
3,3-Disubstituted 3,4-Dihydro-1,2,4-benzotriazines: Chemistry, Biological Activity, and Affinity to Sigma Receptors



Abstract
:1. Introduction
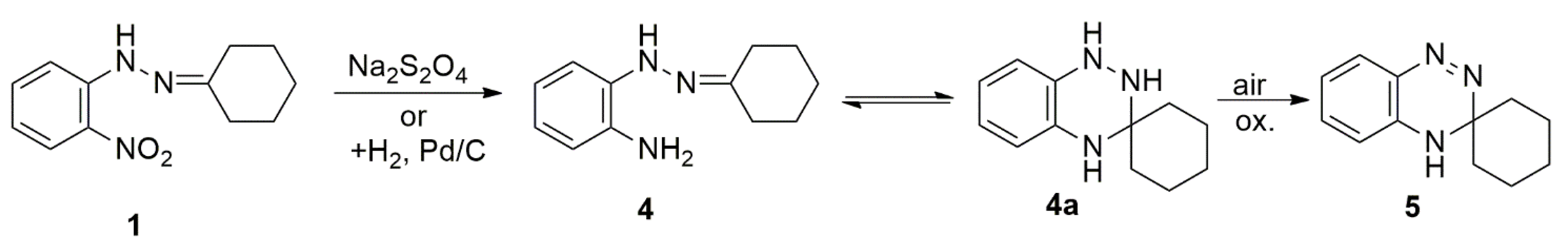
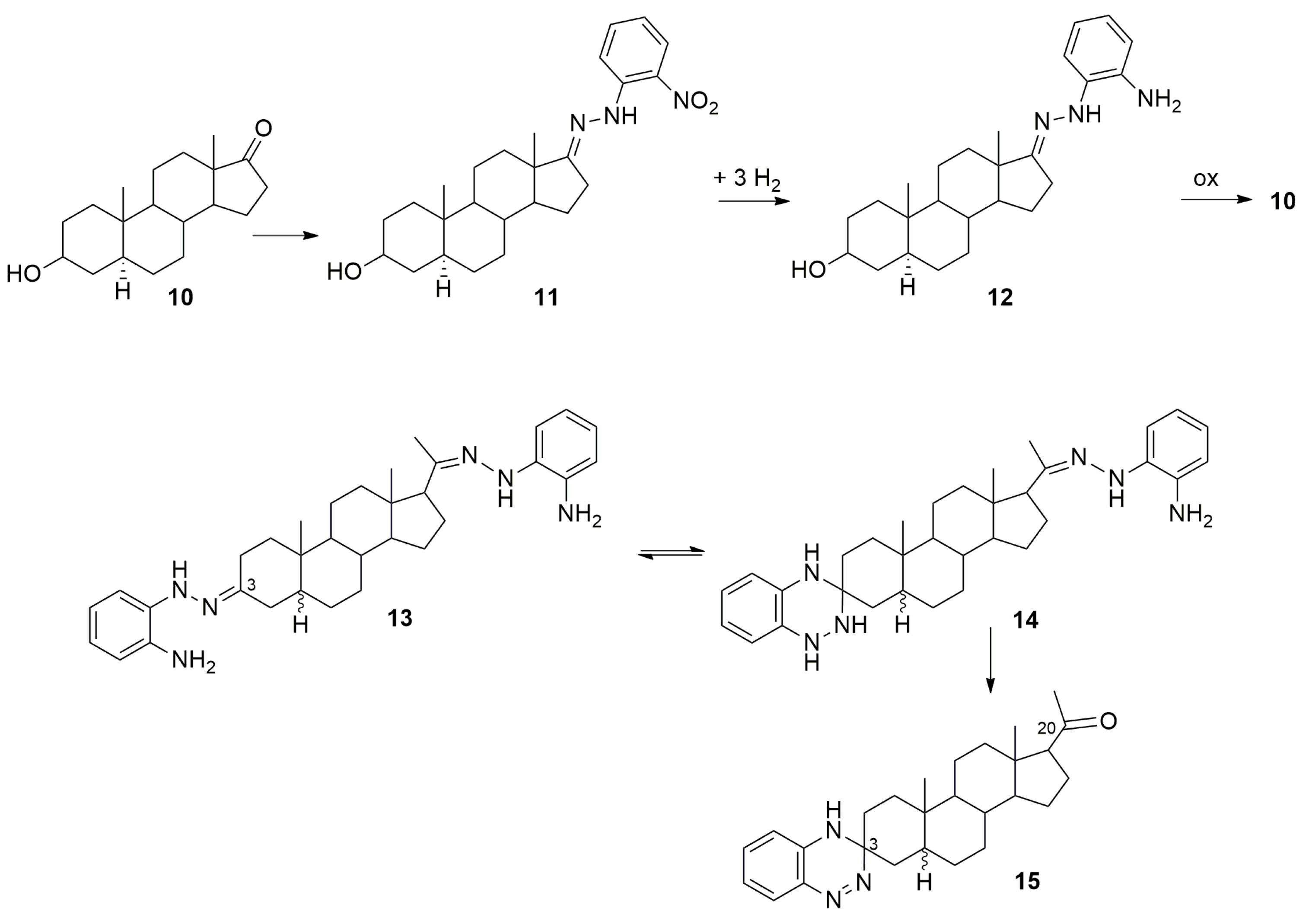
2. Formation of 3,4-Dihydro-(1,2,4)-benzotriazine Derivatives
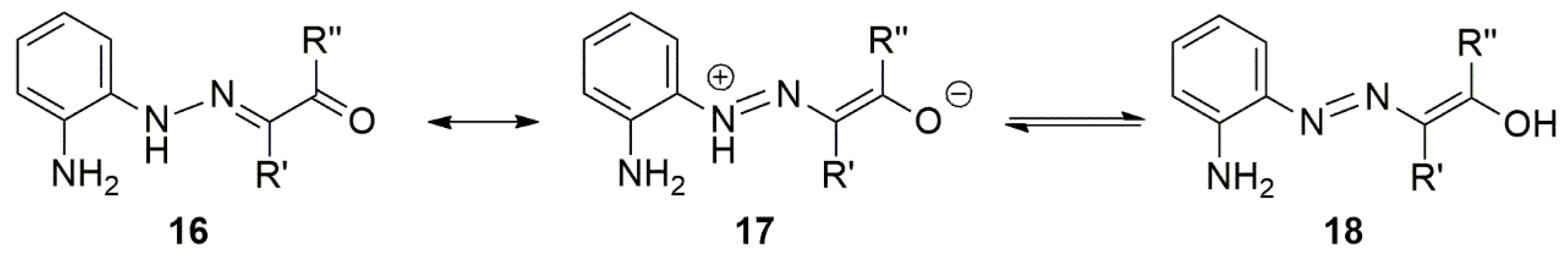
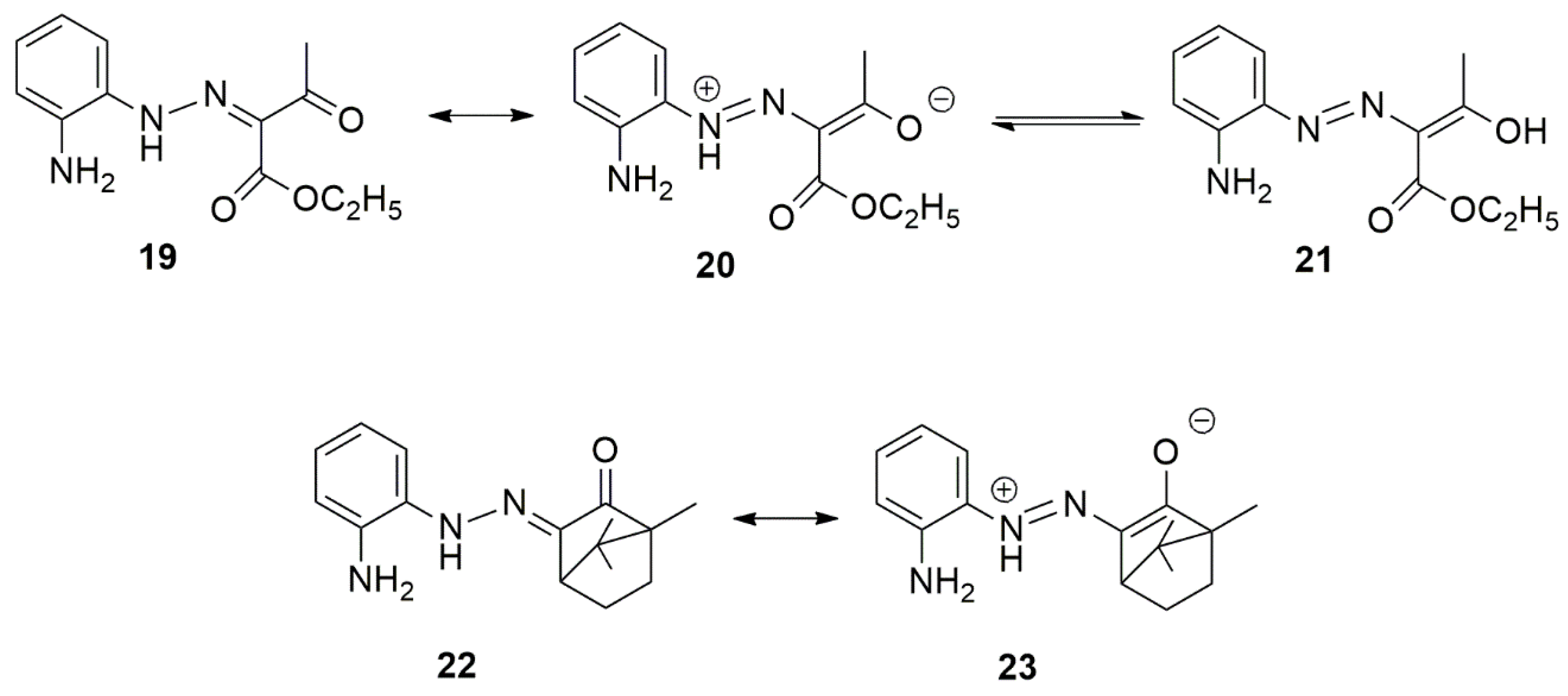
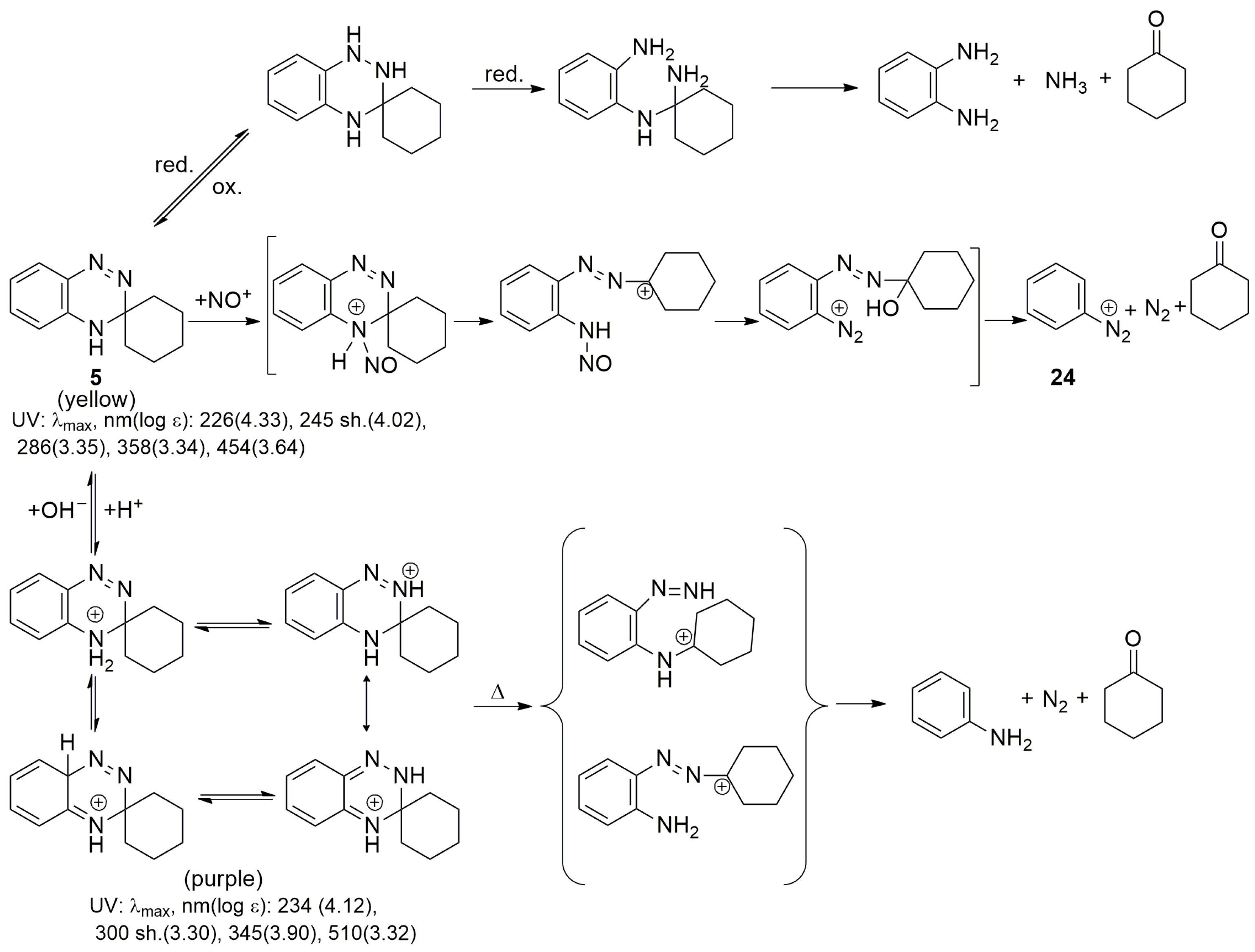
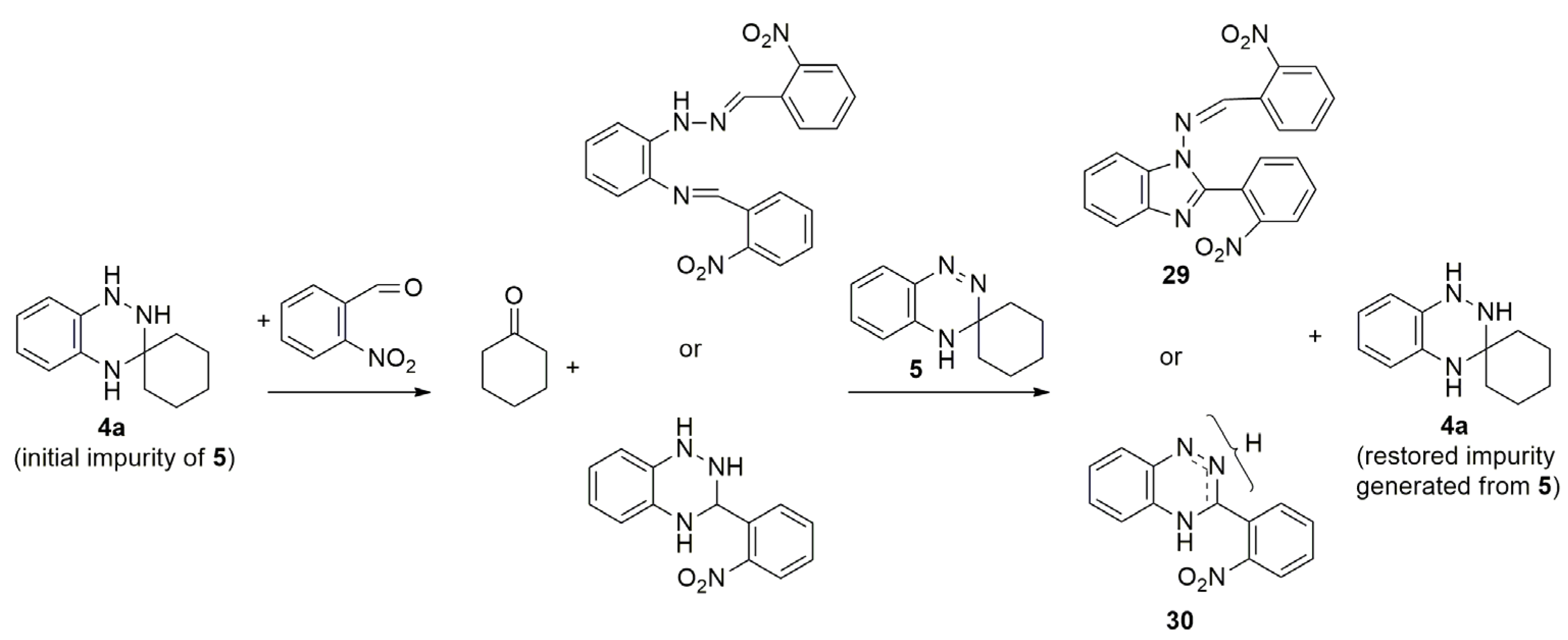
3. Chemical Properties of 3,4-Dihydro-1,2,4-benzotriazines
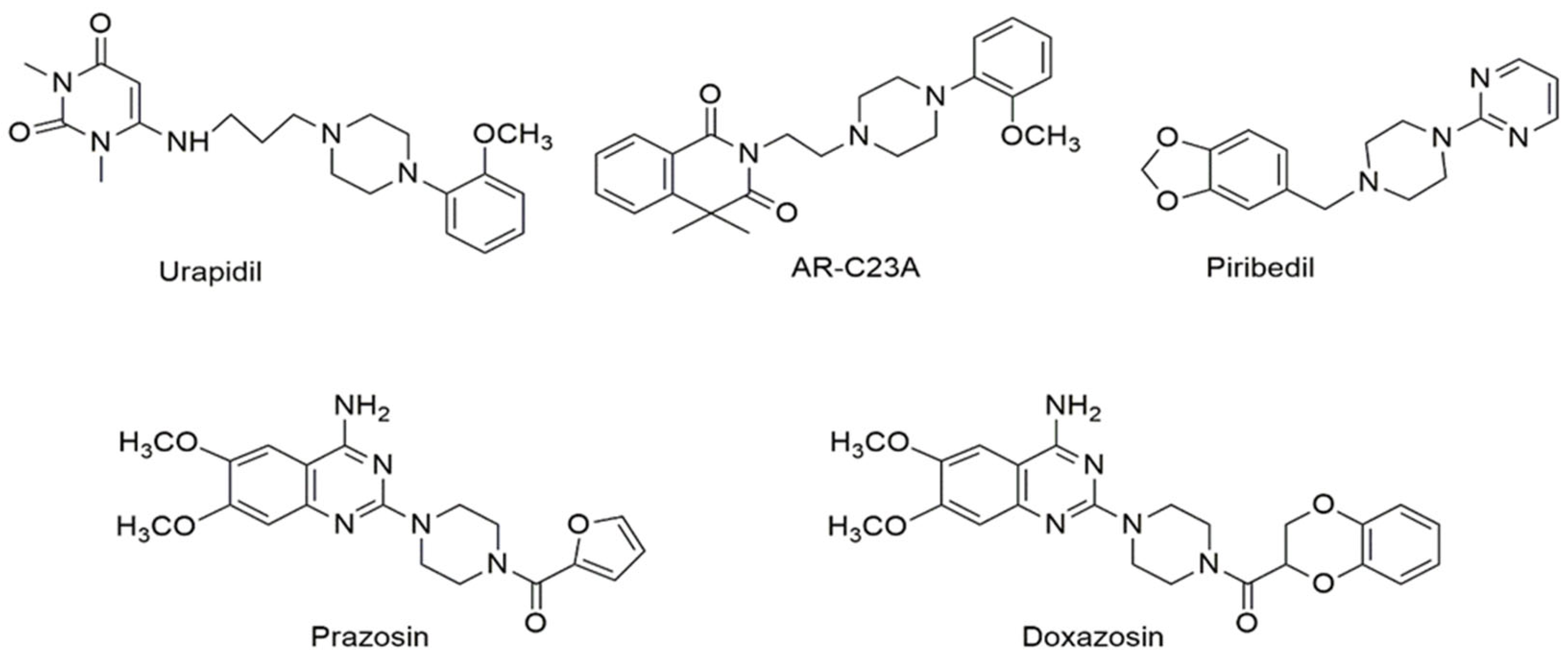
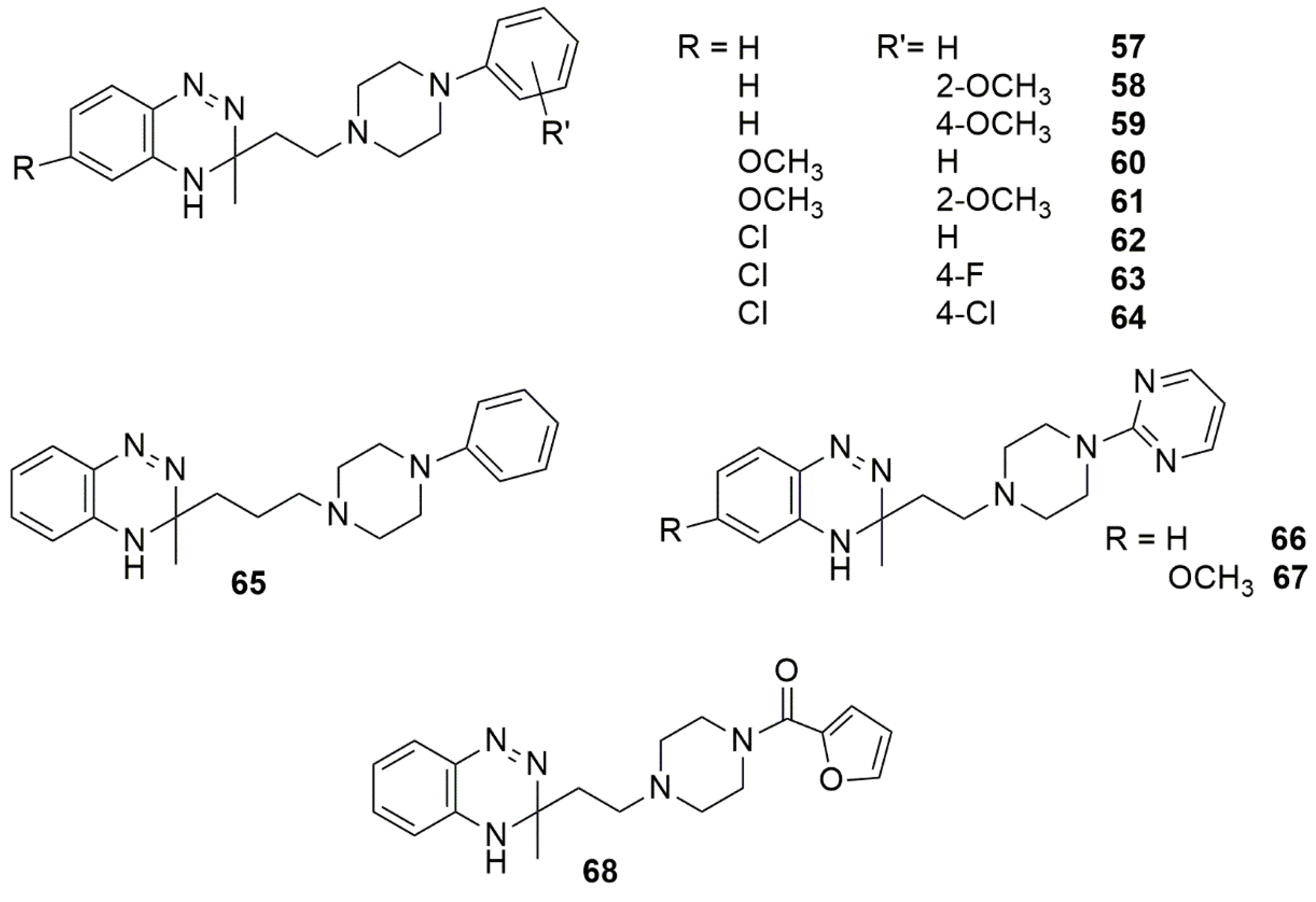
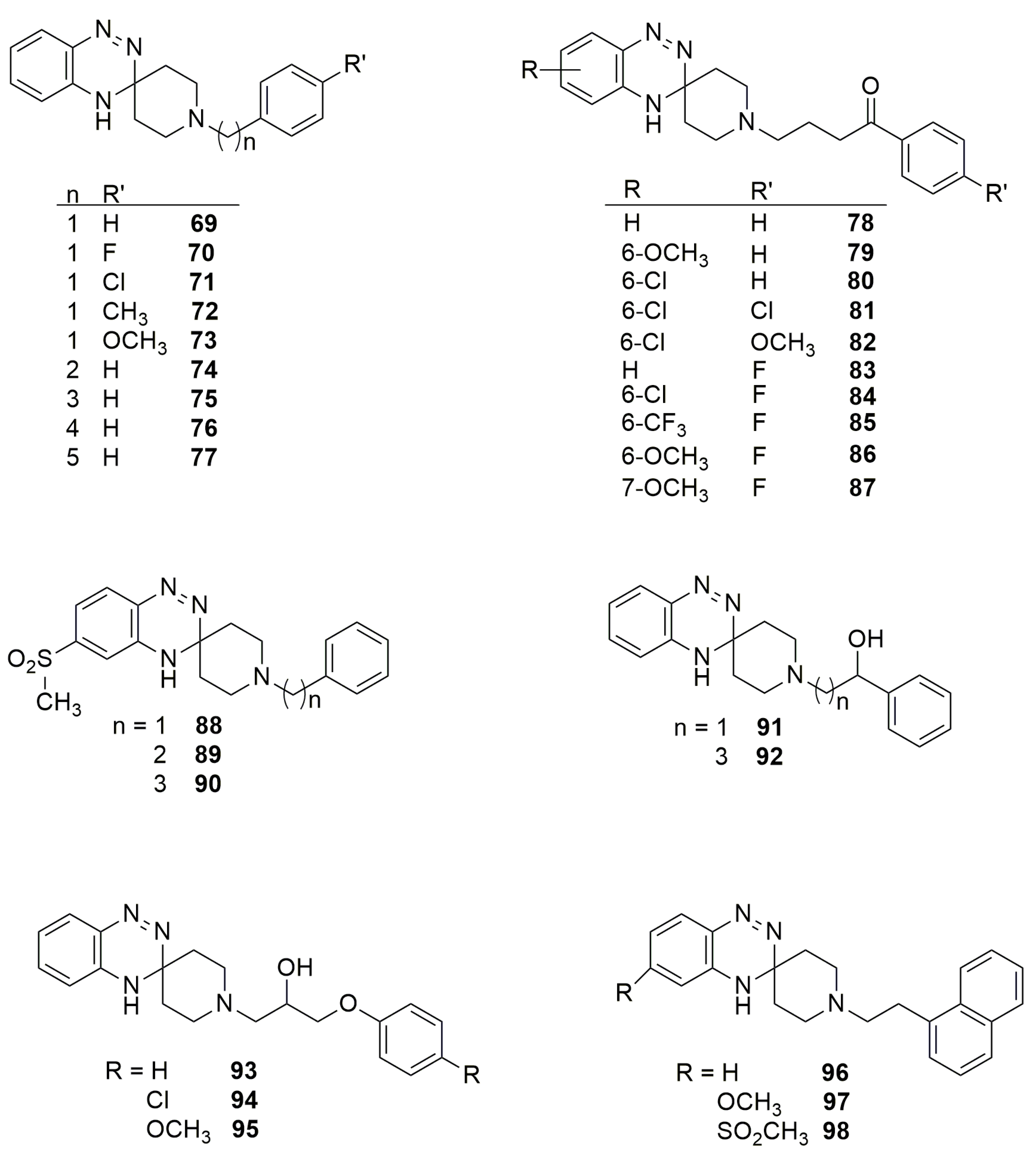
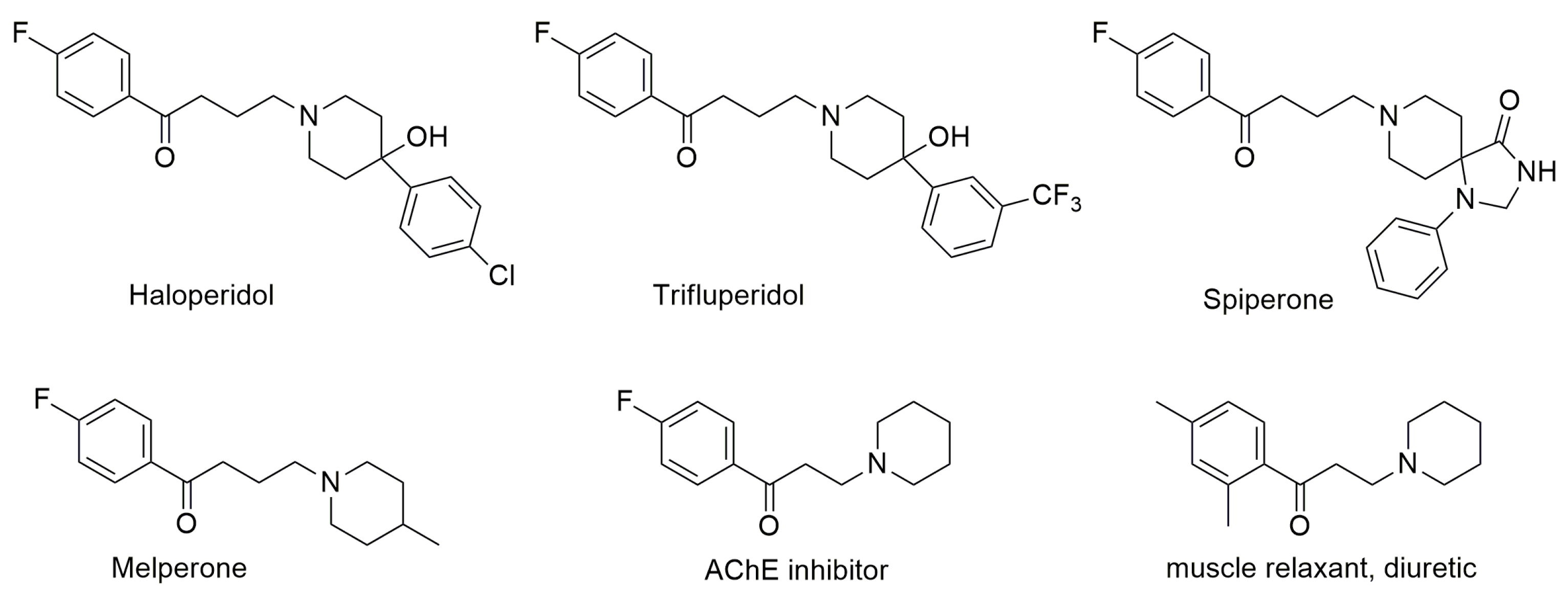
4. Pharmacological Activities
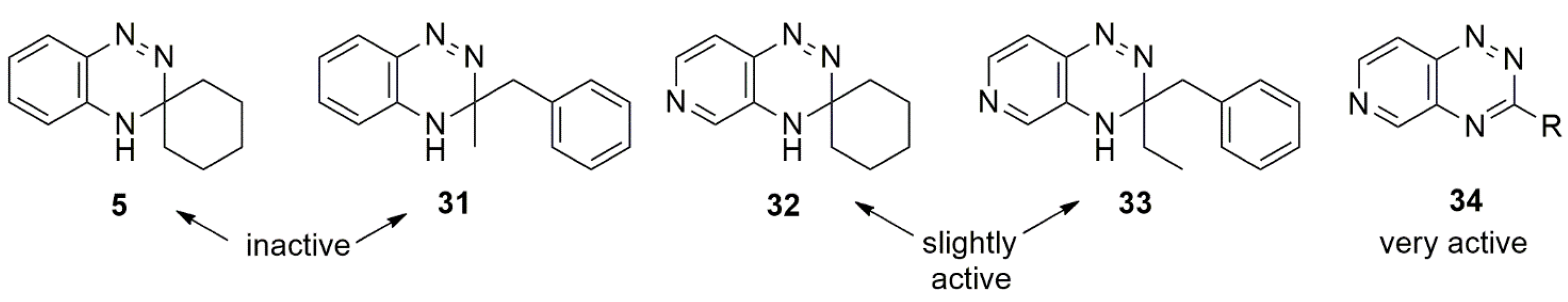
5. Affinity to Sigma Receptors
6. Cytotoxicity and Antiproliferative Activity
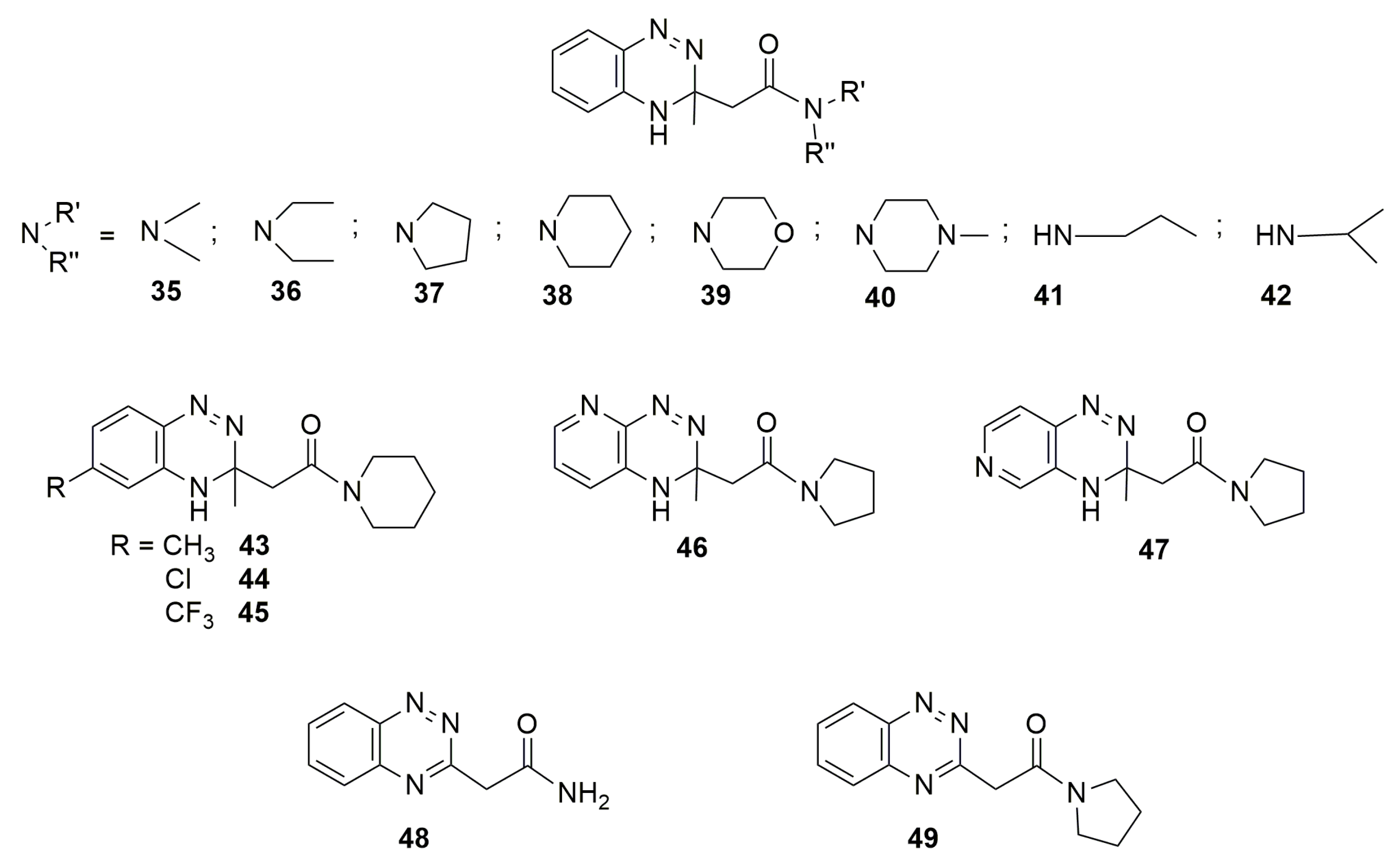
7. Dihydrobenzotriazine Derivatives as Prodrugs: Miscellanea
8. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perkin, W.H., Jr.; Riley, G.C. CCLXXIII—Derivatives of tetrahydrocarbazole. Part IV. J. Chem. Soc. Trans. 1923, 123, 2399–2408. [Google Scholar] [CrossRef]
- Sparatore, F. Ricerche sulle eptatriazine. Nota 1. Riduzione degli o-nitrofenilidrazoni dei ciclanoni. (Heptatriazines. I. Reduction of o-nitrophenylhydrazones of cyclanones). Gazz. Chim. Ital. 1955, 85, 1098–1110. [Google Scholar]
- Sparatore, F.; Cerri, R. Riduzione di o-nitrofenilidrazoni di chetoni. Formazione di derivati 3,4-diidro-benzo-1,2,4-triazinici. (Reduction of o-nitrophenylhydrazones of ketones. Formation of 3,4-dihydro-benzo-1,2,4-triazine derivatives). In Proceedings of the Convegno di Chimica del Farmaco e dei Prodotti Biologicamente Attivi, Roma, Italy, 15–18 December 1969. Book of Abstracts: Abstract N.57. [Google Scholar]
- Sparatore, F.; Cerri, R. Reduction of cyclohexanone 2-nitrophenylhydrazone. Formation of cyclohexane-3-spiro-3,4-dihydro1,2,4-benzotriazine. J. Heterocyclic Chem. 1979, 16, 1001–1003. [Google Scholar] [CrossRef]
- Cerri, R.; Boido, A.; Sparatore, F. Preparazione di 3,4-diidro-1,2,4-benzotriazine-3,3-disostituite. (Preparation of 3,3-disubstituted 3,4-dihydro-1,2,4-benzotriazines). Farmaco Ed. Sci. 1980, 35, 715–724. [Google Scholar]
- Cerri, R.; Boido, A.; Sparatore, F. Oxidation and acid-catalyzed cyclization of aldehyde 2-aminophenylhydrazones. Alternative syntheses for 1,2,4-benzotriazines and benzimidazoles. J. Heterocyclic Chem. 1979, 16, 1005–1008. [Google Scholar] [CrossRef]
- Mohammadi Ziarani, G.; Mostofi, M.; Gholamzadeh, P.; Mohammadi-Khanaposhtani, M.; Yavari, H. The synthesis of 1,2,4-benzotriazines. Arkivoc 2019, 41–105. [Google Scholar] [CrossRef]
