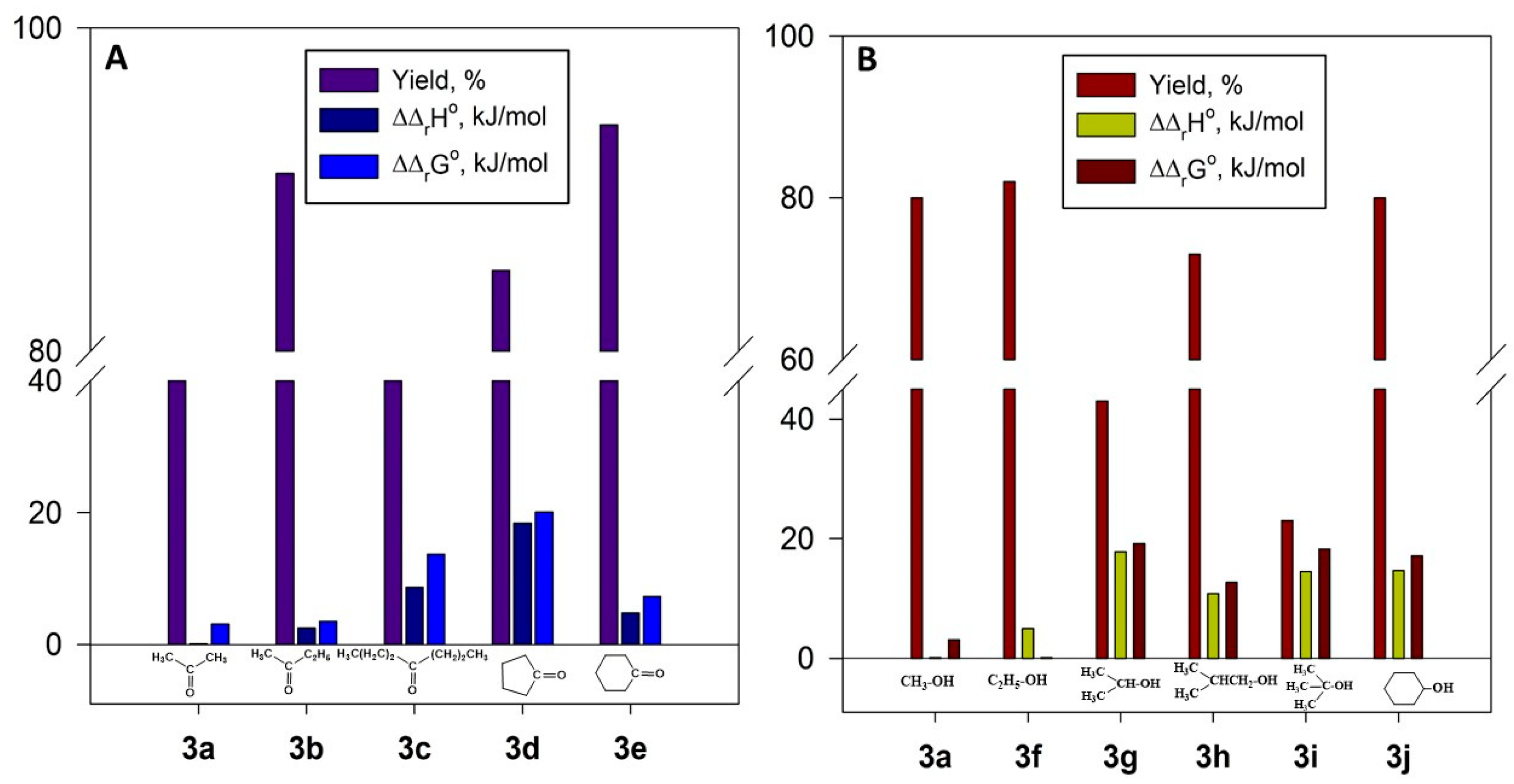
2.4.4. Thermodynamic Parameters of 3a–j Formation
3. Materials and Methods
3.1. Chemistry
Synthesis
- (1)
- A mixture of 0.61 g (0.005 mol) of 1a and 0.73 g (0.006 mol) of 1,3-propanesultone in 5 mL of methanol was refluxed for 3 h, then 4 mL of acetone was added to the hot solution, and the mixture was refluxed for further 1.5 h. On the next day, the volatiles were removed in a vacuum, the residue was stirred with diethyl ether for 2 h, and the crystals formed were filtered and dried to afford 1.27 g (80%) of compound 3a, m.p. 174–177 °C (methanol–acetone, 1:20). IR spectrum (solid, ν/cm−1): 1729 s (C=O), 1637 w (C=Cpyridine), 1208 s, 1160 s, 1033 s (SO3). 1H-NMR (300.1 MHz, D2O, δ, ppm, J/Hz): 1.84 (m, 2H, -H2CCH2CH2-), 1.89 (s, 6H, 2CH3), 2.84 (t, 2H, 3J 7.2, -CH2SO3), 3.25 (s, 3H, 3J = 7.2, CH3O), 3.47 (t, 2H, 3J = 7.2, OCH2-), 8.42 (d, 1H, 3J = 8.1, H3), 8.31 (t, 1H, 3J = 8.1, H4), 8.76 (t, 1H, 3J = 8.1, H5), 9.31 (d, 1H, 3J = 8.1, H6). 13C-NMR (75.5 MHz, D2O, δ, ppm): 24.10, 26.48, 47.65, 57.69, 70.57, 84.15, 123.80, 130.93, 138.48, 141.63, 148.03, 158.99. Anal. calcd. for C13H23.5N2O6.75S (3a ·1.75H2O): C, 44.87; H, 6.80; N, 8.05; S, 9.21. Found: C, 44.89; H, 6.18; N, 8.28; S, 10.53.
- (2)
