
Preparations of trans- and cis-μ-1,2-Peroxodiiron(III) Complexes

 , ,
, , 
Abstract
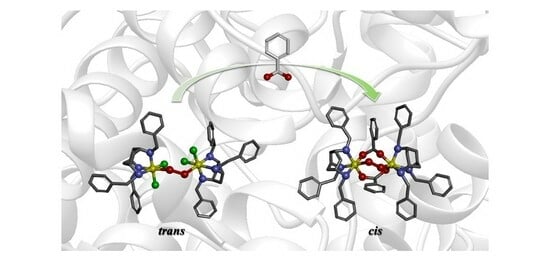
:
1. Introduction
2. Results and Discussion
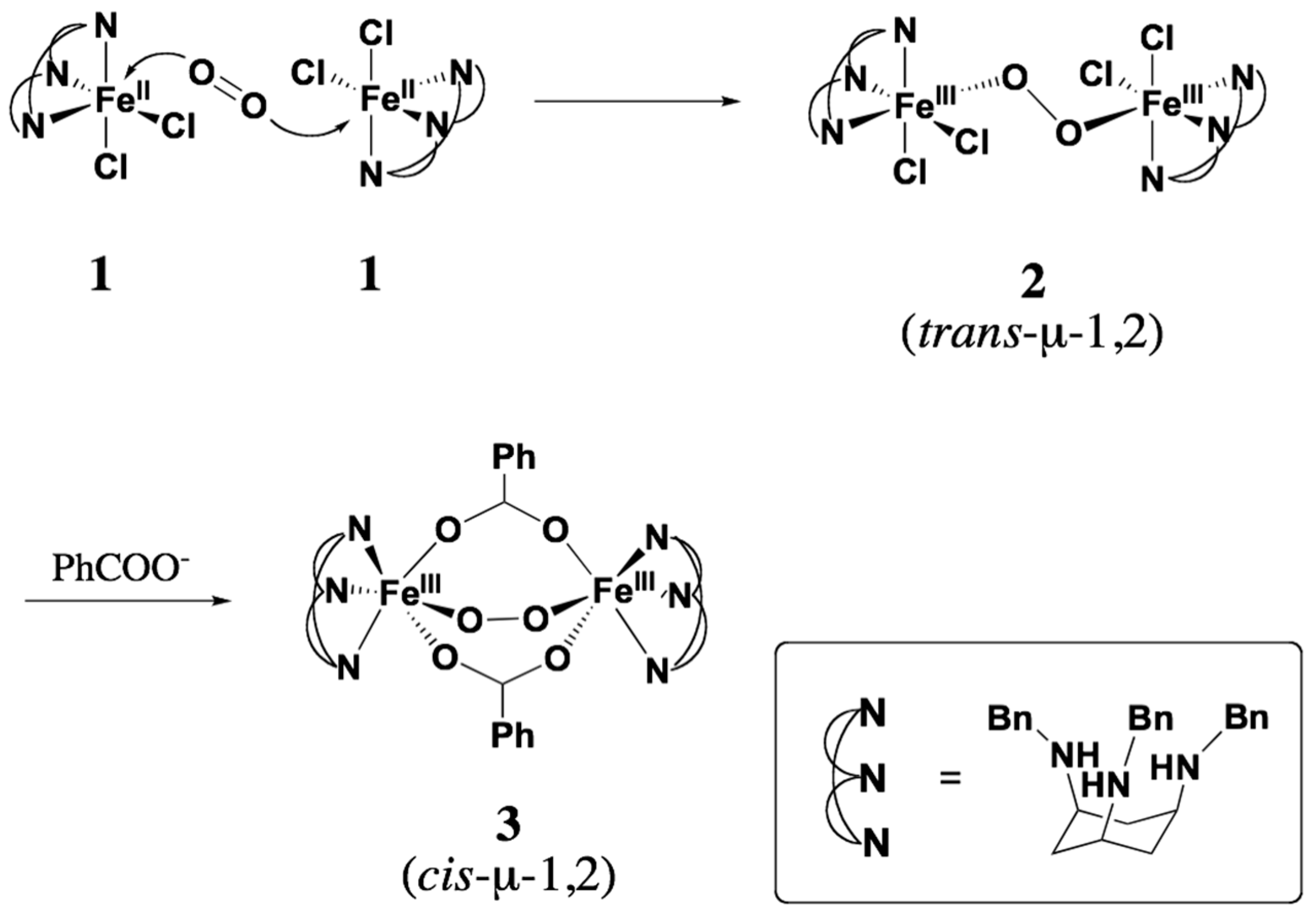
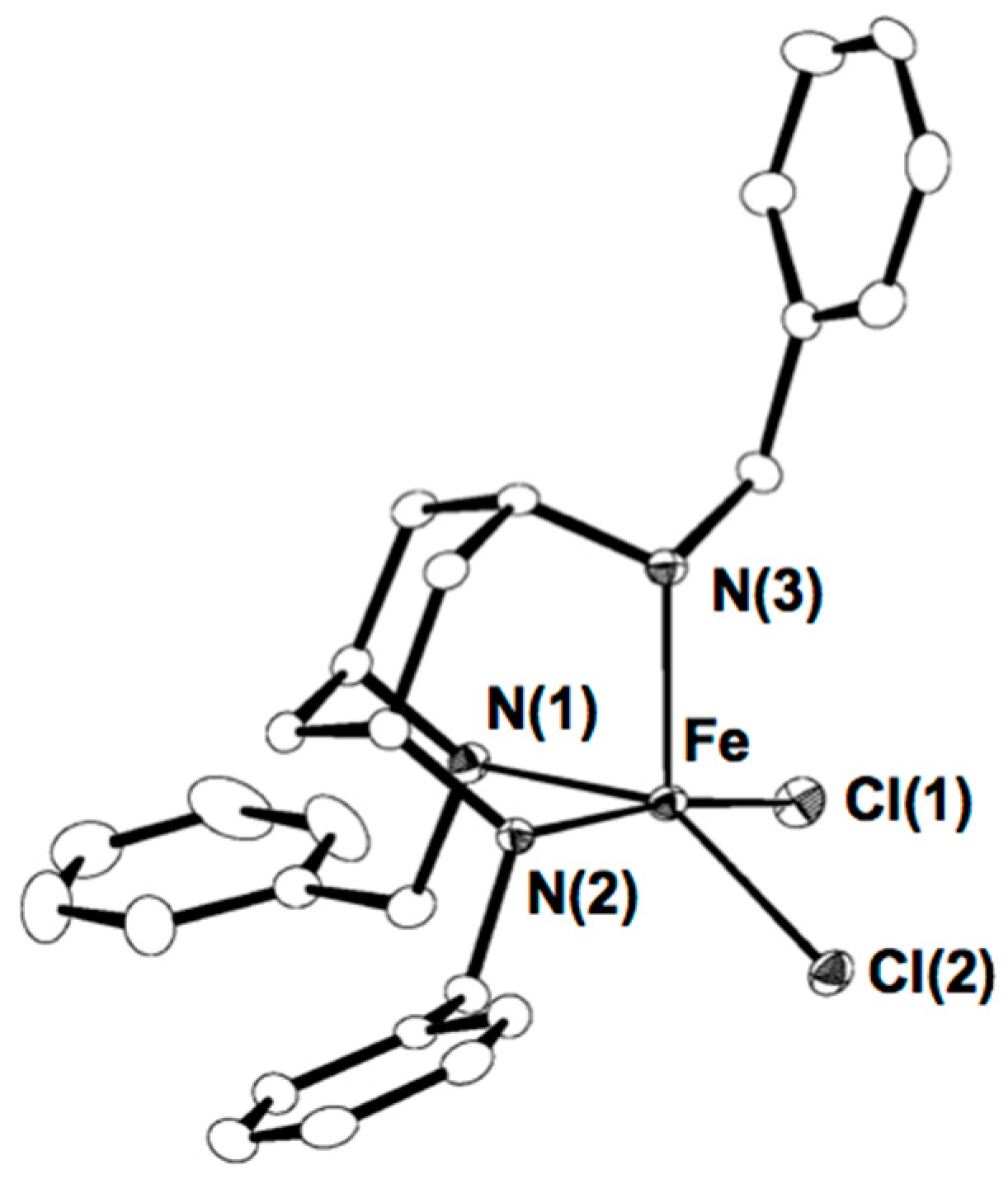
2.1. Synthesis of [FeII(Bn3CY)Cl2] (1)
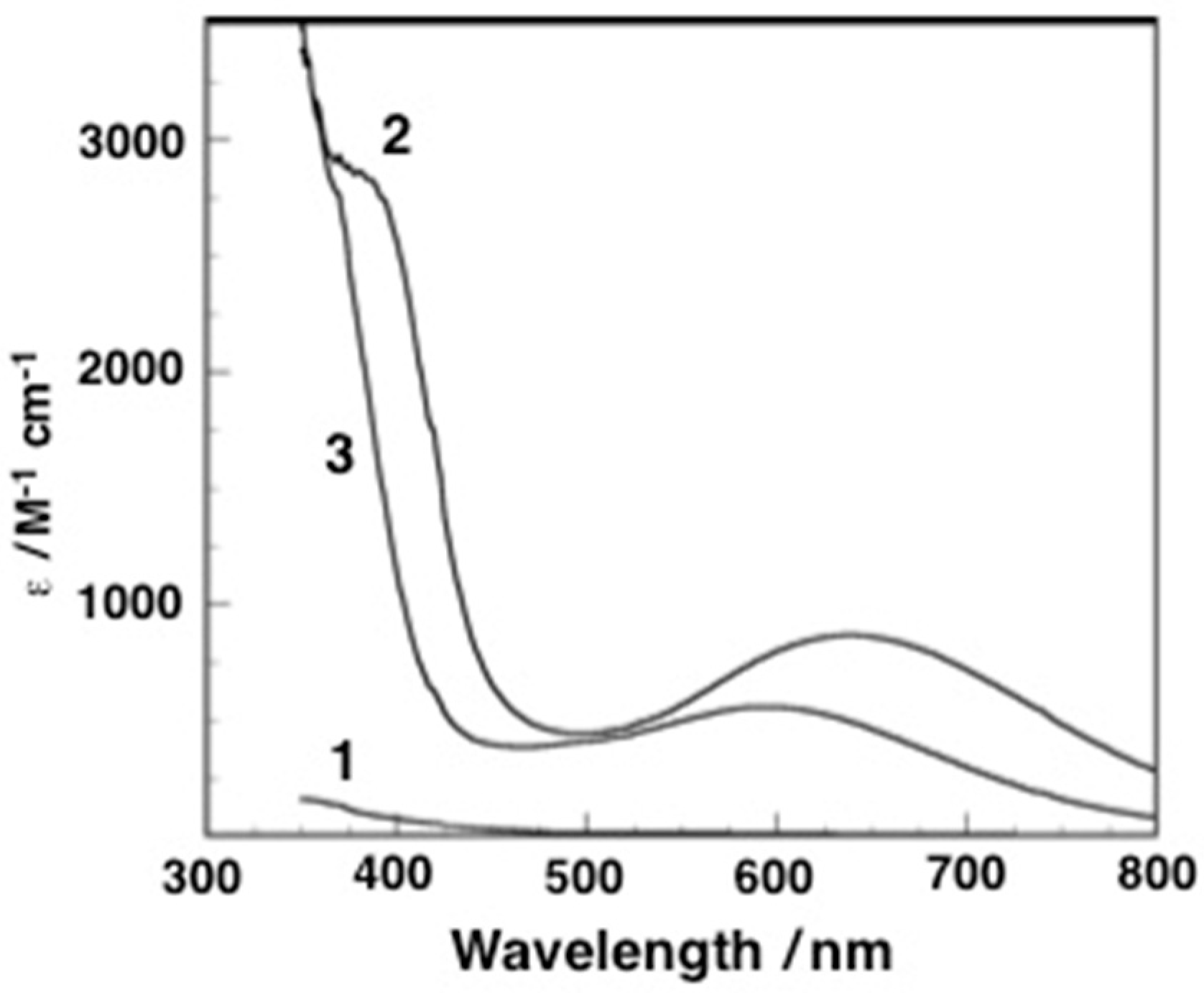
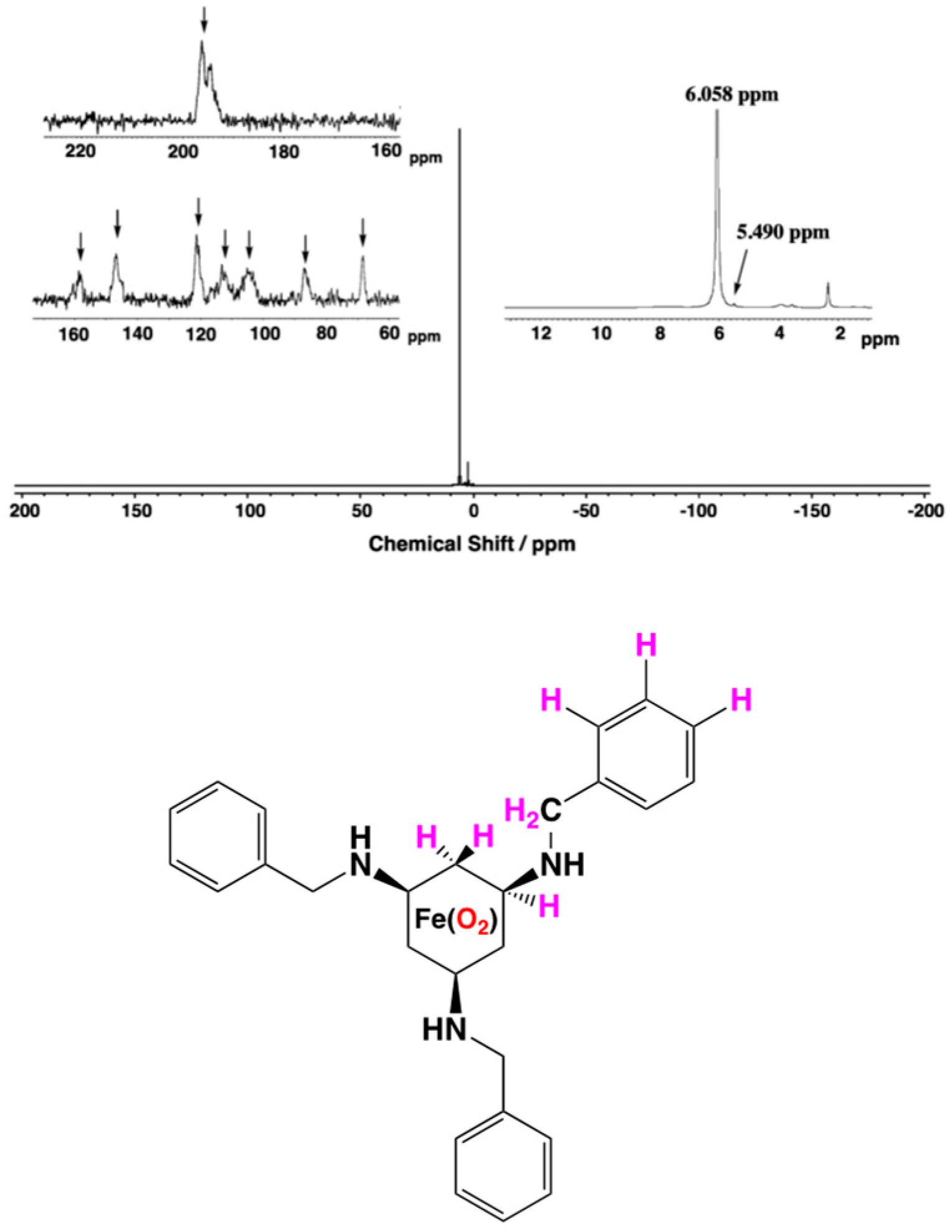
2.2. Formation of the Peroxodiiron Complex 2
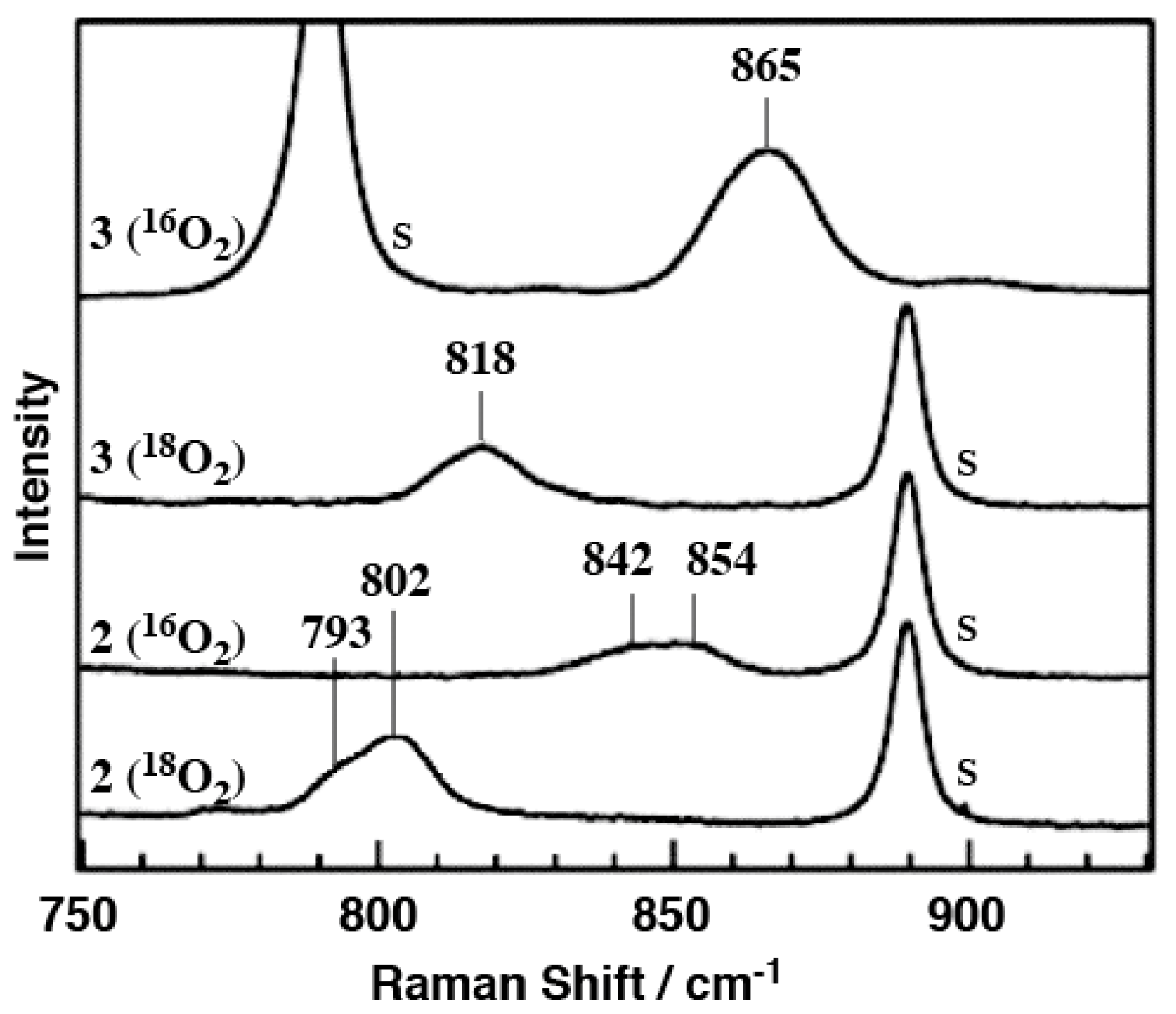
2.3. Resonance Raman Spectroscopy
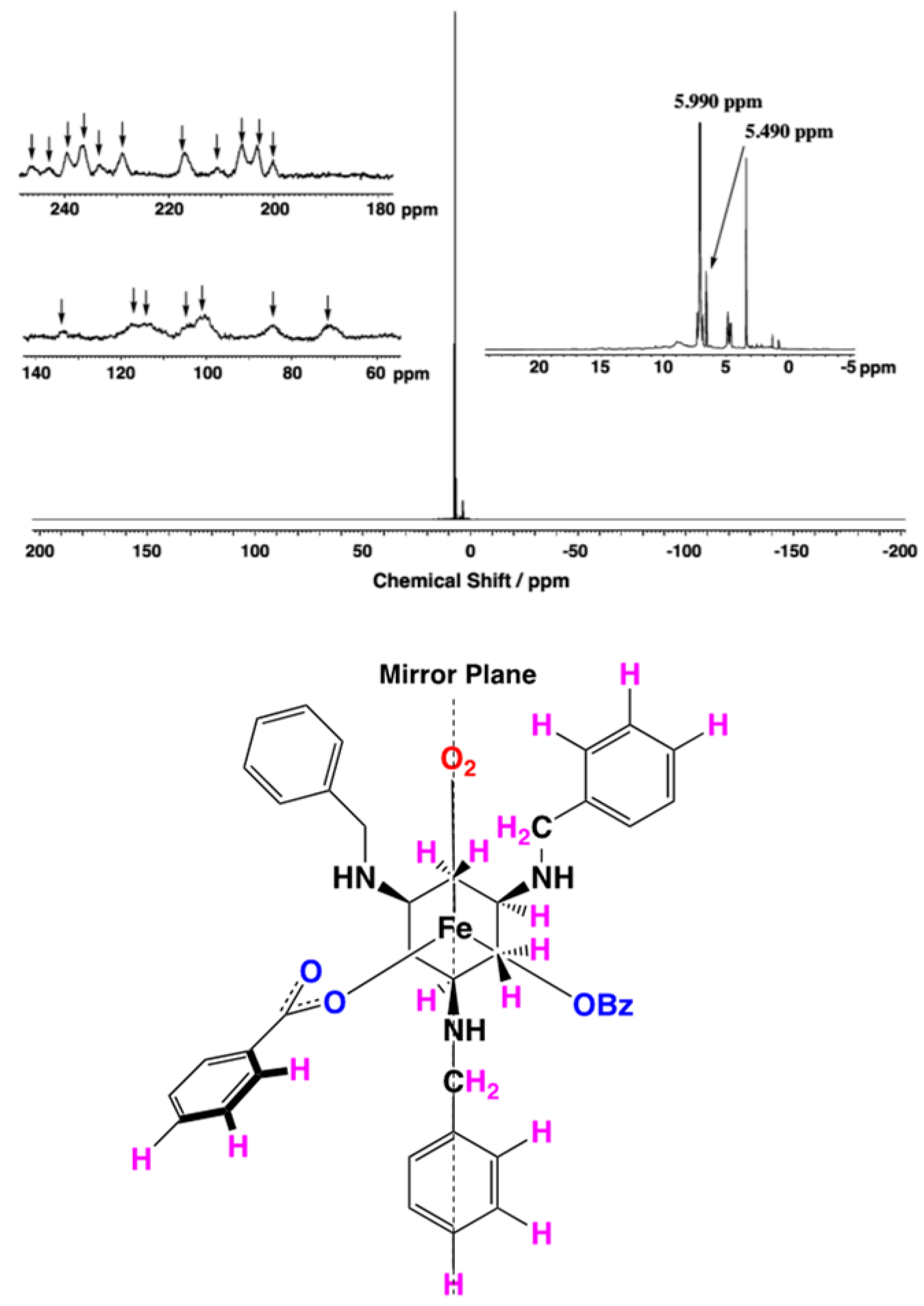
2.4. Conversion of 2 to 3
2.5. 1H-NMR Studies
2.6. DFT Calculations
3. Experimental Section
3.1. Experimental Procedure
3.2. Materials
3.3. Preparation of [FeIICl2(Bn3CY)] (1)
3.4. Preparations of trans- (2) and cis-μ-1,2-Peroxo Diiron(III) Complexes (3)
3.5. Instrumentation
3.6. X-ray Crystallography
3.7. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallar, B.J.; Lipscomb, J.D. Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev. 1996, 96, 2625–2658. [Google Scholar] [CrossRef] [PubMed]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.-K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.-S.; Zhou, J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–350. [Google Scholar] [CrossRef] [PubMed]
- Tshuva, E.Y.; Lippard, S.J. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev. 2004, 104, 987–1012. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.C.-C.; Maji, S.; Chen, P.P.-Y.; Lee, H.K.; Yu, S.S.-F.; Chan, S.I. Alkane Oxidation: Methane Monooxygenases, Related Enzymes, and Their Biomimetics. Chem. Rev. 2017, 117, 8574–8621. [Google Scholar] [CrossRef] [PubMed]
- Que, L., Jr.; Dong, Y. Modeling the Oxygen Activation Chemistry of Methane Monooxygenase and Ribonucleotide Reductase. Acc. Chem. Res. 1996, 29, 190–196. [Google Scholar] [CrossRef]
- Tinberg, C.E.; Lippard, S.J. Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res. 2011, 44, 280–288. [Google Scholar] [CrossRef]
- Wang, W.; Liang, A.D.; Lippard, S.J. Coupling Oxygen Consumption with Hydrocarbon Oxidation in Bacterial Multicomponent Monooxygenases. Acc. Chem. Res. 2015, 48, 2632–2639. [Google Scholar] [CrossRef]
