
Machine-Learning- and Structure-Based Virtual Screening for Selecting Cinnamic Acid Derivatives as Leishmania major DHFR-TS Inhibitors

 , ,
, ,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
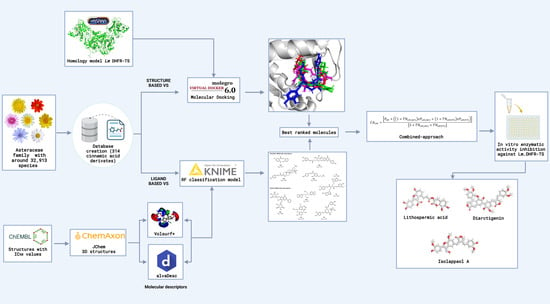
2.1. Combined Ligand-/Structure-Based Virtual Screening Approach Using LmDHFR-TS
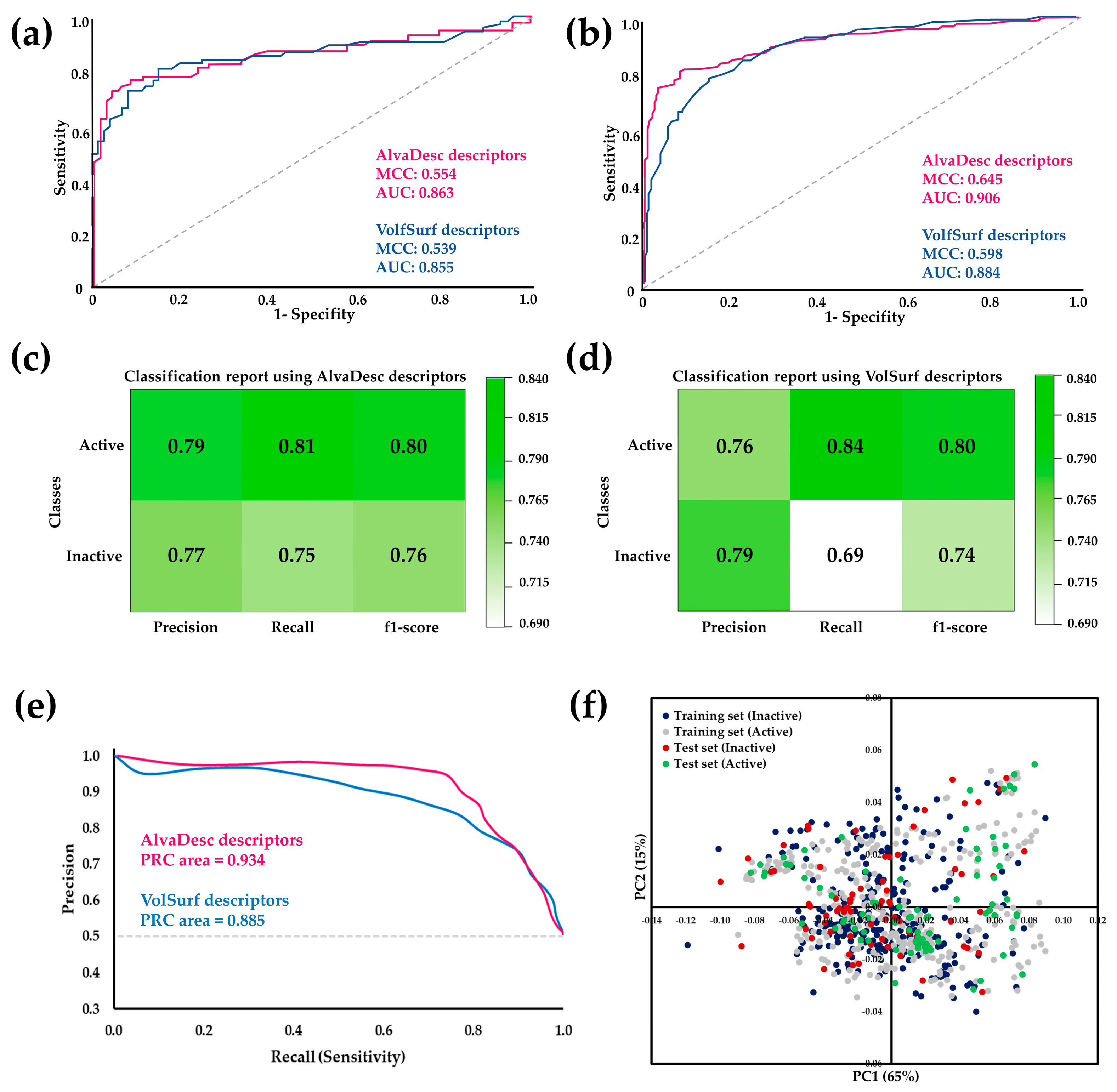
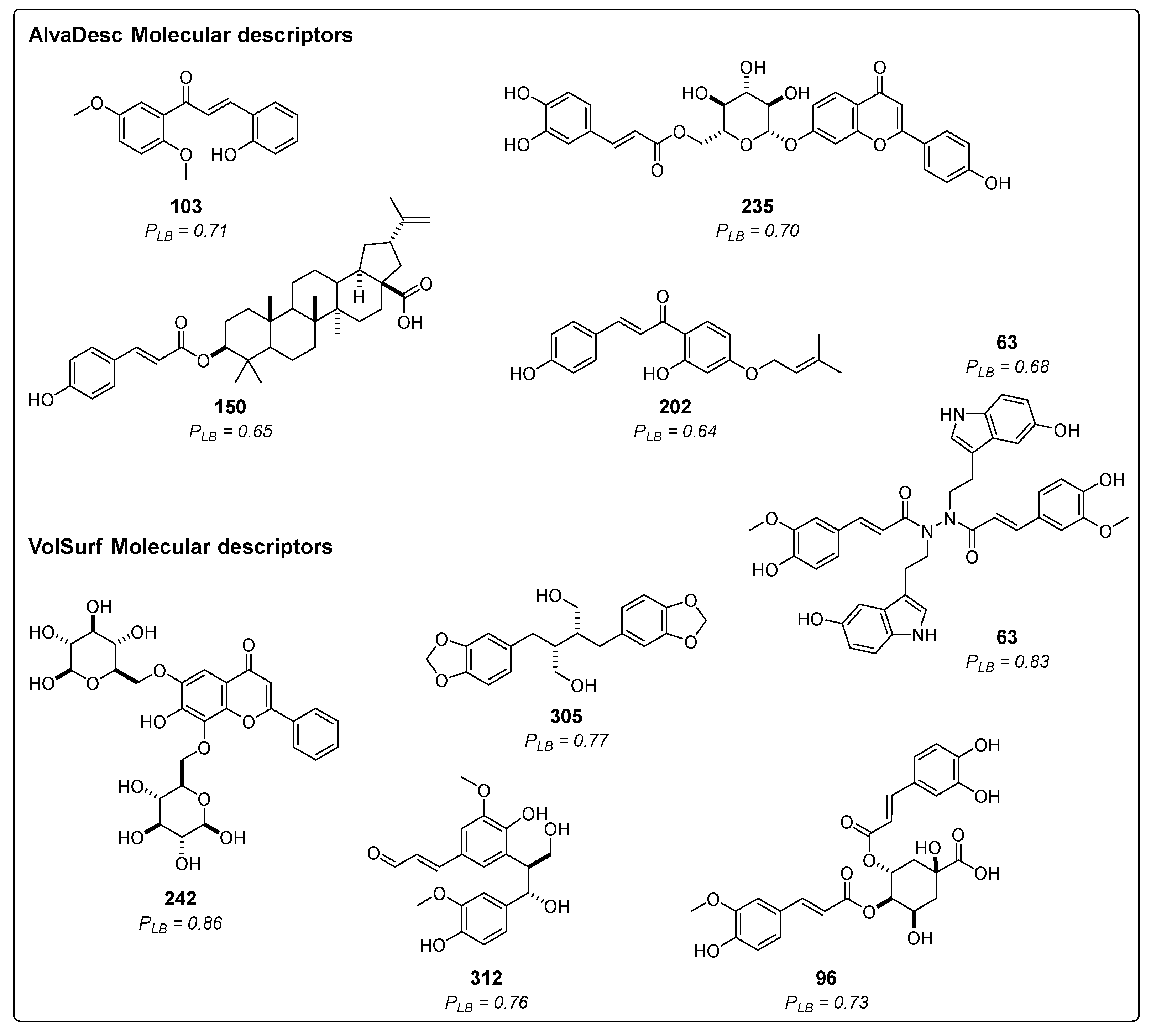
2.1.1. Ligand-Based Virtual Screening
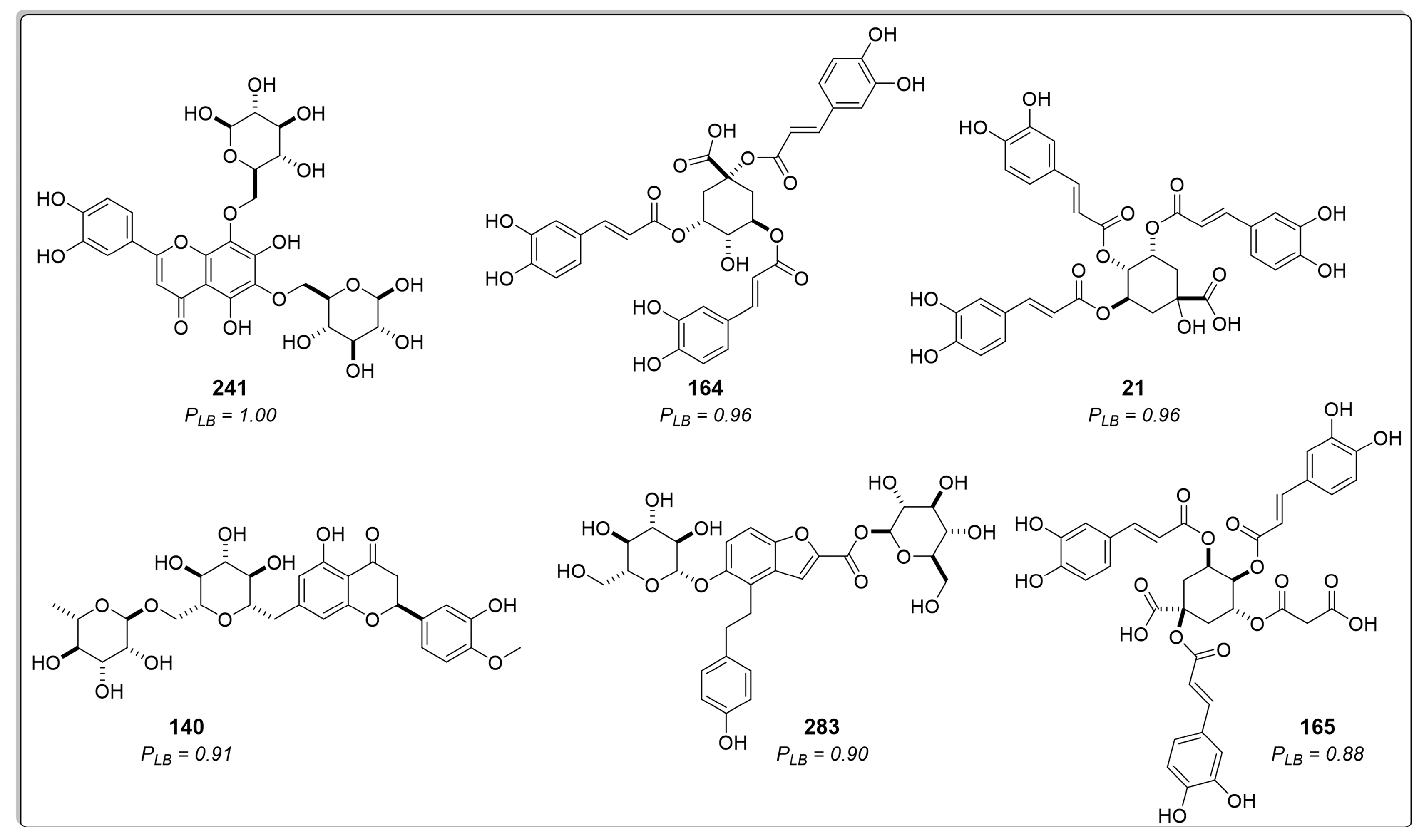
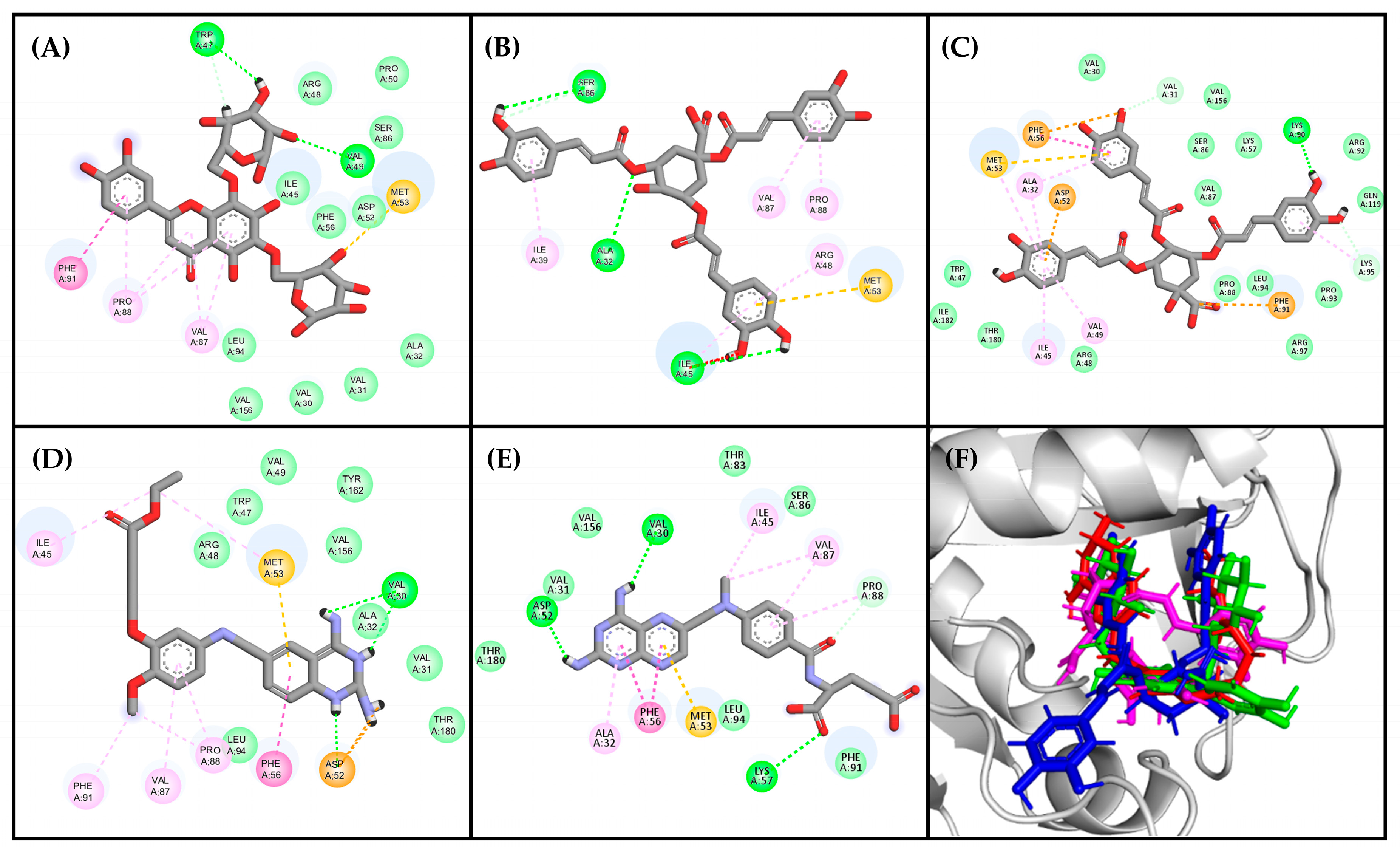
2.1.2. Structure-Based Virtual Screening
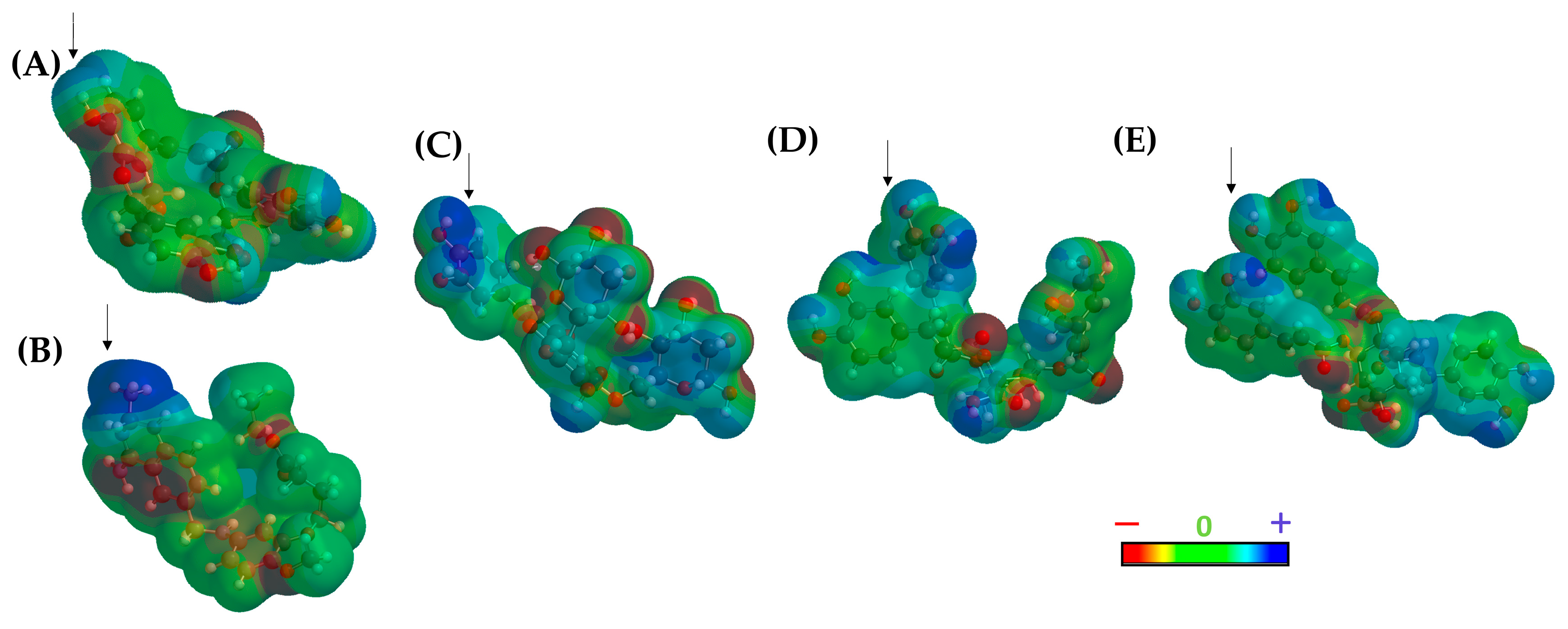
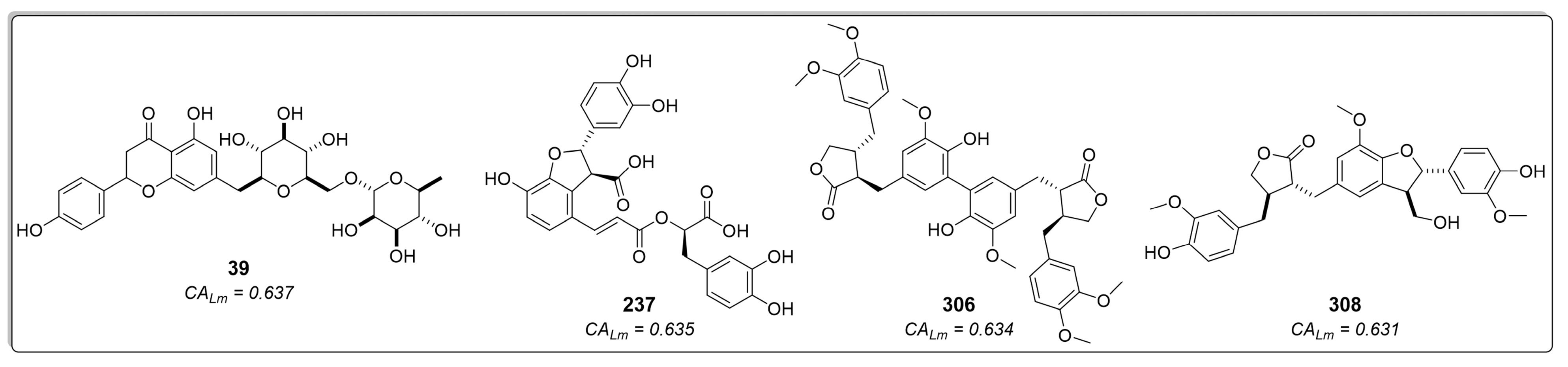
2.1.3. Consensus Analysis of the Two VS Approaches
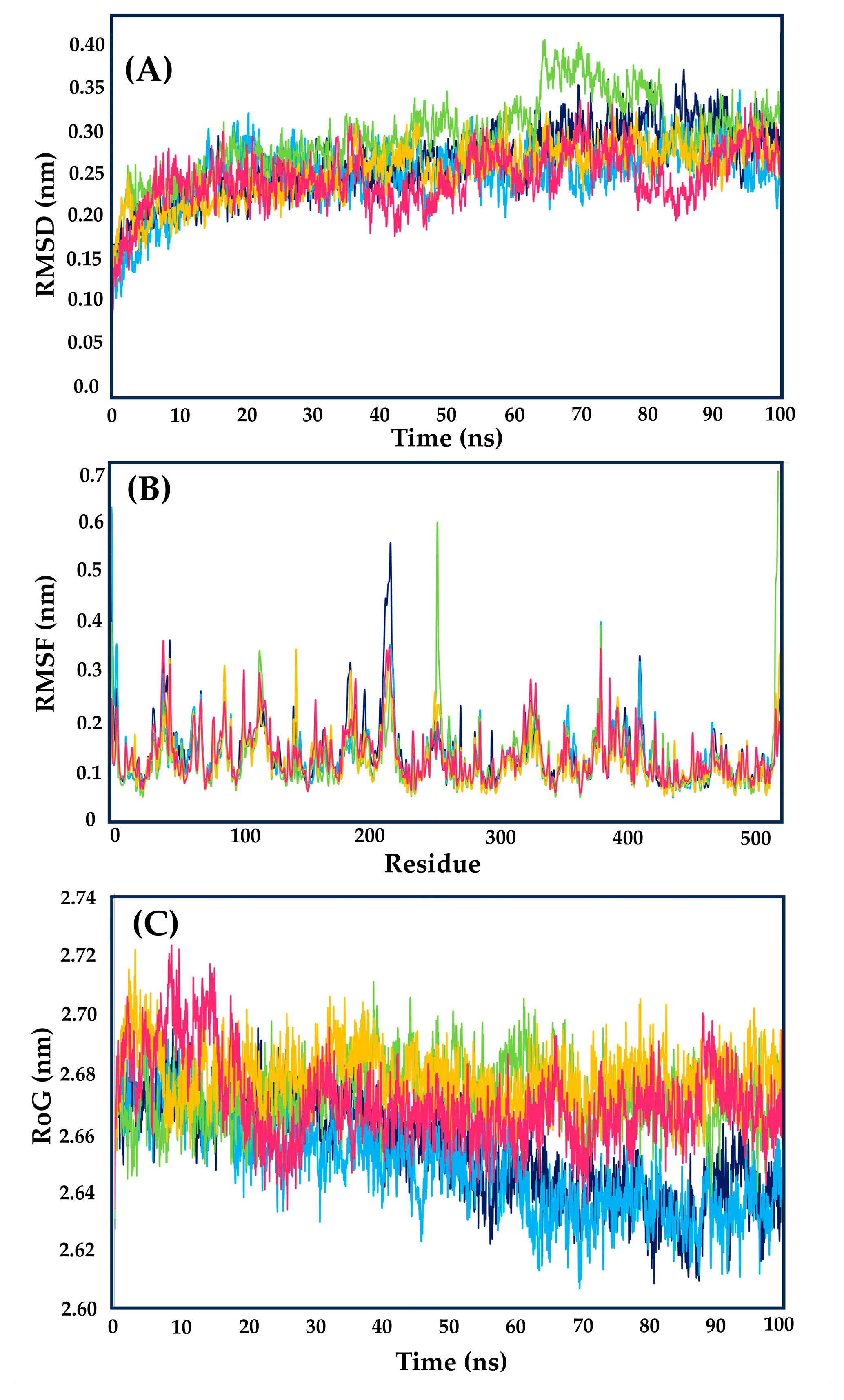
2.2. Molecular Dynamics Simulations