- A mixture of 0.37 g (0.003 mol) of 1a and 0.48 g (0.004 mol) of 1,3-propanesultone in 5 mL of methanol was refluxed for 3 h, then the volatiles were removed in a vacuum, and the remaining crude salt 2a was dissolved in 10 mL of acetone. On the next day, the volatiles were removed in a vacuum, the residue was stirred with diethyl ether for 2 h, and the crystals formed were filtered and dried to ensure 0.51 g (62%) of unreacted compound 2a was obtained. By evaporation, 0.11 g (11.7%) of compound 3a was isolated, m.p. 170–174 °C (methanol–acetone, 1:20). IR spectrum (solid, ν, cm−1): 1738 s (C=O), 1633 w (C=Cpyridine), 1194 s, 1148 s, 1032 s (SO3).
- (a)
- 0.11 g (0.003 mol) of 3a in 3 mL of water was stirred for 7 days at room temperature. After evaporation, 0.10 g (91%) of 3a was obtained.
- (b)
- 0.36 g (0.011 mmol) of 3a in 4 mL of water was refluxed for 4 h. After evaporation and recrystallisation of the residue from CH3CN, 0.10 g (32%) of 2a was obtained.
- (c)
- 0.31 g (0.0097 mol) of 3a in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.17 g (63%) of 2a was obtained.
- (d)
- 0.11 g (0.003 mol) of 3d in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.08 g of a mixture of 3a and 3d was obtained.
- (e)
- 0.11 g (0.003 mol) of 3e in 4 mL of water was stirred at 70–80 °C for 5 h. After evaporation, 0.08 g (97%) of 3a was obtained.