- Whittington, D.A.; Lippard, S.J. Crystal Structures of the Soluble Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath) Demonstrating Geometrical Variability at the Dinuclear Iron Active Site. J. Am. Chem. Soc. 2001, 123, 827–838. [Google Scholar] [CrossRef]
- Kodera, M.; Tsuji, T.; Yasunaga, T.; Kawahara, Y.; Hirano, T.; Hitomi, Y.; Nomura, T.; Ogura, T.; Kobayashi, Y.; Sajith, P.K.; et al. Roles of carboxylate donors in O–O bond scission of peroxodi-iron(iii) to high-spin oxodi-iron(iv) with a new carboxylate-containing dinucleating ligand. Chem. Sci. 2014, 5, 2282–2292. [Google Scholar] [CrossRef]
- Baik, M.H.; Newcomb, M.; Friesner, R.A.; Lippard, S.J. Mechanistic Studies on the Hydroxylation of Methanine by Methane Monooxygenase. Chem. Rev. 2003, 103, 2385–2419. [Google Scholar] [CrossRef]
- Jasniewski, A.J.; Que, L., Jr. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Menage, S.; Brennan, B.A.; Elgren, T.E.; Jang, H.G.; Pearce, L.L.; Que, L., Jr. Dioxygen binding to diferrous centers. Models for diiron-oxo proteins. J. Am. Chem. Soc. 1993, 115, 1851–1859. [Google Scholar] [CrossRef]
- Kim, K.; Lippard, S.J. Structure and Mössbauer Spectrum of a (μ-1,2-Peroxo)bis(μ-carboxylato)diiron(III) Model for the Peroxo Intermediate in the Methane Monooxygenase Hydroxylase Reaction Cycle. J. Am. Chem. Soc. 1996, 118, 4914–4915. [Google Scholar] [CrossRef]
- Ookubo, T.; Sugimoto, H.; Nagayama, T.; Masuda, H.; Sato, T.; Tanaka, K.; Maeda, Y.; Okawa, H.; Hayashi, Y.; Uehara, A.; et al. cis-μ-1,2-Peroxo Diiron Complex: Structure and Reversible Oxygenation. J. Am. Chem. Soc. 1996, 118, 701–702. [Google Scholar] [CrossRef]
- Dong, Y.; Yan, S.; Young, V.G., Jr.; Que, L., Jr. Crystal Structure Analysis of a Synthetic Non-Heme Diiron-O2 Adduct: Insight into the Mechanism of Oxygen Activation. Angew. Chem. Int. Ed. 1996, 35, 618–620. [Google Scholar] [CrossRef]
