

Synthesis and Structural Characterization of p-Carboranylamidine Derivatives †

 , , and
, , and 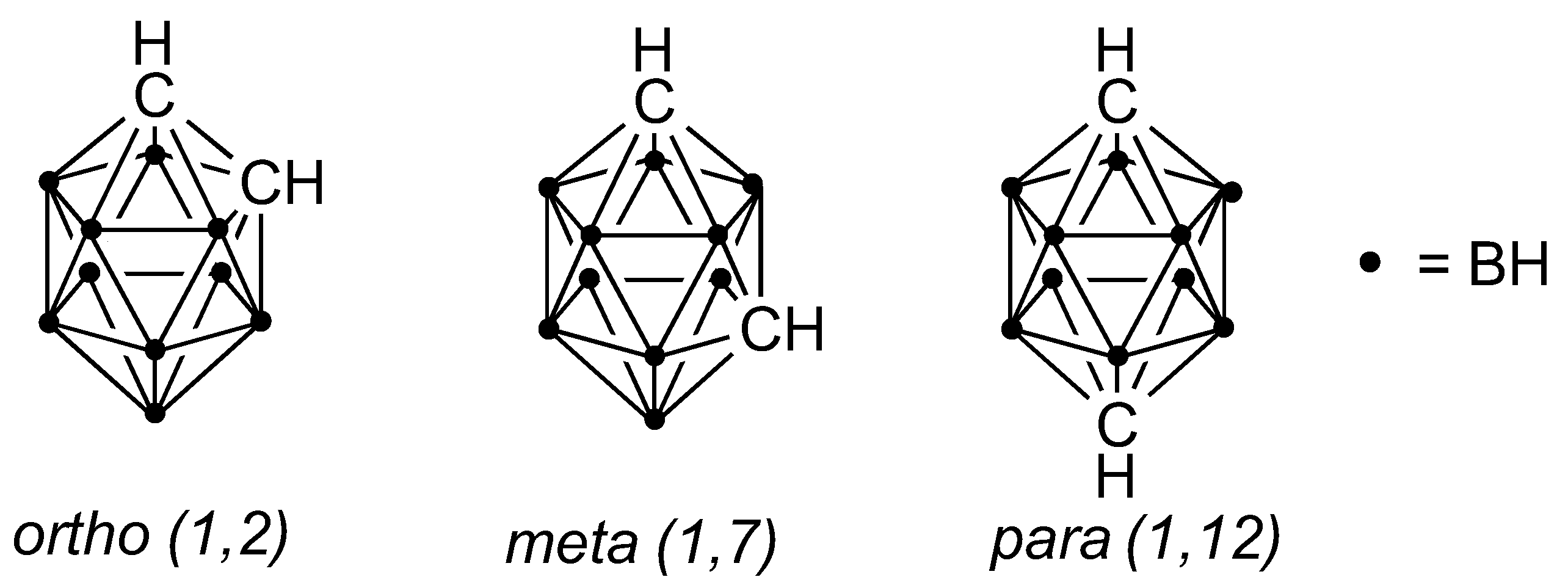{kind=link}
{kind=link}
{kind=link}
{kind=link}
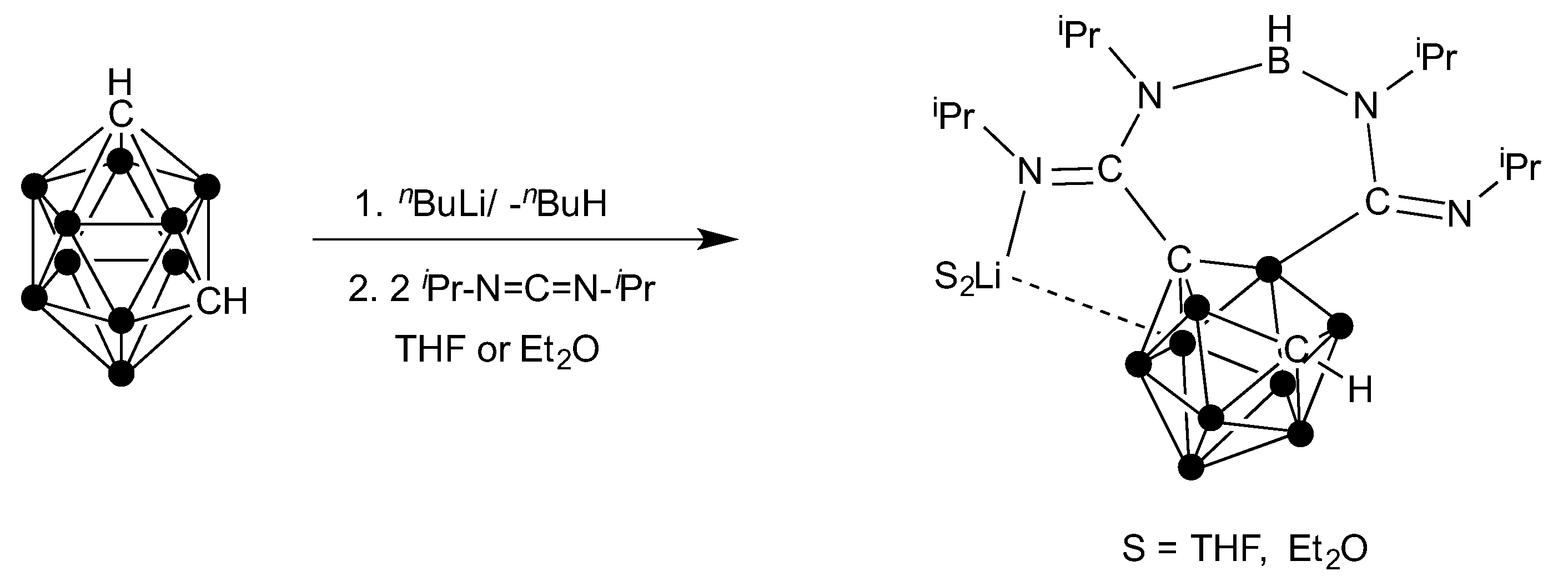{kind=link}
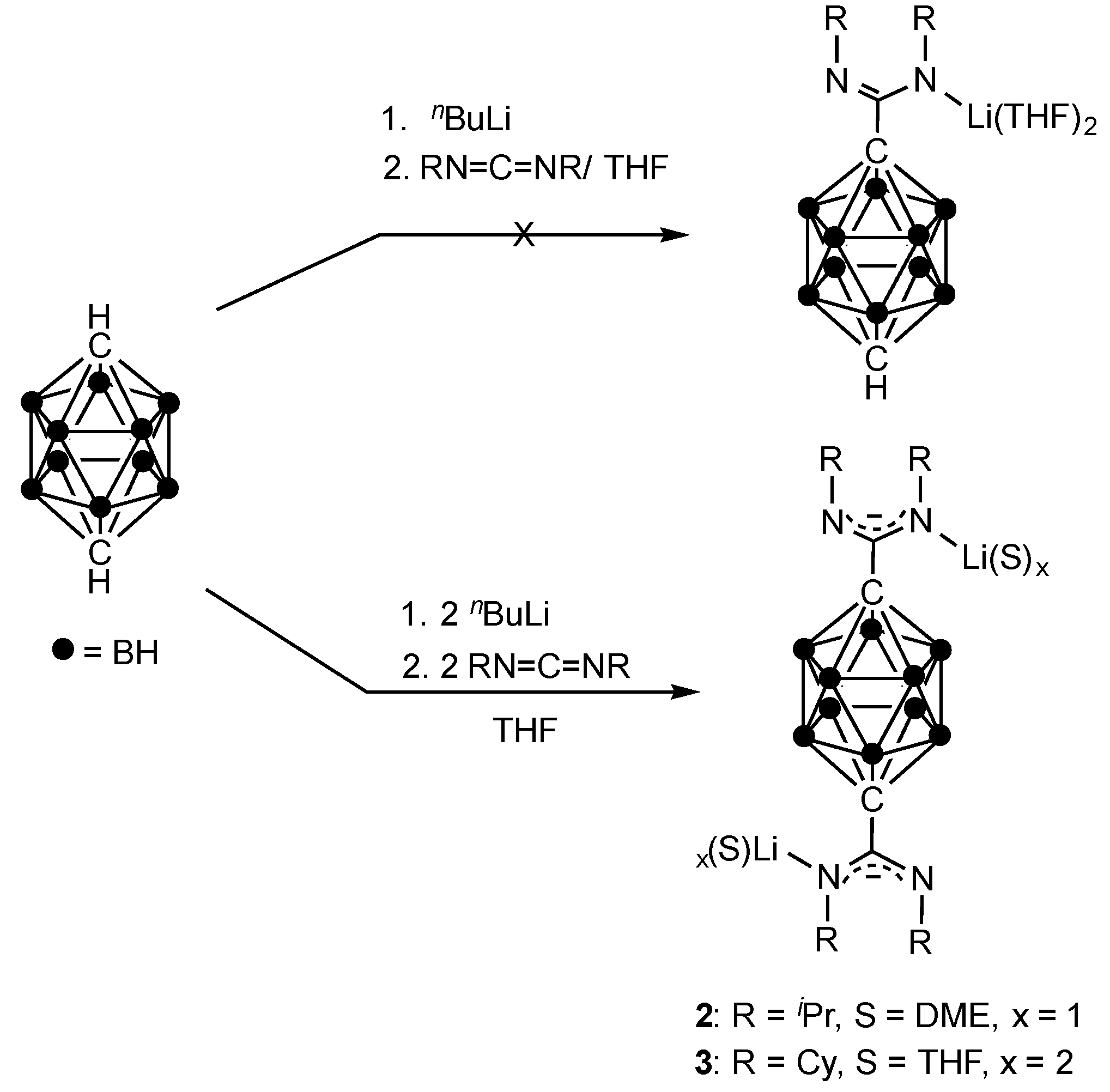{kind=link}
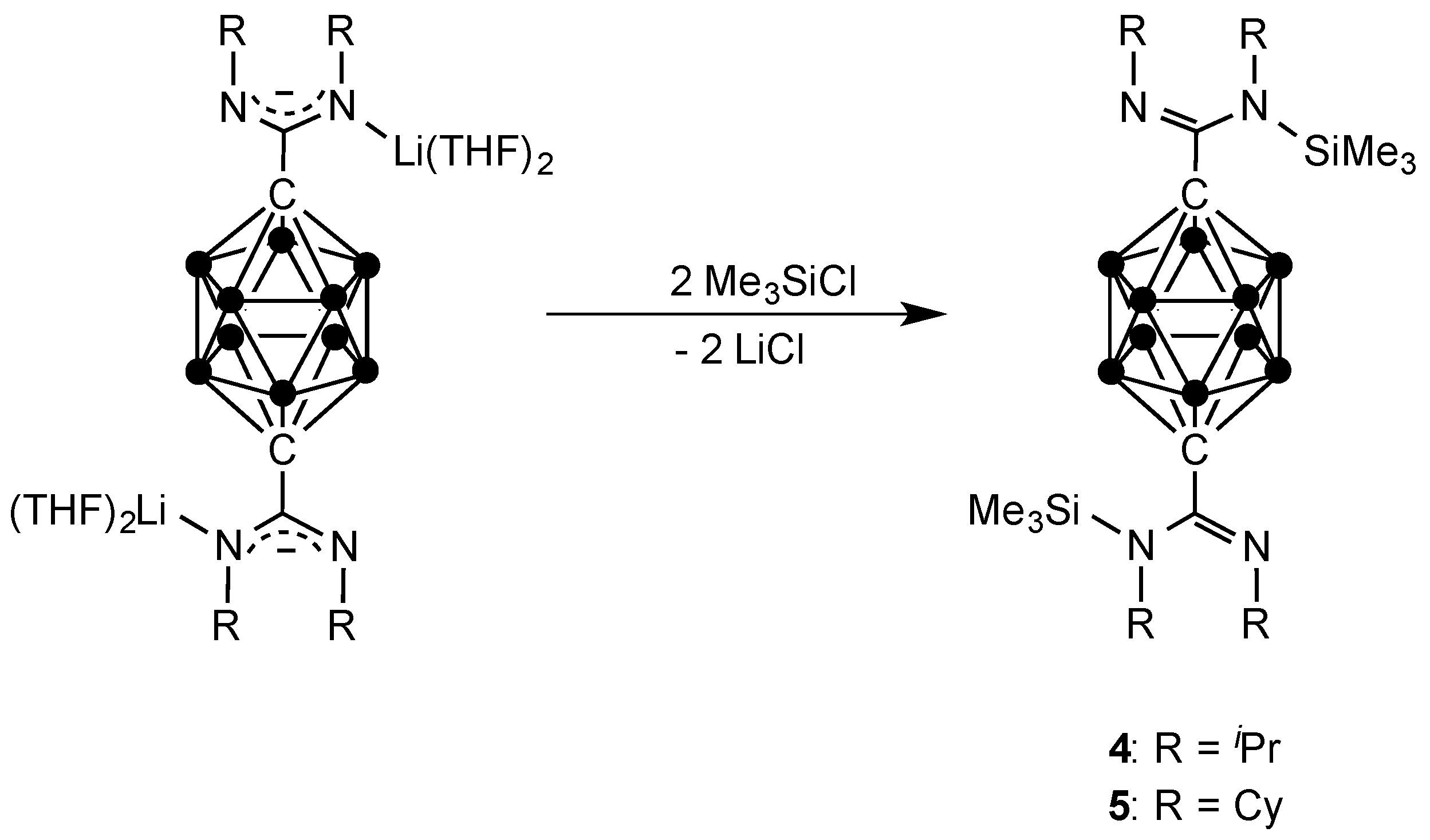{kind=link}
Abstract
1. Introduction
2. Results and Discussion
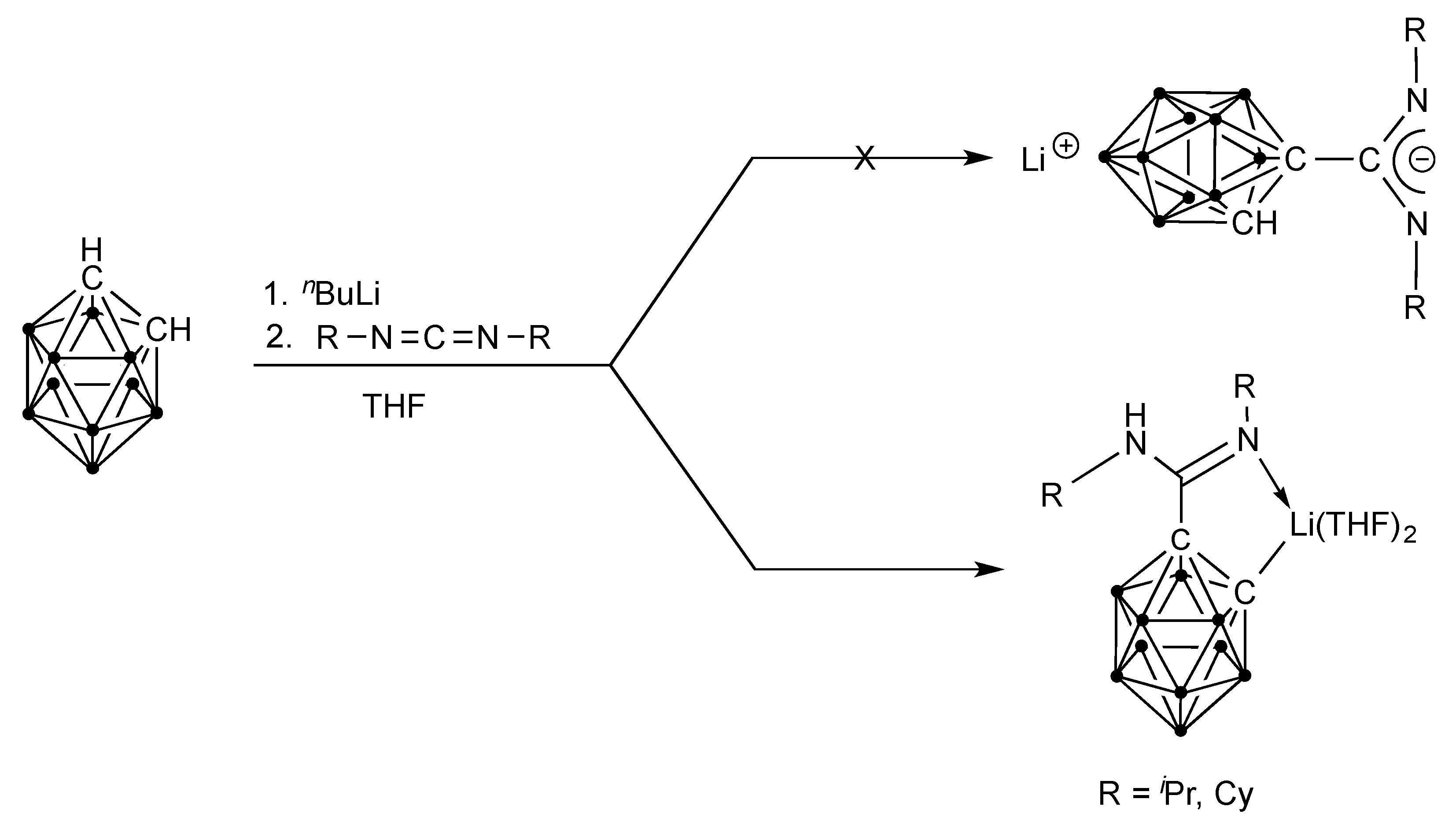
2.1. Synthesis and Characterization
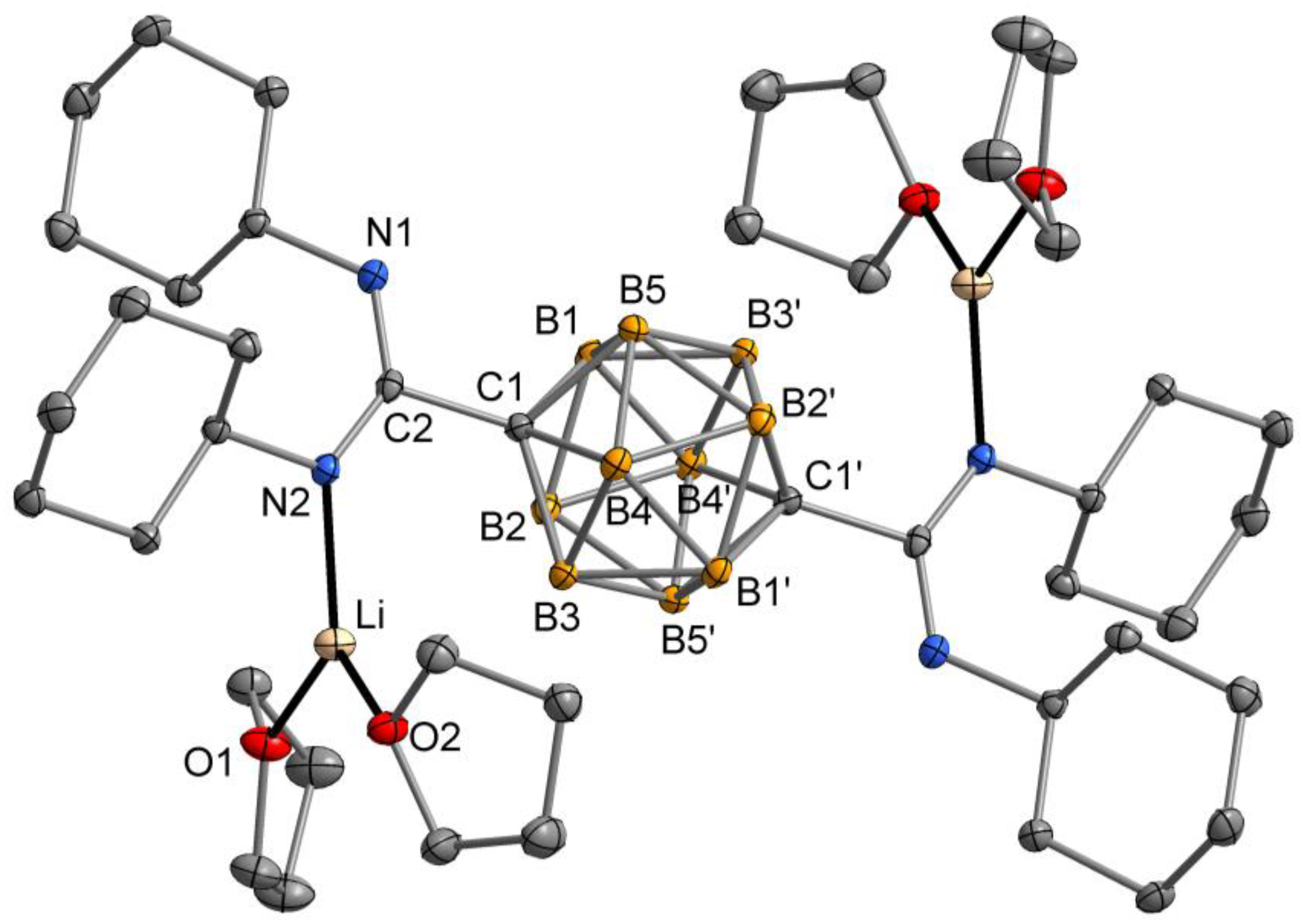
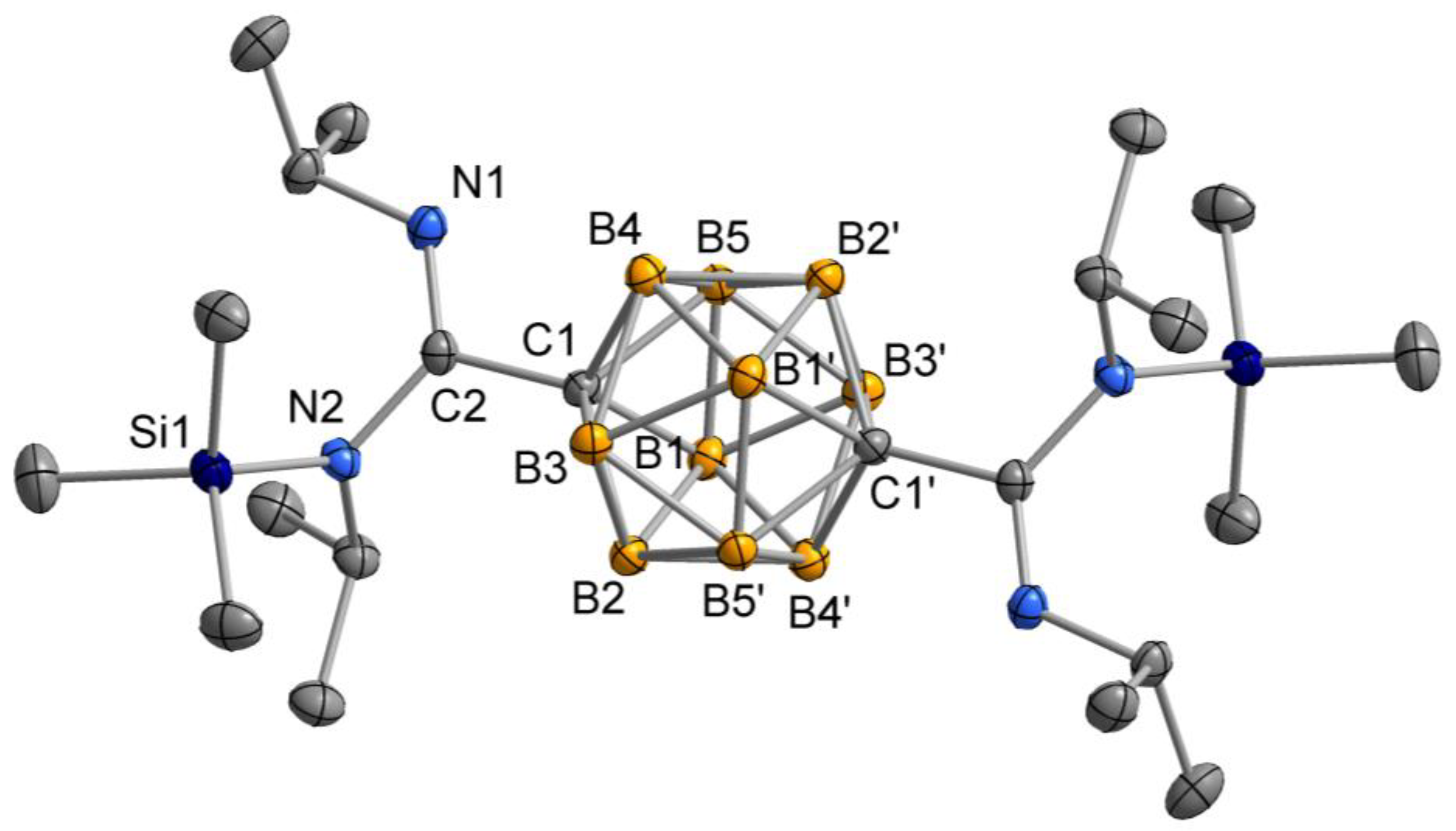
2.2. Crystal and Molecular Structures