- Sparatore, F.; Cerri, R.; Delrio, G.; Rastogi, R.K. Derivati 3,4-diidro-1,2,4-benzotriazin-3,3′-spirosteroidi d′interesse farmacologico. (3,4-Dihydro-1,2,4-benzotriazin-3,3’-spirosteroid derivatives of pharmacological interest). Farmaco Ed. Sci. 1980, 35, 725–734. [Google Scholar]
- Sparatore, F.; Vegetti, P. Ricerche sulle eptatriazine. Nota II. Riduzione dei mono o-nitrofenilidrazoni di composti α-dicarbonilici. (Heptatriazines. II. Reduction of the mono-o-nitrophenylhydrazones of α-dicarbonylic compounds). Gazz. Chim. 1955, 85, 1284–1299. [Google Scholar]
- Sparatore, F.; Ferrarone, F. Riduzione dell’o-nitrofenilidrazone del canfochinone e dell’estere o-nitrofenilazocanfocarbonico. (The reduction of camphorquinone o-nitrophenylhydrazone and ethyl o-nitrophenylazocamphorcarboxylate). Gazz. Chim. 1961, 91, 1282–1293. [Google Scholar]
- Boero, C. Nuovi Derivati Diidrobenzotriazinici (Novel Dihydrobenzotriazine Derivatives). Ph.D. Thesis, University of Genoa, Genova, Italy, 2005. [Google Scholar]
- Palanki, M.S.S.; Cao, J.; Chow, C.P.; Dneprovskaia, E.; Mak, C.C.; McPherson, A.; Pathak, V.P.; Renick, J.; Soll, R.; Zeng, B.; et al. Development of novel benzotriazines for drug discovery. Expert Opin. Drug Discov. 2009, 4, 33–49. [Google Scholar] [CrossRef]
- Kumar, R.; Sirohi, T.S.; Singh, H.; Yadav, R.; Roy, R.K.; Chaudhary, A.; Pandeya, S.N. 1,2,4-triazine analogs as novel class of therapeutic agents. Mini Rev. Med. Chem. 2014, 14, 168–207. [Google Scholar] [CrossRef] [PubMed]
- Misal, S.M.; Gawai, K.R. Azoreductase: A key player of xenobiotic metabolism. Bioresour. Bioprocess. 2018, 5, 17–25. [Google Scholar] [CrossRef]
- Ryan, A. Azoreductases in drug metabolism. Br. J. Pharmacol. 2017, 174, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Reich, M.F.; Fabio, P.F.; Lee, V.J.; Kuck, N.A.; Testa, R.T. Pyrido[3,4-e]-1,2,4-triazines and related heterocycles as potential antifungal agents. J. Med. Chem. 1989, 32, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Metzner, J.; Paintz, M. Analgesic, anti-inflammatory and toxic effects of 3-substituted benzo-1,2,4-triazines. Pharmazie 1981, 36, 714. [Google Scholar] [PubMed]
- Schumann, S.; Mann, G.; Wilde, H.; Ploen, U.; Mager, P.P. Biological effects of 3-substituted acetic acid derivatives of 1,2,4-benzotriazines. Pharmazie 1982, 37, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Boido, A.; Novelli, F.; Savelli, F.; Sparatore, F.; Russo, S.; Filippelli, W.; Susanna, V.; Marmo, E. Synthesis and pharmacological activity of 3-methyl-3,4-dihydrobenzo- and pyrido-1,2,4-triazine-3- acetamides. Farmaco 1989, 44, 279–301. [Google Scholar]