- Hagadorn, J.R.; Que, L., Jr.; Tolman, W.B. A Bulky Benzoate Ligand for Modeling the Carboxylate-Rich Active Sites of Non-Heme Diiron Enzymes. J. Am. Chem. Soc. 1998, 120, 13531–13532. [Google Scholar] [CrossRef] [PubMed]
- LeCloux, D.D.; Barrios, A.M.; Mizoguchi, T.J.; Lippard, S.J. Modeling the Diiron Centers of Non-Heme Iron Enzymes. Preparation of Sterically Hindered Diiron(II) Tetracarboxylate Complexes and Their Reactions with Dioxygen. J. Am. Chem. Soc. 1998, 120, 9001–9014. [Google Scholar] [CrossRef]
- Arii, H.; Nagatomo, S.; Miwa, T.K.T.; Jitsukawa, K.; Einaga, H.; Masuda, H. A novel diiron complex as a functional model for hemerythrin. J. Inorg. Biochem. 2000, 82, 153–162. [Google Scholar] [CrossRef]
- Kryatov, S.V.; Rybak-Akimova, E.V.; MacMurdo, V.L.; Que, L., Jr. A Mechanistic Study of the Reaction between a Diiron(II) Complex [FeII2(μ-OH)2(6-Me3-TPA)2]2+ and O2 to Form a Diiron(III) Peroxo Complex. Inorg. Chem. 2001, 40, 2220–2228. [Google Scholar] [CrossRef]
- Tshuva, E.Y.; Lee, D.; Bu, W.; Lippard, S.J. Catalytic Oxidation by a Carboxylate-Bridged Non-Heme Diiron Complex. J. Am. Chem. Soc. 2002, 124, 2416–2417. [Google Scholar] [CrossRef]
- Enomoto, M.; Aida, T. Iron Complexes Capped with Dendrimer-Appended Triazacyclononanes as the Novel Spatially Encumbered Models of Non-Heme Iron Proteins. J. Am. Chem. Soc. 2002, 124, 6099–6108. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Pierce, B.; Krebs, C.; Hendrich, M.P.; Huynh, B.H.; Lippard, S.J. Functional Mimic of Dioxygen-Activating Centers in Non-Heme Diiron Enzymes: Mechanistic Implications of Paramagnetic Intermediates in the Reactions between Diiron(II) Complexes and Dioxygen. J. Am. Chem. Soc. 2002, 124, 3993–4007. [Google Scholar] [CrossRef] [PubMed]
- Chavez, F.A.; Ho, R.Y.N.; Pink, M.; Young, V.G., Jr.; Kryatov, S.V.; Rybak-Akimova, E.V.; Andres, H.; Münck, E.; Que, L., Jr.; Tolman, W.B. Unusual Peroxo Intermediates in the Reaction of Dioxygen with Carboxylate-Bridged Diiron(II,II) Paddlewheel Complexes. Angew. Chem. Int. Ed. 2002, 41, 149–152. [Google Scholar] [CrossRef]