2.3. In Vitro Enzymatic Activity Inhibition for Selected Cinnamic Acid Derivatives (Compounds 237, 306, and 308) against LmDHFR-TS and HsDHFR
2.4. Pharmacokinetic Properties Predictions
3. Materials and Methods
3.1. Cinnamic Acid Derivatives In-House Dataset
3.2. Classificatory Machine Learning Models
3.3. Molecular Docking Calculations
3.4. Molecular Dynamics Simulations
3.5. LmDHFR-TS and HsDHFR Enzymatic Inhibition Assays
3.6. Pharmacokinetic Properties Predictions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| ADMET | Absorption, distribution, metabolism, excretion, and toxicity |
| APD | Applicability domain |
| AUC | Area under the ROC curve |
| BSA | Bovine serum albumin |
| CL | Cutaneous leishmaniasis |
| CYP | Cytochrome |
| DHFR-TS | Dihydrofolate reductase-thymidylate synthase |
| DNA | Deoxyribonucleic acid |
| Eligand | Ligand energy |
| EDTA | Ethylenediaminetetraacetic acid |
| Ei | Docking energy. |
| Emin | Lowest energy value |
| FAK | Focal adhesion kinase |
| Hs | Homo sapiens |
| HTS | High-throughput screening |
| IC50 | Half-maximal inhibitory concentration |
| Lm | Leishmania major |
| PLb | Ligand-based probability |
| MAPK | Mitogen-activated protein kinases |
| MCC | Matthew’s correlation coefficient |
| MD | Molecular dynamics |
| MLP | Molecular lipophilicity potential |
| MM-PBSA | Molecular mechanics Poisson–Boltzmann surface area |
| MTX | Methotrexate |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NTD | Neglected tropical diseases |
| ns | Nanoseconds |
| PCA | Principal component analysis |
| pKa | Acid dissociation constant values |
| PME | Particle-mesh Ewald |
| PRC | Precision-recall curve |
| PSB | Structure-based probability |
| RF | Random forest |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| ROC | Receiver operating characteristic |
| RoG | Radius of gyration |
| SASA | Solvent-accessible surface area |
| SI | Selective index |
| TES | N-[tris(hydroxymethyl)-methyl]-2-aminoethanesulfonic acid |
| TNBC | Triple-negative breast cancer |
| TPSA | Topological polar surface area |
| Vo | Initial reaction rate |
| VS | Virtual screening |
| WHO | World Health Organization |
References
- Leishmaniasis. Available online: https://www.who.int/health-topics/leishmaniasis (accessed on 30 June 2023).