3.2. Calculation Details
3.3. X-ray Crystallographic Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeBruler, C.; Hu, B.; Moss, J.; Liu, X.; Luo, J.; Sun, Y.; Liu, T.L. Designer Two-Electron Storage Viologen Anolyte Materials for Neutral Aqueous Organic Redox Flow Batteries. Chem 2017, 3, 961–978. [Google Scholar] [CrossRef]
- Ichikawa, T.; Kato, T.; Ohno, H. 3D Continuous Water Nanosheet as a Gyroid Minimal Surface Formed by Bicontinuous Cubic Liquid-Crystalline Zwitterions. J. Am. Chem. Soc. 2012, 134, 11354–11357. [Google Scholar] [CrossRef] [PubMed]
- Fadda, A.A.; El-Mekawy, R.E.-D.; AbdelAal, M.T. Synthesis and antimicrobial evaluation of some new N-pyridinium, quinolinium, and isoquinolinium sulfonate derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1148–1154. [Google Scholar] [CrossRef]
- Frederickson, M.; Vuillard, L.; Abell, C. Novel selenium containing non-detergent sulphobetaines. Tetrahedron Lett. 2003, 44, 7925–7928. [Google Scholar] [CrossRef]
- Bondock, S.; Khalifa, W.; Fadda, A.A. Synthesis and antimicrobial evaluation of some new thiazole, thiazolidinone and thiazoline derivatives starting from 1-chloro-3,4-dihydronaphthalene-2-carboxaldehyde. Eur. J. Med. Chem. 2007, 42, 948–954. [Google Scholar] [CrossRef]
- Mostafa, A.S.; El Bialy, S.A.; Bayoumi, W.A.; Ueda, Y.; Ikeda, M.; Kato, N.; Abdelal, A.M. Synthesis and In Vitro Activity of Pyrrolo [3,4-d]pyrimidine-2,5-diones as Potential Non-nucleoside HCV Inhibitors. Curr. Enz. Inhib. 2016, 12, 170–176. [Google Scholar] [CrossRef]
- Tian, X.; Liu, T.; Fang, B.; Wang, A.; Zhang, M.; Hussain, S.; Luo, L.; Zhang, R.; Zhang, Q.; Wu, J.; et al. NeuN-Specific Fluorescent Probe Revealing Neuronal Nuclei Protein and Nuclear Acids Association in Living Neurons under STED Nanoscopy. ACS Appl. Mater. Interf. 2018, 10, 31959–31964. [Google Scholar] [CrossRef]
- Yan, P.; Acker, C.D.; Loew, L.M. Tethered Bichromophoric Fluorophore Quencher Voltage Sensitive Dyes. ACS Sens. 2018, 3, 2621–2628. [Google Scholar] [CrossRef]
- Willner, I.; Ford, W.E. Preparation of zwitterionic electron acceptors and donors. J. Heteroc. Chem. 1983, 20, 1113–1114. [Google Scholar] [CrossRef]
- Kramarova, E.P.; Borisevich, S.S.; Khamitov, E.M.; Korlyukov, A.A.; Dorovatovskii, P.V.; Shagina, A.D.; Mineev, K.S.; Tarasenko, D.V.; Novikov, R.A.; Lagunin, A.A.; et al. Pyridine Carboxamides Based on Sulfobetaines: Design, Reactivity, and Biological Activity. Molecules 2022, 27, 7542. [Google Scholar] [CrossRef]
- Kramarova, E.P.; Lyakhmun, D.N.; Tarasenko, D.V.; Korlyukov, A.A.; Dorovatovskii, P.V.; Shmigol, T.A.; Bylikin SYu Baukov Yu, I.; Negrebetsky, V.V. An expedient synthesis of a picolinamide-based betain bearing a 3-sulfonatopropyl substituent. Mendeleev Commun. 2024, 34, 126–128. [Google Scholar]