- Costas, M.; Cady, C.W.; Kryatov, S.V.; Ray, M.; Ryan, M.J.; Rybak-Akimova, E.V.; Que, L., Jr. Role of Carboxylate Bridges in Modulating Nonheme Diiron(II)/O2 Reactivity. Inorg. Chem. 2003, 42, 7519–7530. [Google Scholar] [CrossRef]
- Zhang, X.; Furutachi, H.; Fujinami, S.; Nagatomo, S.; Maeda, Y.; Watanabe, Y.; Kitagawa, T.; Suzuki, M. Structural and Spectroscopic Characterization of (μ-Hydroxo or μ-Oxo)(μ-peroxo)diiron(III) Complexes: Models for Peroxo Intermediates of Non-Heme Diiron Proteins. J. Am. Chem. Soc. 2005, 127, 826–827. [Google Scholar] [CrossRef] [PubMed]
- Frisch, J.R.; Vu, V.V.; Martinho, M.; Münck, E.; Que, L., Jr. Characterization of Two Distinct Adducts in the Reaction of a Nonheme Diiron(II) Complex with O2. Inorg. Chem. 2009, 48, 8325–8336. [Google Scholar] [CrossRef] [PubMed]
- Cranswick, M.A.; Meier, K.K.; Shan, X.; Stubna, A.; Kaizer, J.; Mehn, M.P.; Münck, E.; Que, L., Jr. Protonation of a Peroxodiiron(III) Complex and Conversion to a Diiron(III/IV) Intermediate: Implications for Proton-Assisted O–O Bond Cleavage in Nonheme Diiron Enzymes. Inorg. Chem. 2012, 51, 10417–10426. [Google Scholar] [CrossRef]
- Frisch, J.R.; McDonnell, R.; Rybak-Akimova, E.V.; Que, L., Jr. Factors Affecting the Carboxylate Shift Upon Formation of Nonheme Diiron-O2 Adducts. Inorg. Chem. 2013, 52, 2627–2636. [Google Scholar] [CrossRef]
- Gordon, J.B.; Vilbert, A.C.; MacMillan, I.M.D.N.; Lancaster, K.M.; Moënne-Loccoz, P.; Goldberg, D.P. Activation of Dioxygen by a Mononuclear Nonheme Iron Complex: Sequential Peroxo, Oxo, and Hydroxo Intermediates. J. Am. Chem. Soc. 2019, 141, 17533–17547. [Google Scholar] [CrossRef]
- Gordon, J.B.; Albert, T.; Dey, A.; Sabuncu, S.; Siegler, M.A.; Bill, E.; Moënne-Loccoz, P.; Goldberg, D.P. A Reactive, Photogenerated High-Spin (S = 2) FeIV(O) Complex via O2 Activation. J. Am. Chem. Soc. 2021, 143, 21637–21647. [Google Scholar] [CrossRef]
- Messerschmidt, A.; Huber, R.; Poulos, T.; Wieghardt, K. Handbook of Metalloproteins; Messerschmidt, A., Huber, R., Poulos, T., Wieghardt, K., Eds.; John Wiley & Sons: West Sussex, UK, 2001; Volume 1. [Google Scholar]