- La Placa, L. Derivati Diidrobenzotiazinici Quali Ligandi per Recettori Sigma e Quali Potenziali Anestetici Locali e Antiaritmici. (Dihydrobenzotriazine Derivatives as Ligands for Sigma Receptors and as Potential Local Anesthetics and Antiarrhythmics). Ph.D. Thesis, University of Genoa, Genova, Italy, 2000. [Google Scholar]
- Caliendo, G.; Di Carlo, R.; Greco, G.; Grieco, P.; Meli, R.; Novellino, E.; Perissutti, E.; Santagada, V. Synthesis, local anesthetic activity and QSAR studies for a set of N-[2-(alkylamino)ethyl]benzotriazol-x-yl acetamides. Eur. J. Med. Chem. 1995, 30, 603–608. [Google Scholar] [CrossRef]
- Vazzana, I.; Boido, A.; Sparatore, F.; Di Carlo, R.; Mattace-Raso, G.; Pacilio, M. Preparation and local anaesthetic activity of N-[2-(tert-amino)ethyl]- and N-(lupinyl)-benzotriazol-1/2-ylacetamides. Farmaco 1997, 52, 131–139. [Google Scholar]
- Tasso, B.; Budriesi, R.; Vazzana, I.; Ioan, P.; Micucci, M.; Novelli, F.; Tonelli, M.; Sparatore, A.; Chiarini, A.; Sparatore, F. Novel Quinolizidinyl Derivatives as Antiarrhythmic Agents: 2. Further Investigation. J. Med. Chem. 2010, 53, 4668–4677. [Google Scholar] [CrossRef]
- Chimirri, A.; De Sarro, A.; De Sarro, G.; Grasso, S.; Trimarchi, G.R.; Zappalà, M. Synthesis and anticonvulsant properties of 2,3,3a,4-tetrahydro-1H-pyrrolo[1,2-a]benzimidazol-1-ones. J. Med. Chem. 1989, 32, 93–95. [Google Scholar] [CrossRef]
- Caliendo, G.; Santagada, V.; Perissutti, E.; Fiorino, F. Derivatives as 5HT1A receptor ligands--past and present. Curr. Med. Chem. 2005, 12, 1721–1753. [Google Scholar] [CrossRef]
- Novelli, F.; Sparatore, F. Synthesis and preliminary pharmacological evaluation of 6-substituted 3-[2-(4-aryl-1-piperazinyl)ethyl]-3,4-dihydro-3-methyl-1,2,4-benzotriazines. Farmaco 1996, 51, 551–558. [Google Scholar]
- Mina, C. Derivati Dihydrobenzotriazinici Attivi sul Sistema Cardiocircolatorio. (Dihydrobenzotriazine Derivatives Active on Cardiocirculatory System). Ph.D. Thesis, University of Genoa, Genova, Italy, 2002. [Google Scholar]
- Novelli, F.; Sparatore, F. Preparation and pharmacological activities of spiro[3,4-dihydro-6/7-R-1,2,4-benzotriazine-3,4′-(1′-substituted)piperidines]. Farmaco 1996, 51, 541–550. [Google Scholar] [CrossRef]
- Platou, E.S.; Refsum, H.; Myhre, E.S.; Amlie, J.P.; Landmark, K. The mode of antiarrhythmic action of melperone. Acta Pharmacol. Toxicol. 1982, 50, 108–112. [Google Scholar] [CrossRef]