- Koizumi, Y.; Arai, M.; Tomoda, H.; Omura, S. Oxaline, a fungal alkaloid, arrests the cell cycle in M phase by inhibition of tubulin polymerization. Biochim. Biophys. Acta 2004, 1693, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Gottstein, W.J.; Kim, C.U.; Shih, K.M.; McGregor, D.N. Hetacillin (R)- and (S)-sulfoxides. Synthesis and structure-activity relationships. J. Med. Chem. 1978, 21, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Burns, H.D.; Dannals, R.F.; Langström, B.; Ravert, H.T.; Zemyan, S.E.; Duelfer, T.; Wong, D.F.; Frost, J.J.; Kuhar, M.J.; Wagner, H.N. (3-N-[11C]methyl)spiperone, a ligand binding to dopamine receptors: Radiochemical synthesis and biodistribution studies in mice. J. Nucl. Med. 1984, 25, 1222–1227. [Google Scholar]
- Barrow, J.C.; Rittle, K.E.; Ngo, P.L.; Selnick, H.G.; Graham, S.L.; Pitzenberger, S.M.; McGaughey, G.B.; Colussi, D.; Lai, M.T.; Huang, Q.; et al. Design and synthesis of 2,3,5-substituted imidazolidin-4-one inhibitors of BACE-1. Chem. Med. Chem. 2007, 2, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Augelli-Szafran, C.E.; Roth, B.D.; Essenburg, A.; Hamelehle, K.L.; Krause, B.R.; Stanfield, R.L. Imidazolidinones and pyrazolones as novel ACAT inhibitors: Chemistry and biological activity. Bioorg. Med. Chem. Lett. 1994, 4, 1095–1100. [Google Scholar] [CrossRef]
- Rinnová, M.; Nefzi, A.; Houghten, R.A. Opioid activity of 4-imidazolidinone positional analogues of Leu-Enkephalin. Bioorg. Med. Chem. Lett. 2002, 12, 3175–3178. [Google Scholar] [CrossRef]
- Liu, J.; Cui, G.; Zhao, M.; Cui, C.; Ju, J.; Peng, S. Dual-acting agents that possess reversing resistance and anticancer activities: Design, synthesis, MES-SA/Dx5 cell assay, and SAR of Benzyl 1,2,3,5,11,11a-hexahydro-3,3-dimethyl-1-oxo-6H-imidazo[3′,4′:1,2]pyridin[3,4-b]indol-2-substitutedacetates. Bioorg. Med. Chem. 2007, 15, 7773–7788. [Google Scholar] [CrossRef]
- Vale, N.; Matos, J.; Gut, J.; Nogueira, F.; do Rosário, V.; Rosenthal, P.J.; Moreira, R.; Gomes, P. Imidazolidin-4-one peptidomimetic derivatives of primaquine: Synthesis and antimalarial activity. Bioorg. Med. Chem. Lett. 2008, 18, 4150–4153. [Google Scholar] [CrossRef]
- Vale, N.; Prudêncio, M.; Marques, C.A.; Collins, M.S.; Gut, J.; Nogueira, F.; Matos, J.; Rosenthal, P.J.; Cushion, M.T.; do Rosário, V.E.; et al. Imidazoquines as antimalarial and antipneumocystis agents. J. Med. Chem. 2009, 52, 7800–7807. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, M.; Qian, K.; Zhang, X.; Lee, K.H.; Wu, J.; Liu, Y.N.; Peng, S. Benzyl 1,2,3,5,11,11a-hexahydro-3,3-dimethyl-1-oxo-6H-imidazo[3′,4′:1,2]pyridin[3,4-b]indole-2-substituted acetates: One-pot-preparation, anti-tumor activity, docking toward DNA and 3D QSAR analysis. Bioorg. Med. Chem. 2010, 18, 1910–1917. [Google Scholar] [CrossRef] [PubMed]