- Walleck, S.; Zimmermann, T.P.; Hachmeister, H.; Pilger, C.; Huser, T.; Katz, S.; Hildebrandt, P.; Stammler, A.; Bögge, H.; Bill, E.; et al. Generation of a μ-1,2-hydroperoxo FeIIIFeIII and a μ-1,2-peroxo FeIVFeIII Complex. Nature Commun. 2022, 13, 1376. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lippard, S.J. Modeling Dioxygen-Activating Centers in Non-Heme Diiron Enzymes: Carboxylate Shifts in Diiron(II) Complexes Supported by Sterically Hindered Carboxylate Ligands. Inorg. Chem. 2002, 41, 2704–2719. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; DuBois, J.L.; Pierce, B.; Hedman, B.; Hodgson, K.O.; Hendrich, M.P.; Lippard, S.J. Structural and Spectroscopic Studies of Valence-Delocalized Diiron(II,III) Complexes Supported by Carboxylate-Only Bridging Ligands. Inorg. Chem. 2002, 41, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Sekino, M.; Furutachi, H.; Tojo, R.; Hishi, A.; Kajikawa, H.; Suzuki, T.; Suzuki, K.; Fujinami, S.; Akine, S.; Sakata, Y.; et al. New mechanistic insights into intramolecular aromatic ligand hydroxylation and benzyl alcohol oxidation initiated by the well-defined (μ-peroxo)diiron(III) complex. Chem. Commun. 2017, 53, 8838–8841. [Google Scholar] [CrossRef] [PubMed]
- Çelenligil-Çetin, R.; Staples, R.J.; Stavropoulos, P. Synthesis, Characterization, and Reactivity of Ferrous and Ferric Oxo/Peroxo Pivalate Complexes in Relation to Gif-Type Oxygenation of Substrates. Inorg. Chem. 2000, 39, 5838–5846. [Google Scholar] [CrossRef] [PubMed]
- Balland, V.; Banse, F.; Anxolabéhère-Mallart, E.; Nierlich, M.; Girerd, J.-J. Iron Complexes Containing the Ligand N,N′-Bis(6-methyl-2-pyridylmethyl)-N,N′-bis(2-pyridylmethyl)ethane-1,2-diamine: Structural, Spectroscopic, and Electrochemical Studies, Reactivity with Hydrogen Peroxide and the Formation of a Low-Spin Fe−OOH Complex. Eur. J. Inorg. Chem. 2003, 2003, 2529–2535. [Google Scholar] [CrossRef]