- Gul, H.I.; Demirtas, A.; Ucar, G.; Taslimi, P.; Gulcin, I. Synthesis of Mannich Bases by Two Different Methods and Evaluation of their Acetylcholine Esterase and Carbonic Anhydrase Inhibitory Activities. Lett. Drug Des. Discov. 2017, 14, 573–580. [Google Scholar] [CrossRef]
- Chawla, H.P.S.; Gautam, B.C.; Kapil, R.S.; Anand, N.; Patnaik, G.K.; Vohra, O.P.; Shrivastava, M.M. Agents acting on the central nervous system. XII. 3-Tert-aminopropiophenones as central muscle relaxants and diuretics. J. Med. Chem. 1970, 13, 480–488. [Google Scholar] [CrossRef]
- Clark, M.L.; Braun, G.A.; Hewson, J.R.; Serafetinides, E.A.; Colmore, J.P.; Rahhal, D.K. Trifluperidol and cholesterol in man. Clin. Pharmacol. Ther. 1968, 9, 333–340. [Google Scholar] [CrossRef]
- Canfrán-Duque, A.; Casado, M.E.; Pastor, O.; Sánchez-Wandelmer, J.; de la Peña, G.; Lerma, M.; Mariscal, P.; Bracher, F.; Lasunción, M.A.; Busto, R. Atypical antipsychotics alter cholesterol and fatty acid metabolism in vitro. J. Lipid Res. 2013, 54, 310–324. [Google Scholar] [CrossRef]
- Moebius, F.F.; Striessnig, J.; Glossmann, H. The mysteries of sigma receptors: New family members reveal a role in cholesterol synthesis. Trends Pharmacol. Sci. 1997, 18, 67–70. [Google Scholar] [CrossRef]
- Novelli, F.; Sparatore, F. Spiro[1,2,4-benzotriazine-3(4H),4′-(1′-substituted)piperidines] and related compounds as ligands for sigma receptors. Farmaco 2002, 57, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Romano, M. Spiro[3,4-diidro-6-R-1,2,4-benzotriazino-3,x′-azaciclo (o biciclo)alcani] (Spiro[3,4-dihydro-6-R-1,2,4-benzotriazino-3,x′-azacyclo (or bicyclo)alkanes]. Ph.D. Thesis, University of Genoa, Genova, Italy, 1998. [Google Scholar]
- Pergolizzi, J.; Varrassi, G.; Coleman, M.; Breve, F.; Christo, D.K.; Christo, P.J.; Moussa, C. The Sigma Enigma: A Narrative Review of Sigma Receptors. Cureus 2023, 15, e35756. [Google Scholar] [CrossRef] [PubMed]
- Aishwarya, R.; Abdullah, C.S.; Morshed, M.; Remex, N.S.; Bhuiyan, M.S. Sigmar1’s Molecular, Cellular, and Biological Functions in Regulating Cellular Pathophysiology. Front. Physiol. 2021, 12, 705575. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Fallica, A.F.; Pittalà, V.; Modica, M.N.; Salerno, L.; Romeo, G.; Marrazzo, A.; Helal, M.A.; Intagliata, S. Recent Advances in the Development of Sigma Receptor Ligands as Cytotoxic Agents: A Medicinal Chemistry Perspective. J. Med. Chem. 2021, 64, 7926–7962. [Google Scholar] [CrossRef]
- Georgiadis, M.-O.; Karoutzou, O.; Foscolos, A.-S.; Papanastasiou, I. Sigma Receptor (σR) Ligands with Antiproliferative and Anticancer Activity. Molecules 2017, 22, 1408. [Google Scholar] [CrossRef]