- Mostardeiro, M.A.; Ilha, V.; Dahmer, J.; Caro, M.S.; Dalcol, I.I.; da Silva, U.F.; Morel, A.F. Cyclopeptide alkaloids: Stereochemistry and synthesis of the precursors of discarines C and D and myrianthine A. J. Nat. Prod. 2013, 76, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Battista, K.; Bignan, G.C.; Brenneman, D.E.; Connolly, P.J.; Liu, J.; Middleton, S.A.; Orsini, M.; Reitz, A.B.; Rosenthal, D.I.; et al. A selective small molecule NOP (ORL-1 receptor) partial agonist for the treatment of anxiety. Bioorg. Med. Chem. Lett. 2015, 25, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.J.; Kozlowski, M.C.; Song, A.; Wang, W. (5S)-2,2,3-Trimethyl-5-(phenylmethyl)-4-imidazolidinone. In Encyclopedia of Reagents for Organic Synthesis (EROS); Wiley: Hoboken, NJ, USA, 2016; pp. 1–5. [Google Scholar]
- Bedos, P.; Amblard, M.; Subra, G.; Dodey, P.; Luccarini, J.M.; Paquet, J.L.; Pruneau, D.; Aumelas, A.; Martinez, J. A rational approach to the design and synthesis of a new bradykinin B(1) receptor antagonist. J. Med. Chem. 2000, 43, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Nogueira, F.; do Rosário, V.E.; Gomes, P.; Moreira, R. Primaquine dipeptide derivatives bearing an imidazolidin-4-one moiety at the N-terminus as potential antimalarial prodrugs. Eur. J. Med. Chem. 2009, 44, 2506–2516. [Google Scholar] [CrossRef] [PubMed]
- Blackmore, T.R.; Thompson, P.E. ChemInform Abstract: Imidazolidin-4-ones: Their Syntheses and Applications. Heterocyclic 2011, 83, 1953–1975. [Google Scholar] [CrossRef]
- Curtius, T. Hydrazide und Azide organischer Säuren I. Abhandlung. J. Für Prakt. Chem. 1894, 50, 275–294. [Google Scholar] [CrossRef]
- Mailyan, A.K.; Eickhoff, J.A.; Minakova, A.S.; Gu, Z.; Lu, P.; Zakarian, A. Cutting-Edge and Time-Honored Strategies for Stereoselective Construction of C–N Bonds in Total Synthesis. Chem. Rev. 2016, 116, 4441–4557. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Sarkar, A. The Curtius Rearrangement: Applications in Modern Drug Discovery and Medicinal Chemistry. Chem. Med. Chem. 2018, 13, 2351–2373. [Google Scholar] [CrossRef]
- Bariwal, J.; Van der Eycken, E. C–N bond forming cross-coupling reactions: An overview. Chem. Soc. Rev. 2013, 42, 9283–9303. [Google Scholar] [CrossRef]
- Gomez, S.; Peters, J.A.; Maschmeyer, T. The Reductive Amination of Aldehydes and Ketones and the Hydrogenation of Nitriles: Mechanistic Aspects and Selectivity Control. Adv. Synth. Catal. 2002, 344, 1037–1057. [Google Scholar] [CrossRef]
- Swamy, K.C.; Kumar, N.N.; Balaraman, E.; Kumar, K.V. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Magni, L.; Örtengren, B.; Sjöberg, B.; Wahlqvist, S. Stability, Absorption and Excretion Studies with Hetacillin. Scand. J. Clin. and Lab. Invest. 1967, 20, 195–201. [Google Scholar] [CrossRef]