- Mialane, P.; Nivorojkine, A.; Pratviel, G.; Azéma, L.; Slany, M.; Godde, F.; Simaan, A.; Banse, F.; Karger-Grsel, T.; Bouchoux, G.; et al. Structures of Fe(II) Complexes with N,N,N’-Tris(2-pyridylmethyl)ethane-1,2-diamine Type Ligands. Bleomycin-like DNA Cleavage and Enhancement by an Alkylammonium Substituent on the N’ Atom of the Ligand. Inorg. Chem. 1999, 38, 1085–1092. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; Rijn, J.V.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Kojima, T.; Leising, R.A.; Yan, S.; Que, L., Jr. Alkane functionalization at nonheme iron centers. Stoichiometric transfer of metal-bound ligands to alkane. J. Am. Chem. Soc. 1993, 115, 11328–11335. [Google Scholar] [CrossRef]
- Itoh, S.; Nakao, H.; Berreau, L.M.; Kondo, T.; Komatsu, M.; Fukuzumi, S. Mechanistic Studies of Aliphatic Ligand Hydroxylation of a Copper Complex by Dioxygen: A Model Reaction for Copper Monooxygenases. J. Am. Chem. Soc. 1998, 120, 2890–2899. [Google Scholar] [CrossRef]
- Tyeklár, Z.; Jacobson, R.R.; Wei, N.; Murthy, N.N.; Zubieta, J.; Karlin, K.D. Reversible reaction of dioxygen (and carbon monoxide) with a copper(I) complex. X-ray structures of relevant mononuclear Cu(I) precursor adducts and the trans-(μ-1,2-peroxo)dicopper(II) product. J. Am. Chem. Soc. 1993, 115, 2677–2689. [Google Scholar] [CrossRef]
- Komiyama, K.; Furutachi, H.; Nagatomo, S.; Hashimoto, A.; Hayashi, H.; Fujinami, S.; Suzuki, M.; Kitagawa, T. Dioxygen Reactivity of Copper(I) Complexes with Tetradentate Tripodal Ligands Having Aliphatic Nitrogen Donors: Synthesis, Structures, and Properties of Peroxo and Superoxo Complexes. Bull. Chem. Soc. Jpn. 2004, 77, 59–72. [Google Scholar] [CrossRef]
- Jacobson, R.R.; Tyeklar, Z.; Farooq, A.; Karlin, K.D.; Liu, S.; Zubieta, J. A copper-oxygen (Cu2-O2) complex. Crystal structure and characterization of a reversible dioxygen binding system. J. Am. Chem. Soc. 1998, 110, 3690–3692. [Google Scholar] [CrossRef]
- Evans, D.F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003–2005. [Google Scholar] [CrossRef]
- Lide, L.D. (Ed.) CRC Handbook, 90th ed.; CRC Press: Boca Raton, FL, USA, 2010; Chapter 9–24. [Google Scholar]