- Quaglia, W.; Giannella, M.; Piergentili, A.; Pigini, M.; Brasili, L.; Toro, R.; Rossetti, L.; Spampinato, S.; Melchiorre, C. 1-Benzyl-3,4-dihydrospiro[2H-1-benzothiopyran-2,4′-piperidine] (spipethiane), a potent and highly selective σ1 ligand. J. Med. Chem. 1998, 41, 1557–1560. [Google Scholar] [CrossRef]
- Piergentili, A.; Amantini, C.; Del Bello, F.; Giannella, M.; Mattioli, L.; Palmery, M.; Perfumi, M.; Pigini, M.; Santoni, G.; Tucci, P.; et al. Novel highly potent and selective σ1 receptor antagonists related to spipethiane. J. Med. Chem. 2010, 53, 1261–1269. [Google Scholar] [CrossRef]
- Gilligan, P.J.; Kergaye, A.A.; Lewis, B.M.; McElroy, J.F. Piperidinyl tetralin σ ligands. J. Med. Chem. 1994, 37, 364–370. [Google Scholar] [CrossRef]
- Maier, C.A.; Wünsch, B. Novel spiropiperidines as highly potent and subtype selective σ-receptor ligands. Part 1. J. Med. Chem. 2002, 45, 438–448. [Google Scholar] [CrossRef]
- Maier, C.A.; Wünsch, B. Novel σ receptor ligands. Part 2. SAR of spiro[[2]benzopyran-1,4′-piperidines] and spiro[[2]benzofuran-1,4′-piperidines] with carbon substituents in position 3. J. Med. Chem. 2002, 45, 4923–4930. [Google Scholar] [CrossRef]
- Glennon, R.A.; Ablordeppey, S.Y.; Ismaiel, A.M.; El-Ashmawy, M.B.; Fischer, J.B.; Howie, K.B. Structural Features Important for σ1 Receptor Binding. J. Med. Chem. 1994, 37, 1214–1219. [Google Scholar] [CrossRef]
- Ablordeppey, S.Y.; Fisher, J.B.; Glennon, R.A. Is a nitrogen atom an important pharmacophoric element in sigma ligand binding? Bioorg. Med. Chem. 2000, 8, 2105–2111. [Google Scholar] [CrossRef]
- Guareschi, I. Sulla triacetonammina e suoi derivati alchilici. Atti R. Accad. Sci. Torino 1894, 680–692. [Google Scholar]
- Banert, K.; Fink, K.; Hagedorn, M.; Richter, F. Synthesis of triacetonamine N-alkyl derivatives reinvestigated. ARKIVOC 2012, 3, 379–390. [Google Scholar] [CrossRef]
- Ablordeppey, S.Y.; Fischer, J.B.; Burke Howie, K.J.; Glennon, R.A. Design, synthesis and binding of sigma receptor ligands derived from butaclamol. Med. Chem. Res. 1992, 2, 368–375. [Google Scholar]
- Sparatore, A.; Novelli, F.; Sparatore, F. Quinolizidine derivatives as ligands for sigma receptors. Med. Chem. Res. 2002, 11, 1–11. [Google Scholar]
- Sparatore, A.; Novelli, F.; Sparatore, F. 1-(Arylalkyl)quinolizidine derivatives and thio-isosteric analogs as ligands for sigma receptors. Helv. Chim. Acta 2004, 87, 580–591. [Google Scholar] [CrossRef]
- Berardi, F.; Ferorelli, S.; Colabufo, N.A.; Leopoldo, M.; Perrone, R.; Tortorella, V. A multireceptorial binding reinvestigation on an extended class of σ ligands: N-[ω-(Indan-1-yl and tetralin-1-yl)alkyl] derivatives of 3,3-dimethylpiperidine reveal high affinities towards σ1 and EBP sites. Bioorg. Med. Chem. 2001, 9, 1325–1335. [Google Scholar] [CrossRef]