- Brown, D.M.; Hannan, D.P.; Langley, P.F. Biotransformation of hetacillin to ampicillin in man. Toxicol. Appl. Pharmacol. 1969, 15, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheesem Information from the website:an, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhurko, G.A. ChemCraft, Version 1.6 (Build 332).
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theor. Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 15410. [Google Scholar] [CrossRef] [PubMed]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using atoms in molecules. Chem. Phys. Lett. 1992, 194, 1–8. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Bruker. APEX3, RLATT, CELL_NOW, TWINABS, SAINT-Plus and SADABS. 2016. [Google Scholar]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sec. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sec. 2015, 71, 3–8. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard JA, K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]

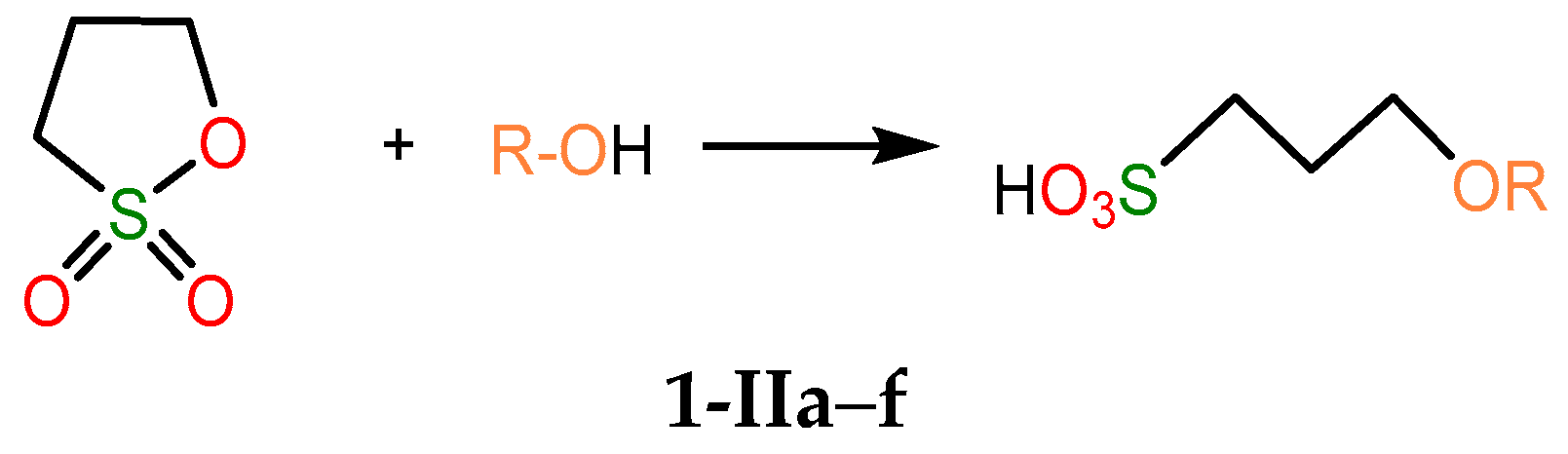


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent ID | Reagent Structure | Product ID | Product Structure | Yield, % |
|---|---|---|---|---|
| 1a |  | 2a | 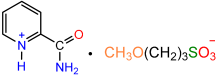 | 65 |
| 1a |  | 2b | 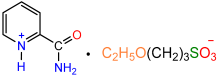 | 85 |
| 1a |  | 2c | 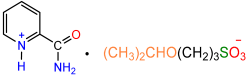 | 59 |
| 1a |  | 2d | 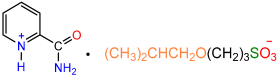 | 57 |
| 1a |  | 2e | 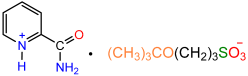 | 25 |
| 1a |  | 2f | 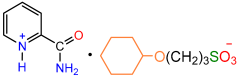 | 89 |
| 1b | 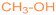 | 2g [11] | 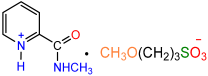 | 81 |
| Reagent ID | Reagent Structure | Product ID | Product Structure | Yield, % |
|---|---|---|---|---|
| 2a | 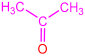 | 3a |  | 80 |
| 2a | 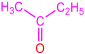 | 3b | 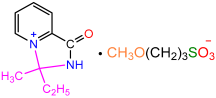 | 91 |
| 2a | 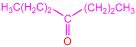 | 3c |  | 75 |
| 2a | 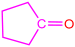 | 3d | 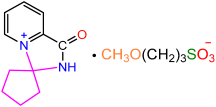 | 85 |
| 2a | 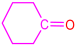 | 3e | 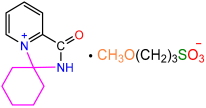 | 94 |
| 2b | 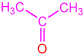 | 3f | 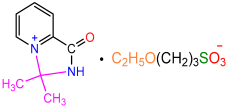 | 82 |
| 2c | 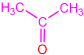 | 3g | 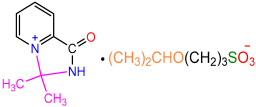 | 43 |
| 2d | 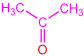 | 3h | 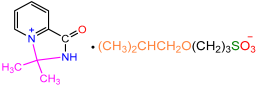 | 73 |
| 2e | 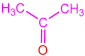 | 3i | 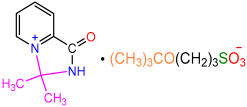 | 23 |
| 2f | 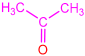 | 3j | 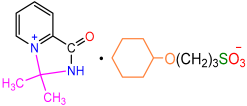 | 80 |
| Datablock | 2a | 3a |
|---|---|---|
| Formula moiety | C4H9O4S, C6H7N2O | 2(C4H9O4S), 2(C9H11N2O) |
| Brutto formula | C10H16N2O5S | C26H40N4O10S2 |