- Abadías-Granado, I.; Diago, A.; Cerro, P.A.; Palma-Ruiz, A.M.; Gilaberte, Y. Cutaneous and Mucocutaneous Leishmaniasis. Actas Dermosifiliogr. (Engl. Ed.) 2021, 112, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Wijerathna, T.; Gunathilaka, N. Diurnal Adult Resting Sites and Breeding Habitats of Phlebotomine Sand Flies in Cutaneous Leishmaniasis Endemic Areas of Kurunegala District, Sri Lanka. Parasit. Vectors 2020, 13, 284. [Google Scholar] [CrossRef]
- Salgado-Almario, J.; Hernández, C.A.; Ovalle, C.E. Geographical Distribution of Leishmania Species in Colombia, 1985–2017. Biomedica 2019, 39, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Ovalle-Bracho, C.; Londoño-Barbosa, D.; Salgado-Almario, J.; González, C. Evaluating the Spatial Distribution of Leishmania Parasites in Colombia from Clinical Samples and Human Isolates (1999 to 2016). PLoS ONE 2019, 14, e0214124. [Google Scholar] [CrossRef] [PubMed]
- Boletín Epidemiológico. Available online: https://www.ins.gov.co/buscador-eventos/BoletinEpidemiologico/2022_Boletín_epidemiologico_semana_25.pdf (accessed on 28 May 2023).
- Leishmaniasis—OPS/OMS|Organización Panamericana de la Salud. Available online: https://www.paho.org/es/temas/leishmaniasis (accessed on 5 June 2023).
- Wilairatana, P.; Chanmol, W.; Rattaprasert, P.; Masangkay, F.R.; Milanez, G.D.J.; Kotepui, K.U.; Kotepui, M. Prevalence and Characteristics of Malaria Co-Infection among Individuals with Visceral Leishmaniasis in Africa and Asia: A Systematic Review and Meta-Analysis. Parasit. Vectors 2021, 14, 545. [Google Scholar] [CrossRef]
- Herrera Acevedo, C.; Scotti, L.; Feitosa Alves, M.; Formiga Melo Diniz, M.D.F.; Scotti, M.T. Computer-Aided Drug Design Using Sesquiterpene Lactones as Sources of New Structures with Potential Activity against Infectious Neglected Diseases. Molecules 2017, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Kumar, V.; Tiwari, R.K.; Ravidas, V.; Pandey, K.; Kumar, A. Amphotericin B: A Drug of Choice for Visceral Leishmaniasis. Acta Trop. 2022, 235, 106661. [Google Scholar] [CrossRef]
- Palić, S.; Beijnen, J.H.; Dorlo, T.P.C. An Update on the Clinical Pharmacology of Miltefosine in the Treatment of Leishmaniasis. Int. J. Antimicrob. Agents 2022, 59, 106459. [Google Scholar] [CrossRef]
- Rafiq, M.; Naveed, M.; Khan, Z.U.; Raza, A.; Awrejcewicz, J.; Soori, A.H.; Ul Haq, I.; Mohsin, M. Modeling the Spread of Leishmaniasis Disease via Delayed Analysis. Alex. Eng. J. 2022, 61, 11197–11209. [Google Scholar] [CrossRef]
- Sánchez-Suárez, J.; Bernal, F.A.; Coy-Barrera, E. Colombian Contributions Fighting Leishmaniasis: A Systematic Review on Antileishmanials Combined with Chemoinformatics Analysis. Molecules 2020, 25, 5704. [Google Scholar] [CrossRef]
- Hevener, K.E.; Pesavento, R.; Ren, J.; Lee, H.; Ratia, K.; Johnson, M.E. Chapter Twelve—Hit-to-Lead: Hit Validation and Assessment. In Methods in Enzymology; Lesburg, C.A., Ed.; Modern Approaches in Drug Discovery; Academic Press: Cambridge, MA, USA, 2018; Volume 610, pp. 265–309. [Google Scholar] [CrossRef]
- Bhattacharjee, A.K. 10—Pharmacophore-Based Virtual Screening of Large Compound Databases Can Aid “Big Data” Problems in Drug Discovery. In Big Data Analytics in Chemoinformatics and Bioinformatics; Basak, S.C., Vračko, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 231–246. [Google Scholar] [CrossRef]
- Shreya; Shweta; Dagur, P.; Rakshit, G.; Ghosh, M. Chapter 7—Virtual Screening of Phytochemicals for Drug Discovery. In Phytochemistry, Computational Tools and Databases in Drug Discovery; Egbuna, C., Rudrapal, M., Tijjani, H., Eds.; Drug Discovery Update; Elsevier: Amsterdam, The Netherlands, 2023; pp. 149–179. [Google Scholar] [CrossRef]
- McGibbon, M.; Money-Kyrle, S.; Blay, V.; Houston, D.R. SCORCH: Improving Structure-Based Virtual Screening with Machine Learning Classifiers, Data Augmentation, and Uncertainty Estimation. J. Adv. Res. 2023, 46, 135–147. [Google Scholar] [CrossRef]
- Joshi, T.; Sharma, P.; Joshi, T.; Mathpal, S.; Pandey, S.C.; Pandey, A.; Chandra, S. Chapter 4—Recent Advances on Computational Approach towards Potential Drug Discovery against Leishmaniasis. In Pathogenesis, Treatment and Prevention of Leishmaniasis; Samant, M., Chandra Pandey, S., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 63–84. [Google Scholar] [CrossRef]
- Acevedo, C.H.; Scotti, L.; Scotti, M.T. In Silico Studies Designed to Select Sesquiterpene Lactones with Potential Antichagasic Activity from an In-House Asteraceae Database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Acevedo, C.; Dos Santos Maia, M.; Cavalcanti, É.B.V.S.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Selection of Antileishmanial Sesquiterpene Lactones from SistematX Database Using a Combined Ligand-/Structure-Based Virtual Screening Approach. Mol. Divers. 2021, 25, 2411–2427. [Google Scholar] [CrossRef] [PubMed]
- Ayeleso, T.B.; Matumba, M.G.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef] [PubMed]