3. Experimental Section
3.1. General Procedures and Instrumentation
3.2. Synthesis of Compound p-C2H10B10[C(NiPr)2Li(DME)]2 (2)
3.3. Synthesis of Compound p-C2H10B10[C(NCy)2Li(THF)2]2 (3)
3.4. Synthesis of Compound p-C2H10B10[C(iPrN(SiMe3)(=NiPr)]2 (4)
3.5. Synthesis of Compound p-C2H10B10[C(CyN(SiMe3)(=NCy)]2 (5)
3.6. X-ray Crystallography
4. Conclusions and Future Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. π Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin? Angew. Chem. Int. Ed. 2014, 53, 12191–12195. [Google Scholar] [CrossRef]
- Brown, A.D.; Colquhoun, H.M.; Daniels, A.J.; MacBride, J.A.H.; Stephenson, I.R.; Wade, K. Polymers and ceramics based on icosahedral carboranes. Model studies of the formation and hydrolytic stability of aryl ether, ketone, amide and borane linkages between carborane units. J. Mater. Chem. 1992, 2, 793–804. [Google Scholar] [CrossRef]
- Murphy, D.M.; Mingos, D.M.P.; Haggitt, J.L.; Poell, H.R.; Westcott, S.A.; Marder, T.B.; Taylor, N.J.; Kanis, D.R. Synthesis of icosahedral carboranes for second-harmonic generation. Part 2. J. Mater. Chem. 1993, 3, 139–148. [Google Scholar] [CrossRef]