- Hamama, W.S.; Abd El-Magid, O.M.; Zoorob, H.H. Chemistry of Quinuclidines as Nitrogen Bicyclic Bridged-Ring Structures. J. Heterocyclic Chem. 2006, 43, 1397–1420. [Google Scholar] [CrossRef]
- Peters, D.; Olsen, G.M.; Nielsen, S.F.; Nielsen, E. Spiro-Quinuclidine Derivatives, Their Preparation and Use. U.S. Patent No 6352995, 5 March 2002. [Google Scholar]
- Froestl, W.; Muhs, A.; Pfeifer, A. Cognitive enhancers (nootropics). Part 1: Drugs interacting with receptors. J. Alzheimers Dis. 2012, 32, 793–887. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Vercouillie, J.; Ouach, A.; Mavel, S.; Gulhan, Z.; Chicheri, G.; Jarry, C.; Massip, S.; Deloye, J.-B.; Guilloteau, D.; et al. Design of α7 nicotinic acetylcholine receptor ligands in quinuclidine, tropane and quinazoline series. Chemistry, molecular modeling, radiochemistry, in vitro and in rats evaluations of a [18F] quinuclidine derivative. Eur. J. Med. Chem. 2014, 82, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Neremberg, J.B.; Erb, J.M.; Bergman, J.M.; O’Malley, S.; Chang, R.S.; Scott, A.L.; Broten, T.P.; Bock, M.G. 4-Oxospiro[benzopyran-2,4’-piperidines] as selective α1a-adrenergic receptor antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-S.; Lu, H.-L.; Zhang, L.-J.; Wu, Z. Sigma-2 receptor ligands and their perspectives in cancer diagnosis and therapy. Med. Res. Rev. 2014, 34, 532–566. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, F.; Sparatore, A.; Alama, A.; Novelli, F.; Bruzzo, C.; Sparatore, F. Novel sigma binding site ligands as inhibitors of cell proliferation in breast cancer. Oncology Res. 2003, 13, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Vilner, B.J.; John, C.S.; Bowen, W.D. Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res. 1995, 55, 408–413. [Google Scholar]
- Crawford, K.W.; Bowen, W.D. Sigma-2 receptor agonists activate a novel apoptotic pathway and potentiate antineoplastic drugs in breast tumor cell lines. Cancer Res. 2002, 62, 313–322. [Google Scholar]
- Rupprecht, R. Neuroactive steroids: Mechanisms of action and neuropsychopharmacological properties. Psychoneuroendocrinology 2003, 28, 139–168. [Google Scholar] [CrossRef]
- Reddy, D.S.; Kulkarni, S.K. Development of neurosteroid-based novel psychotropic drugs. Prog. Med. Chem. 2000, 37, 135–175. [Google Scholar] [CrossRef]