- Majid Shah, S.; Ullah, F.; Ayaz, M.; Sadiq, A.; Hussain, S.; Ali Shah, A.-U.-H.; Adnan Ali Shah, S.; Wadood, A.; Nadhman, A. β-Sitosterol from Ifloga Spicata (Forssk.) Sch. Bip. as Potential Anti-Leishmanial Agent against Leishmania Tropica: Docking and Molecular Insights. Steroids 2019, 148, 56–62. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Flores-Gaspar, A.; Scotti, L.; Mendonça-Junior, F.J.B.; Scotti, M.T.; Coy-Barrera, E. Identification of Kaurane-Type Diterpenes as Inhibitors of Leishmania Pteridine Reductase I. Molecules 2021, 26, 3076. [Google Scholar] [CrossRef] [PubMed]
- Gouri, V.; Upreti, S.; Samant, M. Evaluation of Target-Specific Natural Compounds for Drug Discovery against Leishmaniasis. Parasitol. Int. 2022, 91, 102622. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.P.; Tomaz, D.C.; Ângelo de Souza, L.; Onofre, T.S.; Aquiles de Menezes, W.; Almeida-Silva, J.; Suarez-Fontes, A.M.; Rogéria de Almeida, M.; Manoel da Silva, A.; Bressan, G.C.; et al. Synthesis of Cinnamic Acid Derivatives and Leishmanicidal Activity against Leishmania Braziliensis. Eur. J. Med. Chem. 2019, 183, 111688. [Google Scholar] [CrossRef]
- Monzote, L.; Perera Córdova, W.H.; García, M.; Piñón, A.; Setzer, W.N. In-Vitro and in-Vivo Activities of Phenolic Compounds against Cutaneous Leishmaniasis. Rec. Nat. Prod. 2016, 10, 269–276. [Google Scholar]
- Kabir, E.; Uzzaman, M. A Review on Biological and Medicinal Impact of Heterocyclic Compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Citarella, A.; Moi, D.; Pedrini, M.; Pérez-Peña, H.; Pieraccini, S.; Dimasi, A.; Stagno, C.; Micale, N.; Schirmeister, T.; Sibille, G.; et al. Synthesis of SARS-CoV-2 Mpro Inhibitors Bearing a Cinnamic Ester Warhead with in Vitro Activity against Human Coronaviruses. Org. Biomol. Chem. 2023, 21, 3811–3824. [Google Scholar] [CrossRef]
- Jiang, Y.-Y.; Wu, S.; Wu, Y.-W.; Gao, Y.; Chong, D.; Sun, C.; Wei, M.-Y.; Gu, Y.-C.; Shao, C.-L.; Gu, Y. New Brefeldin A-Cinnamic Acid Ester Derivatives as Potential Antitumor Agents: Design, Synthesis and Biological Evaluation. Eur. J. Med. Chem. 2022, 240, 114598. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xu, K.; Zhang, S.; Zhang, J.; Qiu, Y.; Luo, J.; Tan, G.; Zou, Z.; Wang, W.; Kang, F. Discovery of novel chloropyramine-cinnamic acid hybrids as potential FAK inhibitors for intervention of metastatic triple-negative breast cancer. Bioorg. Med. Chem. 2022, 66, 116809. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, Y.; He, B.; Li, L.; Tian, Y.; Xiao, Y.; Shang, H.; Zou, Z. Design, Synthesis and Evaluation of Ursodeoxycholic Acid-Cinnamic Acid Hybrids as Potential Anti-Inflammatory Agents by Inhibiting Akt/NF-κB and MAPK Signaling Pathways. Eur. J. Med. Chem. 2023, 260, 115785. [Google Scholar] [CrossRef]
- Sabt, A.; Eldehna, W.M.; Ibrahim, T.M.; Bekhit, A.A.; Batran, R.Z. New Antileishmanial Quinoline Linked Isatin Derivatives Targeting DHFR-TS and PTR1: Design, Synthesis, and Molecular Modeling Studies. Eur. J. Med. Chem. 2023, 246, 114959. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Acevedo, C.; de Menezes, R.P.B.; de Sousa, N.F.; Scotti, L.; Scotti, M.T.; Coy-Barrera, E. Kaurane-Type Diterpenoids as Potential Inhibitors of Dihydrofolate Reductase-Thymidylate Synthase in New World Leishmania Species. Antibiotics 2023, 12, 663. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Pu, D.; Tassa, C.; Weissleder, R.; Shaw, S.Y.; Mumper, R.J.; Tropsha, A. Quantitative nanostructure–activity relationship modeling. ACS Nano 2010, 4, 5703–5712. [Google Scholar] [CrossRef]
- Cruciani, G.; Pastor, M.; Guba, W. VolSurf: A New Tool for the Pharmacokinetic Optimization of Lead Compounds. Eur. J. Pharm. Sci. 2000, 11 (Suppl. S2), S29–S39. [Google Scholar] [CrossRef]
- Cruciani, G.; Crivori, P.; Carrupt, P.-A.; Testa, B. Molecular Fields in Quantitative Structure–Permeation Relationships: The VolSurf Approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Mauri, A. AlvaDesc: A Tool to Calculate and Analyze Molecular Descriptors and Fingerprints. In Ecotoxicological QSARs; Roy, K., Ed.; Methods in Pharmacology and Toxicology; Springer: New York, NY, USA, 2020; pp. 801–820. [Google Scholar] [CrossRef]
- Mauri, A.; Bertola, M. Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef]
- Shi, L.; Yan, F.; Liu, H. Screening Model of Candidate Drugs for Breast Cancer Based on Ensemble Learning Algorithm and Molecular Descriptor. Expert Syst. Appl. 2023, 213, 119185. [Google Scholar] [CrossRef]
- Boyd, K.; Eng, K.H.; Page, C.D. Area under the Precision-Recall Curve: Point Estimates and Confidence Intervals. In Proceedings of the Machine Learning and Knowledge Discovery in Databases: European Conference, ECML PKDD 2013, Prague, Czech Republic, 23–27 September 2013; Springer: Berlin/Heidelberg, Germany, 2013; Volume 13. Part III. [Google Scholar]
- Ticha, L.A.; Klaasen, J.A.; Green, I.R.; Naidoo, S.; Baker, B.; Pietersen, R.-D. Phytochemical and Antimicrobial Screening of Flavanones and Chalcones from Galenia Africana and Dicerothamnus Rhinocerotis. Nat. Prod. Commun. 2015, 10, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Repčák, M.; Krausová, T. Phenolic Glucosides in the Course of Ligulate Flower Development in Diploid and Tetraploid Matricaria Chamomilla. Food Chem. 2009, 116, 19–22. [Google Scholar] [CrossRef]