- Koshino, M.; Tanaka, T.; Solin, N.; Suenaga, K.; Isobe, H.; Nakamura, E. Imaging of Single Organic Molecules in Motion. Science 2007, 316, 853. [Google Scholar] [CrossRef]
- Villagómez, C.J.; Sasaki, T.; Tour, J.M.; Grill, L. Bottom-up Assembly of Molecular Wagons on a Surface. J. Am. Chem. Soc. 2010, 132, 16848–16954. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Gaillard, E.R.; Maguire, J.A.; Hosmane, N.S. Synthesis and Properties of Carborane-Appended C3-Symmetrical Extended π Systems. J. Am. Chem. Soc. 2010, 132, 6578–6587. [Google Scholar] [CrossRef]
- Bauduin, P.; Prevost, S.; Farràs, P.; Teixidor, F.; Diat, O.; Zemb, T. A Theta-Shaped Amphiphilic Cobaltabisdicarbollide Anion: Transition From Monolayer Vesicles to Micelles. Angew. Chem. Int. Ed. 2011, 50, 52998–55300. [Google Scholar]
- Cioran, A.M.; Musteti, A.D.; Teixidor, F.; Krpetic, Ž.Č.; Prior, I.A.; He, Q.; Kiely, C.J.; Brust, M.; Viñas, C. Mercaptocarborane-Capped Gold Nanoparticles: Electron Pools and Ion Traps with Switchable Hydrophilicity. J. Am. Chem. Soc. 2012, 134, 212–221. [Google Scholar] [CrossRef]
- Schwartz, J.J.; Mendoza, A.M.; Wattanatorn, N.; Zhao, Y.; Nguyen, V.T.; Spokoyny, A.M.; Mirkin, C.A.; Baše, T.; Weisse, P.S. Surface