- Izzo, N.J.; Xu, J.; Zeng, C.; Kirk, M.J.; Mozzoni, K.; Silky, C.; Rehak, C.; Yurko, R.; Look, G.; Rishton, G.; et al. Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers II: Sigma-2/PGRMC1 Receptors Mediate Abeta 42 Oligomer Binding and Synaptotoxicity. PLoS ONE 2014, 9, e111899. [Google Scholar] [CrossRef]
- Morales-Lázaro, S.L.; González-Ramírez, R.; Rosenbaum, T. Molecular Interplay Between the Sigma-1 Receptor, Steroids, and Ion Channels. Front. Pharmacol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Ojala, K.S.; Ginebaugh, S.P.; Wu, M.; Miller, E.W.; Ortiz, G.; Covarrubias, M.; Meriney, S.D. A high-affinity, partial antagonist effect of 3,4-diaminopyridine mediates action potential broadening and enhancement of transmitter release at NMJs. J. Biol. Chem. 2021, 296, 100302. [Google Scholar] [CrossRef] [PubMed]
- Ceccanti, M.; Libonati, L.; Ruffolo, G.; Cifelli, P.; Moret, F.; Frasca, V.; Palma, E.; Inghilleri, M.; Cambieri, C. Effects of 3,4-diaminopyridine on myasthenia gravis: Preliminary results of an open-label study. Front. Pharmacol. 2022, 13, 982434. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Cintron, E.; Machamer, J.; Ondeck, C.; Pagarigan, K.; Winner, B.; Bodner, P.; Kelly, K.; Pennington, M.R.; McNutt, P. Symptomatic treatment of botulism with a clinically approved small molecule. JCI Insight 2020, 5, e132891. [Google Scholar] [CrossRef]
- Bruno, O. Preparazione di Derivati benzo- e pirido-3,4-diidro-1,2,4-triazinici di Potenziale Interesse Farmacologico. (Preparation of benzo- and pyrido-3,4-dihydro-1,2,4-triazine Derivatives of Potential Pharmacological Interest). Ph.D. Thesis, University of Genoa, Genova, Italy, 1981. [Google Scholar]
- Loiacono, E. Composti Eterociclici di Vario Interesse Farmacologico. (Heterocyclic Compounds Endowed with Various Pharmacological Interest). Ph.D. Thesis, University of Genoa, Genova, Italy, 2006. [Google Scholar]
- Epishina, M.A.; Kulikov, A.S.; Fershtat, L.L. Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines. Molecules 2022, 27, 2575. [Google Scholar] [CrossRef]
- Ji, Y.; Long, L.; Zheng, Y. Recent advances of stable Blatter radicals: Synthesis, properties and applications. Mater. Chem. Front. 2020, 4, 3433–3443. [Google Scholar] [CrossRef]





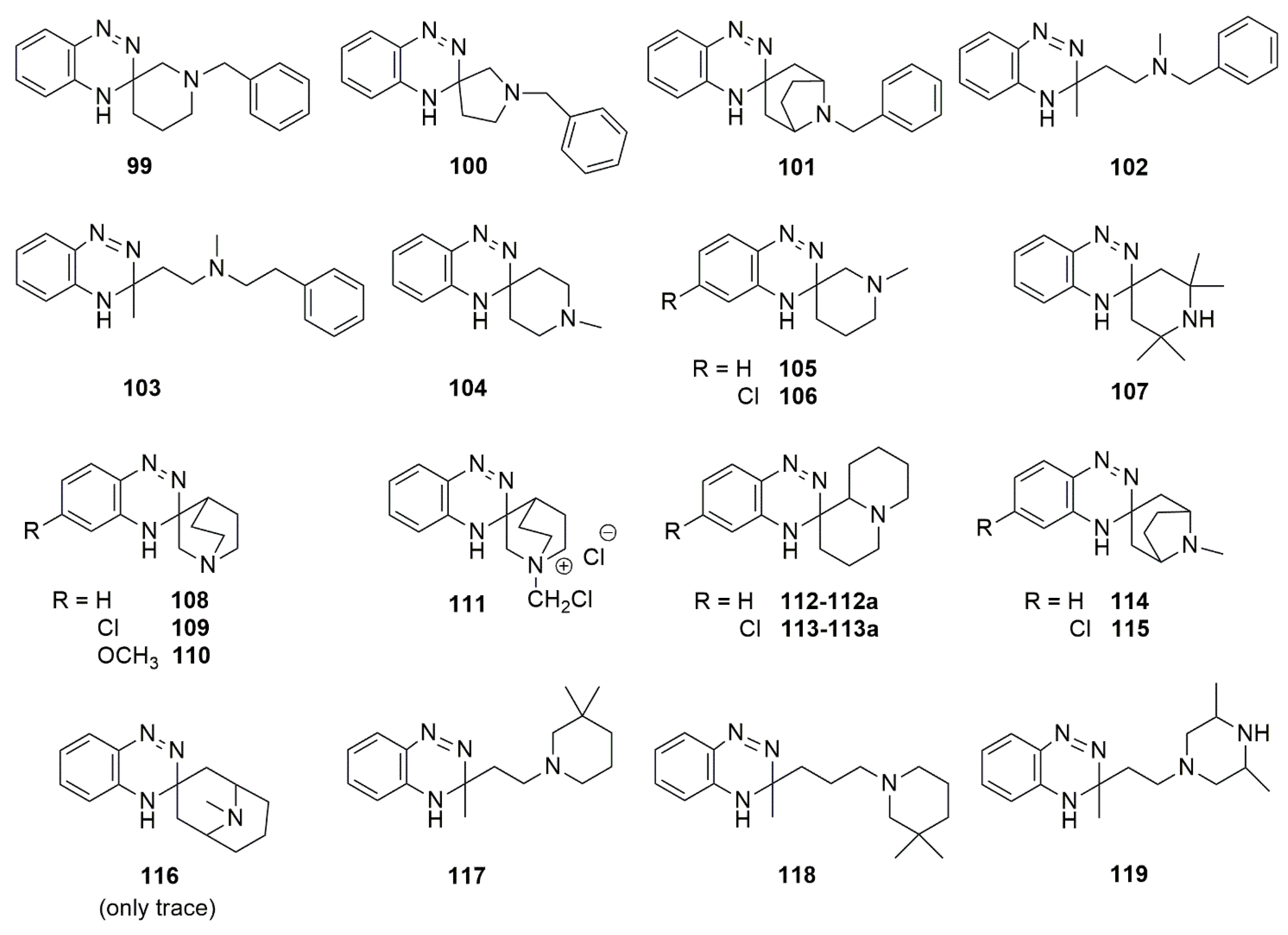
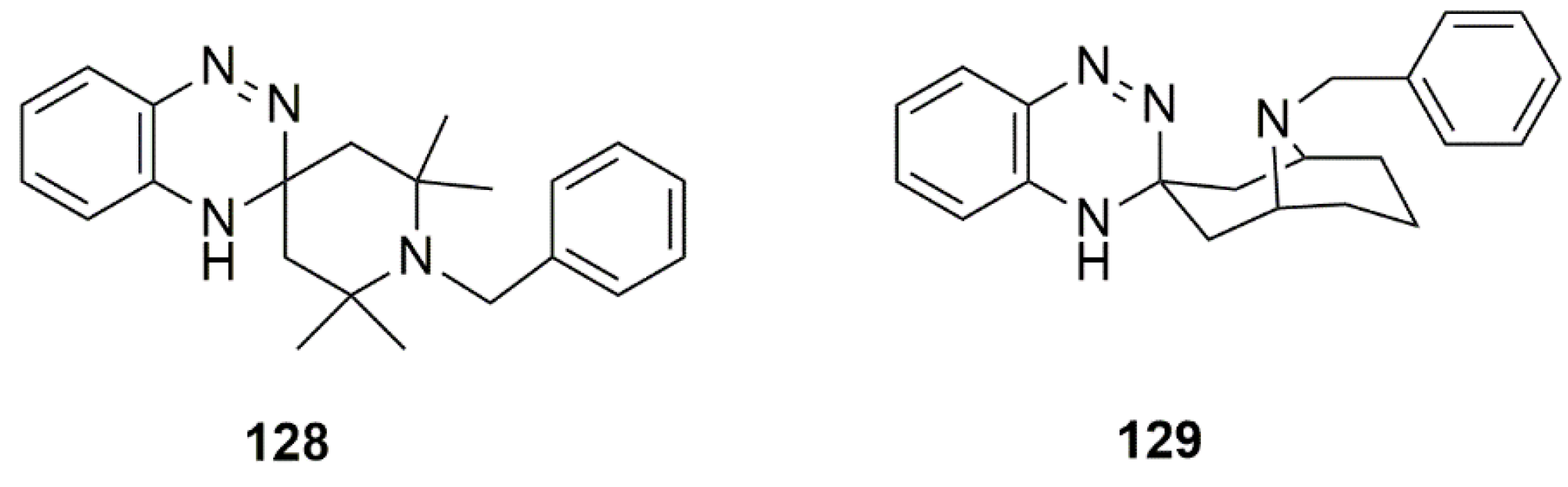
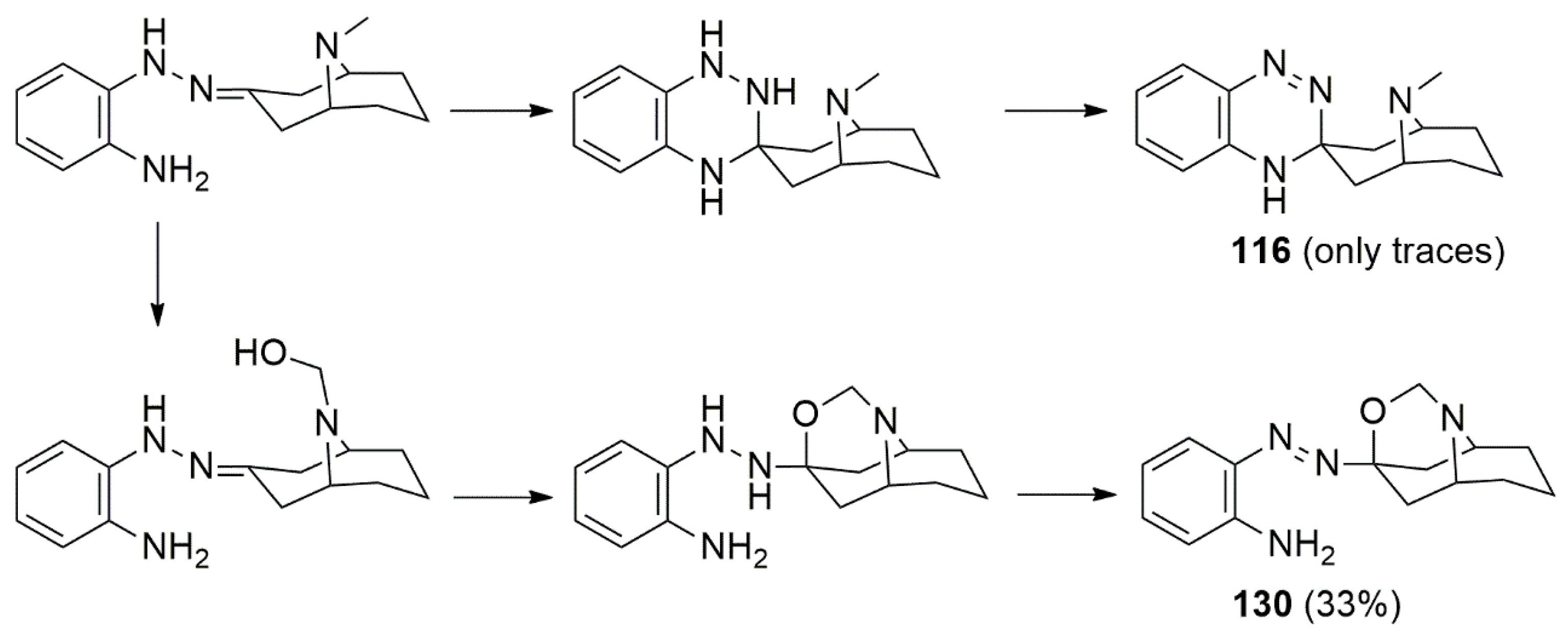
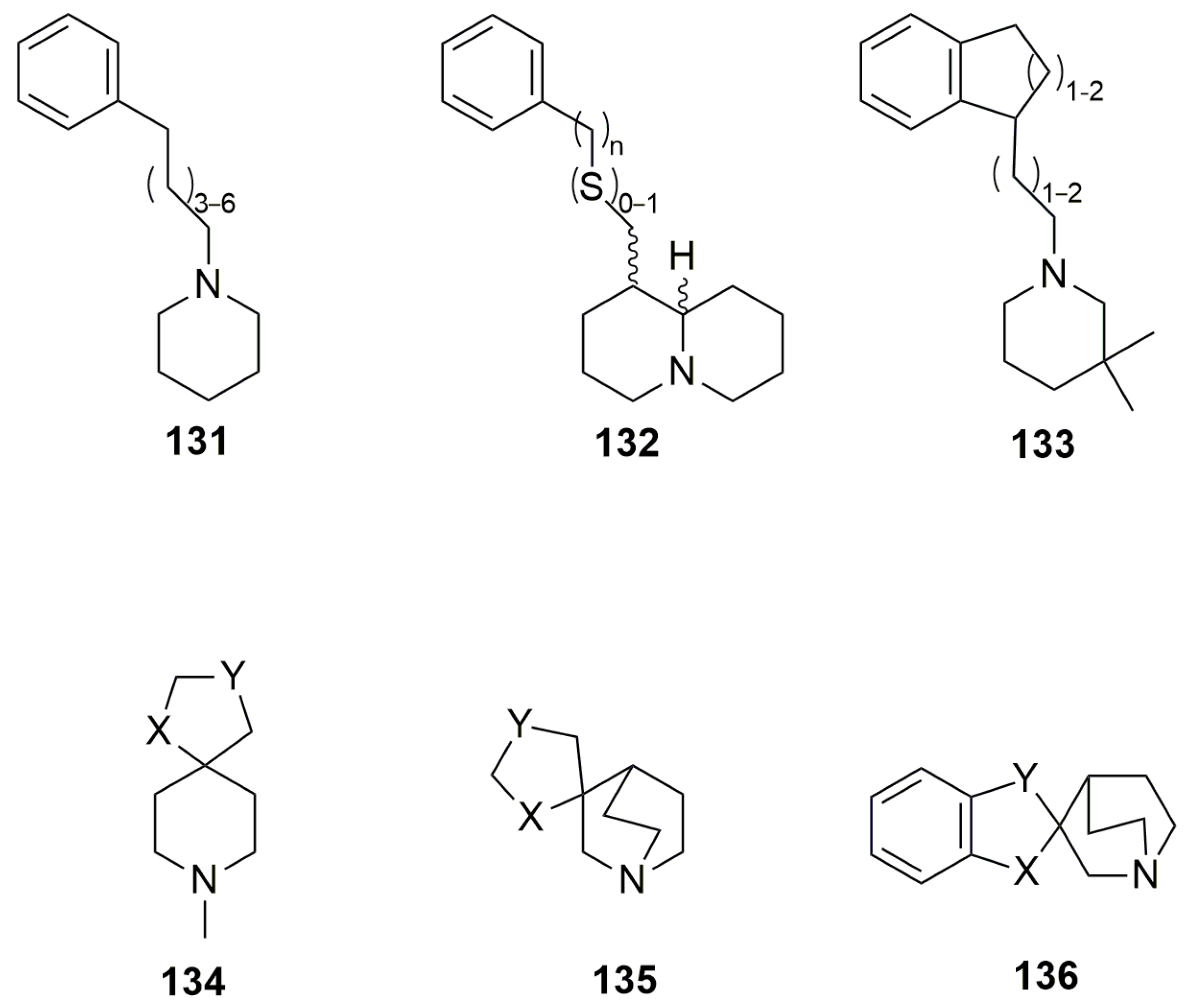


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | Ki (nM) | Ratio | References | ||
| σ1 | σ2 | σ2/σ1 | |||
| 120 (spipethiane) | X = S | 0.50 | 416 | 832 | [42] |
| 121 | O | 0.62 | 22 | 35.5 | [42] |
| 122 | CH2 | 0.44 | 14 | 31.8 | [43] |
| 123 | C=O | 1.40 | 10 | 7.14 | [42,44] |