- Shoeb, M.; MacManus, S.M.; Jaspars, M.; Trevidu, J.; Nahar, L.; Kong-Thoo-Lin, P.; Sarker, S.D. Montamine, a Unique Dimeric Indole Alkaloid, from the Seeds of Centaurea montana (Asteraceae), and Its in Vitro Cytotoxic Activity against the CaCo2 Colon Cancer Cells. Tetrahedron 2006, 62, 11172–11177. [Google Scholar] [CrossRef]
- Zofou, D.; Kuete, V.; Titanji, V.P.K. 17—Antimalarial and Other Antiprotozoal Products from African Medicinal Plants. In Medicinal Plant Research in Africa; Kuete, V., Ed.; Elsevier: Oxford, UK, 2013; pp. 661–709. [Google Scholar] [CrossRef]
- Genovese, S.; Epifano, F.; Curini, M.; Dudra-Jastrzebska, M.; Luszczki, J.J. Prenyloxyphenylpropanoids as a Novel Class of Anticonvulsive Agents. Bioorg. Med. Chem. Lett. 2009, 19, 5419–5422. [Google Scholar] [CrossRef] [PubMed]
- Gobbo-Neto, L.; Lopes, N.P. Online Identification of Chlorogenic Acids, Sesquiterpene Lactones, and Flavonoids in the Brazilian Arnica Lychnophora ericoides Mart. (Asteraceae) Leaves by HPLC-DAD-MS and HPLC-DAD-MS/MS and a Validated HPLC-DAD Method for Their Simultaneous Analysis. J. Agric. Food Chem. 2008, 56, 1193–1204. [Google Scholar] [CrossRef]
- Borsato, M.L.C.; Grael, C.F.F.; Souza, G.E.P.; Lopes, N.P. Analgesic Activity of the Lignans from Lychnophora Ericoides. Phytochemistry 2000, 55, 809–813. [Google Scholar] [CrossRef]
- Könye, R.; Tóth, G.; Sólyomváry, A.; Mervai, Z.; Zürn, M.; Baghy, K.; Kovalszky, I.; Horváth, P.; Molnár-Perl, I.; Noszál, B.; et al. Chemodiversity of Cirsium Fruits: Antiproliferative Lignans, Neolignans and Sesquineolignans as Chemotaxonomic Markers. Fitoterapia 2018, 127, 413–419. [Google Scholar] [CrossRef]
- de Athayde, A.E.; Richetti, E.; Wolff, J.; Lusa, M.G.; Biavatti, M.W. “Arnicas” from Brazil: Comparative Analysis among Ten Species. Rev. Bras. Farmacogn. 2019, 29, 401–424. [Google Scholar] [CrossRef]
- Amoa Onguéné, P.; Ntie-Kang, F.; Lifongo, L.L.; Ndom, J.C.; Sippl, W.; Mbaze, L.M.A. The potential of anti-malarial compounds derived from African medicinal plants. Part I: A pharmacological evaluation of alkaloids and terpenoids. Malar. J. 2013, 12, 1–26. [Google Scholar] [CrossRef]
- Ahmad, Q.Z.; Jahan, N.; Ahmad, G. Nephroprotective effect of Kabab chini (Piper cubeba) in gentamycin-induced nephrotoxicity. Saudi J. Kidney Dis. Transpl. 2012, 23, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Ostad, S.N.; Rajabi, A.; Khademi, R.; Farjadmand, F.; Eftekhari, M.; Hadjiakhoondi, A.; Khanavi, M. Cytotoxic potential of Centaurea bruguierana ssp. belangerana: The MTT assay. Acta Med. Iran. 2016, 54, 583–589. [Google Scholar] [PubMed]
- Maganti, L.; Manoharan, P.; Ghoshal, N. Probing the Structure of Leishmania Donovani Chagasi DHFR-TS: Comparative Protein Modeling and Protein–Ligand Interaction Studies. J. Mol. Model. 2010, 16, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Vickers, T.J.; Beverley, S.M. Folate metabolic pathways in Leishmania. Essays Biochem. 2011, 51, 63–80. [Google Scholar] [CrossRef]
- Zuccotto, F.; Martin, A.C.R.; Laskowski, R.A.; Thornton, J.M.; Gilbert, I.H. Dihydrofolate Reductase: A Potential Drug Target in Trypanosomes and Leishmania. J. Comput. Aided Mol. Des. 1998, 12, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Desam, N.R.; Al-Rajab, A.J. Chapter 19—Herbal Biomolecules: Anticancer Agents. In Herbal Biomolecules in Healthcare Applications; Mandal, S.C., Nayak, A.K., Dhara, A.K., Eds.; Academic Press: Cambridge, MA, USA, 2022; pp. 435–474. [Google Scholar] [CrossRef]
- Raj, S.; Sasidharan, S.; Tripathi, T.; Saudagar, P. Biofunctionalized Chrysin-Conjugated Gold Nanoparticles Neutralize Leishmania Parasites with High Efficacy. Int. J. Biol. Macromol. 2022, 205, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Onawole, A.T.; Sulaiman, K.O.; Kolapo, T.U.; Akinde, F.O.; Adegoke, R.O. COVID-19: CADD to the rescue. Virus Res. 2020, 285, 198022. [Google Scholar] [CrossRef]
- Shulha, O.; Çiçek, S.S.; Wangensteen, H.; Kroes, J.; Mäder, M.; Girreser, U.; Sendker, J.; Jöhrer, K.; Greil, R.; Schühly, W.; et al. Lignans and sesquiterpene lactones from Hypochaeris radicata subsp. neapolitana (Asteraceae, Cichorieae). Phytochemistry 2019, 165, 112047. [Google Scholar] [CrossRef]
- Begum, T.; Prakash, R.; Duarah, G.; Gogoi, S. Utilization of Rh-carbenoid CH insertion reactions for the synthesis of bioactive natural products. Stud. Nat. Prod. Chem. 2020, 65, 349–380. [Google Scholar] [CrossRef]
- Liu, X.; Chen, R.; Shang, Y.; Jiao, B.; Huang, C. Lithospermic acid as a novel xanthine oxidase inhibitor has anti-inflammatory and hypouricemic effects in rats. Chem. Biol. Interact. 2008, 176, 137–142. [Google Scholar] [CrossRef]
- Tabrez, S.; Rahman, F.; Ali, R.; Akand, S.K.; Alaidarous, M.A.; Banawas, S.; Bin Dukhyil, A.A.; Rub, A. Hesperidin targets Leishmania donovani sterol C-24 reductase to fight against leishmaniasis. ACS Omega 2021, 6, 8112–8118. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Xu, L.; Li, D.; Hou, T. Recent developments in computational prediction of HERG blockage. Curr. Top. Med. Chem. 2013, 13, 1317–1326. [Google Scholar] [CrossRef]