| Formula weight | 276.31 | 632.74 |
| Diffractometer | Bruker QUEST | Bruker QUEST |
| Scan mode | ω and ϕ scans | ω and ϕ scans |
| Anode [Wavelength, Å] | MoKα [0.71073] microfocus sealed X-ray tube | MoKα [0.71073] microfocus sealed X-ray tube |
| Crystal Dimensions, mm | 0.04 × 0.07 × 0.14 | 0.03 × 0.05 × 0.1 |
| Crystal colour | colourless | colourless |
| Crystal system | monoclinic | monoclinic |
| a, Å | 9.9079(5) | 12.422(2) |
| b, Å | 12.6799(6) | 8.7237(15) |
| c, Å | 19.8194(10) | 14.699(2) |
| α, ° | 90 | 90 |
| β, ° | 92.227(3) | 109.106(6) |
| γ, ° | 90 | 90 |
| Volume, Å3 | 2488.1(2) | 1505.1(4) |
| Density, g cm−3 | 1.475 | 1.396 |
| Temperature, K | 100 | 100 |
| Tmin/Tmax | 0.497553/0.746069 | 0.5954/0.7461 |
| μ, mm−¹ | 0.276 | 0.238 |
| Space group | P1211 | P121/n1 |
| Z | 8 | 2 |
| F(000) | 1168 | 672 |
| Reflections collected | 29400 | 13353 |
| Independent reflections | 29400 | 3544 |
| Reflections (I > 2σ(I)) | 27603 | 3030 |
| Parameters | 654 | 193 |
| Rint | 0.00 | 0.0598 |
| 2θmin − 2θmax, ° | 3.814 − 55.750 | 5.226 − 55.752 |
| wR2 (all reflections) | 0.1531 | 0.2011 |
| R1(I > σ(I)) | 0.0572 | 0.0730 |
| GOF | 1.055 | 0.972 |
| ρmin/ρmax, e Å−3 | −0.748/1.719 | −0.445/0.676 |
| Restraints | 1 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kramarova, E.P.; Lyakhmun, D.N.; Tarasenko, D.V.; Borisevich, S.S.; Khamitov, E.M.; Yusupova, A.R.; Korlyukov, A.A.; Romanenko, A.R.; Shmigol, T.A.; Bylikin, S.Y.; et al. Reaction of Picolinamides with Ketones Producing a New Type of Heterocyclic Salts with an Imidazolidin-4-One Ring. Molecules 2024, 29, 206. https://doi.org/10.3390/molecules29010206
Kramarova EP, Lyakhmun DN, Tarasenko DV, Borisevich SS, Khamitov EM, Yusupova AR, Korlyukov AA, Romanenko AR, Shmigol TA, Bylikin SY, et al. Reaction of Picolinamides with Ketones Producing a New Type of Heterocyclic Salts with an Imidazolidin-4-One Ring. Molecules. 2024; 29(1):206. https://doi.org/10.3390/molecules29010206
Chicago/Turabian StyleKramarova, Eugenia P., Dmitry N. Lyakhmun, Dmitry V. Tarasenko, Sophia S. Borisevich, Edward M. Khamitov, Alfia R. Yusupova, Alexander A. Korlyukov, Alexander R. Romanenko, Tatiana A. Shmigol, Sergey Yu. Bylikin, and et al. 2024. "Reaction of Picolinamides with Ketones Producing a New Type of Heterocyclic Salts with an Imidazolidin-4-One Ring" Molecules 29, no. 1: 206. https://doi.org/10.3390/molecules29010206
APA StyleKramarova, E. P., Lyakhmun, D. N., Tarasenko, D. V., Borisevich, S. S., Khamitov, E. M., Yusupova, A. R., Korlyukov, A. A., Romanenko, A. R., Shmigol, T. A., Bylikin, S. Y., Baukov, Y. I., & Negrebetsky, V. V. (2024). Reaction of Picolinamides with Ketones Producing a New Type of Heterocyclic Salts with an Imidazolidin-4-One Ring. Molecules, 29(1), 206. https://doi.org/10.3390/molecules29010206