| 124 | 0.21 | 302 | 1440 | [43] | |
| 125 | R = H | 0.69 | 99.7 | 146 | [45] |
| 126 | OCH3 | 1.20 | 3500 | 2710 | [45] |
| 127 | C≡N | 1.54 | 1590 | 1030 | [46] |
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
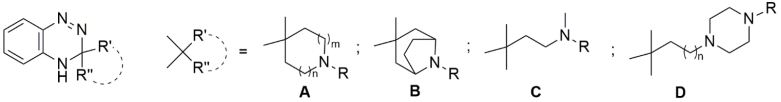
| Compounds | Structure | m | n | R | Ki (nM) | Ratio | |||
| σ1 a | σ2 b | σ2/σ2 | |||||||
| 69 | A | 1 | 1 | CH2-C6H5 | 2.8 0.6 c | 640 4220 | cd | 229 7033 | |
| 70 | A | 1 | 1 | CH2-C6H4-F(4) | 2.8 | ||||
| 71 | A | 1 | 1 | CH2-C6H4-Cl(4) | 1.25 | 346 | 277 | ||
| 72 | e | A | 1 | 1 | CH2-C6H4-CH3(4) | 2.5 | |||
| 73 | A | 1 | 1 | CH2-C6H4-OCH3(4) | 4.0 | ||||
| 74 | A | 1 | 1 | CH2-CH2-C6H5 | 4.0 c | ||||
| 76 | e | A | 1 | 1 | (CH2)4-C6H5 | 3.8 | 43 | 11.3 | |
| 77 | e | A | 1 | 1 | (CH2)5-C6H5 | 0.96 | 52 | 55.3 | |
| 78 | e | A | 1 | 1 | (CH2)3-CO-C6H5 | 14.0 | 277 | 19.8 | |
| 83 | A | 1 | 1 | (CH2)3-CO-C6H4-F(4) | 12.0 c | ||||
| 86 | A f | 1 | 1 | (CH2)3-CO-C6H4-F(4) | 10.8 | 656 | 60.7 | ||
| 99 | A | 0 | 2 | CH2-C6H5 | 10.6 | ||||
| 100 | A | 0 | 1 | CH2-C6H5 | 47.0 | ||||
| 101 | B | CH2-C6H5 | 2.6 | ||||||
| 102 | e | C | CH2-C6H5 | 35.0 | |||||
| 103 | e | C | CH2-CH2-C6H5 | 37.5 | |||||
| 104 | A | 1 | 1 | CH3 | 3410 | ||||
| 57 | D | 1 | C6H5 | 110.0 | |||||
| 65 | D | 2 | C6H5 | 25.0 | |||||
| haloperidol | 6.0 | 59 | 9.8 | ||||||
| haloperidol | c | 2.0 | |||||||
 | |||||
|---|---|---|---|---|---|
| Compounds | R | Ki (nM) | IC50 (µM) | ||
| σ1 | σ2 | MCF-7 | MDA-B231 | ||
| 69 | CH2-C6H5 | 2.8 | 640 | 340 | 148 |
| 76 | (CH2)4-C6H5 | 3.8 | 43 | 83 | 36 |
| 77 | (CH2)5-C6H5 | 0.9 | 52 | 90 | 33 |
| 78 | (CH2)3-CO-C6H5 | 14.0 | 277 | 169 | 85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparatore, F.; Sparatore, A. 3,3-Disubstituted 3,4-Dihydro-1,2,4-benzotriazines: Chemistry, Biological Activity, and Affinity to Sigma Receptors. Molecules 2024, 29, 132. https://doi.org/10.3390/molecules29010132
Sparatore F, Sparatore A. 3,3-Disubstituted 3,4-Dihydro-1,2,4-benzotriazines: Chemistry, Biological Activity, and Affinity to Sigma Receptors. Molecules. 2024; 29(1):132. https://doi.org/10.3390/molecules29010132
Chicago/Turabian StyleSparatore, Fabio, and Anna Sparatore. 2024. "3,3-Disubstituted 3,4-Dihydro-1,2,4-benzotriazines: Chemistry, Biological Activity, and Affinity to Sigma Receptors" Molecules 29, no. 1: 132. https://doi.org/10.3390/molecules29010132
APA StyleSparatore, F., & Sparatore, A. (2024). 3,3-Disubstituted 3,4-Dihydro-1,2,4-benzotriazines: Chemistry, Biological Activity, and Affinity to Sigma Receptors. Molecules, 29(1), 132. https://doi.org/10.3390/molecules29010132