- Sun, L.; Yang, H.; Li, J.; Wang, T.; Li, W.; Liu, G.; Tang, Y. In silico prediction of compounds binding to human plasma proteins by QSAR models. ChemMedChem 2018, 13, 572–581. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner: Version 2.0 and beyond. SIGKDD Explor. 2009, 11, 26–31. [Google Scholar] [CrossRef]
- Hanley, J.A.; McNeil, B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 1982, 143, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta 1975, 405, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef] [PubMed]
- Hehre, W.J.; Ohlinger, W.A. Spartan ’14; Wavefunction Inc.: Irvine, CA, USA, 2014; pp. 429–507. ISBN 978-1-890661-42-2. [Google Scholar]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 43–67. ISBN 1064-3745. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Angarita-Rodríguez, A.; Quiroga, D.; Coy-Barrera, E. Indole-Containing Phytoalexin-Based Bioisosteres as Antifungals: In Vitro and In Silico Evaluation against Fusarium oxysporum. Molecules 2020, 25, 45. [Google Scholar] [CrossRef] [PubMed]
- Grumont, R.; Sirawaraporn, W.; Santi, D.V. Heterologous Expression of the Bifunctional Thymidylate Synthase-Dihydrofolate Reductase from Leishmania major. Biochemistry 1988, 27, 3776–3784. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef]
- Park, S.Y.; Hong, S.S.; Han, X.H.; Hwang, J.S.; Lee, D.; Ro, J.S.; Hwang, B.Y. Lignans from Arctium lappa and Their Inhibition of LPS-Induced Nitric Oxide Production. Chem. Pharm. Bull. 2007, 55, 150–152. [Google Scholar] [CrossRef]
- Umehara, K.; Sugawa, A.; Kuroyanagi, M.; Ueno, A.; Taki, T. Studies on Differentiation-Inducers from Arctium Fructus. Chem. Pharm. Bull. 1993, 41, 1774–1779. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Ligand | Docking Score (kJ/mol) | SD | RMSD |
|---|---|---|---|---|
| 1 | 241 | −182.8 | 5.4 | 1.0 |
| 2 | 164 | −175.6 | 7.1 | 1.8 |
| 3 | 21 | −175.5 | 11.2 | 1.0 |
| 4 | 242 | −169.6 | 1.9 | 1.2 |
| 5 | 140 | −167.0 | 3.3 | 0.4 |
| 6 | 283 | −165.4 | 4.8 | 1.7 |
| 7 | 165 | −161.8 | 7.4 | 1.2 |
| 8 | 235 | −161.4 | 5.9 | 0.9 |
| 9 | 285 | −160.9 | 8.8 | 1.2 |
| 10 | 63 | −160.1 | 5.2 | 1.1 |
| Redocking | MTX | −114.2 | 2.2 | 0.3 |
| DQ1 | −134.4 | 2.5 | 0.3 |
| Rank | Ligand | PLB(AD) | PLB(VS) | PSB | CALm |
|---|---|---|---|---|---|
| 1 | 63 | 0.68 | 0.83 | 0.88 | 0.78 |
| 2 | 242 | 0.52 | 0.86 | 0.93 | 0.74 |
| 3 | 96 | 0.55 | 0.73 | 0.77 | 0.67 |
| 4 | 241 | 0.53 | 0.55 | 1.00 | 0.64 |
| 5 | 39 | 0.57 | 0.64 | 0.77 | 0.64 |
| 6 | 237 | 0.61 | 0.55 | 0.84 | 0.64 |
| 7 | 306 | 0.63 | 0.53 | 0.83 | 0.63 |
| 8 | 165 | 0.53 | 0.60 | 0.88 | 0.63 |
| 9 | 140 | 0.59 | 0.51 | 0.91 | 0.63 |
| 10 | 308 | 0.57 | 0.59 | 0.81 | 0.63 |
| 237 | 306 | 308 | MTX | |||||
|---|---|---|---|---|---|---|---|---|
| Energy Contribution | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD | kJ/mol | SD |
| van der Waals | −218.3 | 6.2 | −209.7 | 4.6 | −217.6 | 6.2 | −239.5 | 8.2 |
| Electrostatic | −31.3 | 4.1 | −38.0 | 3.9 | −29.0 | 4.6 | −57.3 | 4.3 |
| Polar solvation | 181.5 | 6.5 | 157.6 | 6.3 | 185.6 | 6.5 | 194.6 | 8.5 |
| SASA | −23.6 | 1.8 | −21.0 | 1.9 | −20.0 | 1.2 | −22.4 | 2.2 |
| Binding energy | −91.6 | 4.7 | −111.1 | 4.2 | −81 | 4.6 | −124.5 | 5.8 |
| Compound | LmDHFR-TS | HsDHFR | SI | ||
|---|---|---|---|---|---|
| IC50 (µM) | CI (95%) | IC50 (µM) | CI (95%) | ||
| hesperidin | 21.6 | 20.2–23.1 | 86.5 | 82.3–87.2 | 4.0 |
| lithospermic acid (237) | 7.5 | 6.8–7.9 | 22.6 | 21.3–24.7 | 3.0 |
| diarctigenin (306) | 6.1 | 5.7–6.4 | 27.9 | 26.8–28.6 | 4.6 |
| isolappaol A (308) | 10.1 | 9.7–10.3 | 44.8 | 42.4–45.9 | 4.4 |
| isovitexin 4′-O-glucoside | 53.2 | 51.1–54.1 | 125.7 | 122.8–127.8 | 2.4 |
| rutin | 41.7 | 40.3–43.1 | 188.9 | 186.2–190.6 | 4.5 |
| MTX | 1.4 | 1.1–1.5 | 4.9 | 4.7–5.1 | 3.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Vega, M.C.; López-Hernández, S.; Sierra-Chavarro, A.; Scotti, M.T.; Scotti, L.; Coy-Barrera, E.; Herrera-Acevedo, C. Machine-Learning- and Structure-Based Virtual Screening for Selecting Cinnamic Acid Derivatives as Leishmania major DHFR-TS Inhibitors. Molecules 2024, 29, 179. https://doi.org/10.3390/molecules29010179
Muñoz-Vega MC, López-Hernández S, Sierra-Chavarro A, Scotti MT, Scotti L, Coy-Barrera E, Herrera-Acevedo C. Machine-Learning- and Structure-Based Virtual Screening for Selecting Cinnamic Acid Derivatives as Leishmania major DHFR-TS Inhibitors. Molecules. 2024; 29(1):179. https://doi.org/10.3390/molecules29010179
Chicago/Turabian StyleMuñoz-Vega, Maria Camila, Sofía López-Hernández, Adrián Sierra-Chavarro, Marcus Tullius Scotti, Luciana Scotti, Ericsson Coy-Barrera, and Chonny Herrera-Acevedo. 2024. "Machine-Learning- and Structure-Based Virtual Screening for Selecting Cinnamic Acid Derivatives as Leishmania major DHFR-TS Inhibitors" Molecules 29, no. 1: 179. https://doi.org/10.3390/molecules29010179
APA StyleMuñoz-Vega, M. C., López-Hernández, S., Sierra-Chavarro, A., Scotti, M. T., Scotti, L., Coy-Barrera, E., & Herrera-Acevedo, C. (2024). Machine-Learning- and Structure-Based Virtual Screening for Selecting Cinnamic Acid Derivatives as Leishmania major DHFR-TS Inhibitors. Molecules, 29(1), 179. https://doi.org/10.3390/molecules29010179