- Kajita, Y.; Arii, H.; Saito, T.; Saito, Y.; Nagatomo, S.; Kitagawa, T.; Funahashi, Y.; Ozawa, T.; Masuda, H. Syntheses, Characterization, and Dioxygen Reactivities of Cu(I) Complexes with cis,cis-1,3,5-Triaminocyclohexane Derivatives: A Cu(III)2O2 Intermediate Exhibiting Higher C−H Activation. Inorg. Chem. 2007, 46, 3322–3335. [Google Scholar] [CrossRef]
- Live, D.H.; Chan, S.I. Bulk susceptibility corrections in nuclear magnetic resonance experiments using superconducting solenoids. Anal. Chem. 1970, 42, 791–792. [Google Scholar] [CrossRef]
- Ostfeld, D.; Cohen, I.A. A cautionary note on the use of the Evans method for magnetic moments. J. Chem. Educ. 1972, 49, 829. [Google Scholar] [CrossRef]
- Schubert, E.M. Utilizing the Evans method with a superconducting NMR spectrometer in the undergraduate laboratory. J. Chem. Educ. 1992, 69, 62. [Google Scholar] [CrossRef]
- Cromer, D.T.; Waber, J.T. International Tables for X-ray Crystallography; Kynoch Press: Birmingham, UK, 1974; Volume 4. [Google Scholar]
- Ibers, J.A.; Hamilton, W.C. Dispersion corrections and crystal structure refinements. Acta Crystallogr. 1964, 17, 781. [Google Scholar] [CrossRef]
- Creagh, D.C.; McAuley, W.J. International Tables for X-ray Crystallography; Table 4.2.6.8.; Kluwer: Boston, MA, USA, 1992; Volume C, pp. 219–222. [Google Scholar]
- Creagh, D.C.; Hubbell, J.H. International Tables for Crystallography; Table 4.2.4.3.; Kluwer: Boston, MA, USA, 1992; Volume C, pp. 200–206. [Google Scholar]
- CrystalStructure 3.7.0: Crystal Structure Analysis Package; Rigaku and Rigaku/MSC (2000–2005): The Woodlands, TX, USA, 2018.
- Prout, C.K.; Carruthers, J.R.; Betteridge, P.W.; Cooper, R.I. CRYSTALS Issue 10; Chemical Crystallography Laboratory: Oxford, UK, 1996. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian Basis Set for Molecular Wavefunctions Containing Third-Row Atoms. J. Chem. Phys. 1970, 52, 1033–1036. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W. Highly correlated systems. Excitation energies of first row transition metals Sc–Cu. J. Chem. Phys. 1989, 91, 1062–1065. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; People, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; People, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Scott, A.P.; Radom, L. Harmonic Vibrational Frequencies: An Evaluation of Hartree−Fock, Møller−Plesset, Quadratic Configuration Interaction, Density Functional Theory, and Semiempirical Scale Factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | 2 | 3 | ||
|---|---|---|---|---|
| S | 0 | 5 | 0 | 5 |
| S (calculated) | 1.78 | 5.00 | 1.77 | 5.00 |
| Energy (kcal/mol) a | 0.0 | 1.3 | 0.0 | 1.7 |
| O–O (Å) | 1.383 | 1.389 | 1.378 | 1.407 |
| Fe–Fe (Å) | 4.646 | 4.668 | 3.792 | 3.734 |
| Fe–O–O–Fe (deg.) | 180.0 | 180.0 | –65.3 | –72.1 |
| ν(O–O) (cm−1) | 938, 940 | 917, 926 | 908, 920 | 894 |
| Raman activ. b | 2.2, 0.9 (×104) | 1.1, 2.1 (×104) | 5.2, 1.8 (×103) | 1.6 × 103 |
| Experiment (cm−1) | 842, 854 | 865 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kajita, Y.; Kubo, M.; Arii, H.; Ishikawa, S.; Saito, Y.; Wasada-Tsutsui, Y.; Funahashi, Y.; Ozawa, T.; Masuda, H. Preparations of trans- and cis-μ-1,2-Peroxodiiron(III) Complexes. Molecules 2024, 29, 205. https://doi.org/10.3390/molecules29010205
Kajita Y, Kubo M, Arii H, Ishikawa S, Saito Y, Wasada-Tsutsui Y, Funahashi Y, Ozawa T, Masuda H. Preparations of trans- and cis-μ-1,2-Peroxodiiron(III) Complexes. Molecules. 2024; 29(1):205. https://doi.org/10.3390/molecules29010205
Chicago/Turabian StyleKajita, Yuji, Masaki Kubo, Hidekazu Arii, Shinya Ishikawa, Yamato Saito, Yuko Wasada-Tsutsui, Yasuhiro Funahashi, Tomohiro Ozawa, and Hideki Masuda. 2024. "Preparations of trans- and cis-μ-1,2-Peroxodiiron(III) Complexes" Molecules 29, no. 1: 205. https://doi.org/10.3390/molecules29010205
APA StyleKajita, Y., Kubo, M., Arii, H., Ishikawa, S., Saito, Y., Wasada-Tsutsui, Y., Funahashi, Y., Ozawa, T., & Masuda, H. (2024). Preparations of trans- and cis-μ-1,2-Peroxodiiron(III) Complexes. Molecules, 29(1), 205. https://doi.org/10.3390/molecules29010205