Dipole Control of Liquid Crystal Alignment. J. Am. Chem. Soc. 2016, 138, 5957–5967. [Google Scholar] [CrossRef]
- Li, Z.; Núñes, R.; Light, M.E.; Ruiz, E.; Teixidor, F.; Viñas, C.; Ruiz-Molina, D.; Roscini, C.; Planas, J.G. Water-Stable Carborane-Based Eu3+/Tb3+ Metal–Organic Frameworks for Tunable Time-Dependent Emission Color and Their Application in Anticounterfeiting Bar-Coding. Chem. Mater. 2022, 34, 4795–4808. [Google Scholar] [CrossRef]
- Zhang, K.; Song, R.; Qi, J.; Zhang, Z.; Yu, C.; Li, K.; Zhang, Z.; Li, B. Colossal Barocaloric Effect in Carboranes as a Performance Tradeoff. Adv. Funct. Mater. 2022, 32, 2112622. [Google Scholar] [CrossRef]
- Minnyaylo, E.O.; Kudryavtseva, A.I.; Zubova, V.Y.; Anisimov, A.A.; Zaitsev, A.V.; Ol’shevskaya, V.A.; Dolgushin, F.M.; Peregudov, A.S.; Muzafarov, A.M. Synthesis of mono- and polyfunctional organosilicon derivatives of polyhedral carboranes for the preparation of hybrid polymer materials. New J. Chem. 2022, 46, 11143–11148. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, J.; Shi, Q.; Ding, L.; Kiu, T.; Fang, Y. Precise Manipulation of Excited State Intramolecular Proton Transfer via Incorporating Charge Transfer toward High-Performance Film-Based Fluorescence Sensing. J. Am. Chem. Soc. 2023, 145, 7408–7415. [Google Scholar] [CrossRef]
- Belmont, J.A.; Soto, J.; King III, R.E.; Donaldson, A.J.; Hewes, J.D.; Hawthorne, M.F. Metallacarboranes in catalysis. 8. I: Catalytic hydrogenolysis of alkenyl acetates. II: Catalytic alkene isomerization and hydrogenation revisited. J. Am. Chem. Soc. 1989, 111, 7475–7486. [Google Scholar] [CrossRef]
- Teixidor, F.; Flores, M.A.; Viñas, C.; Kivekäs, R.; Sillanpää, R. [Rh(7-SPh-8-Me-7,8-C2B9H10)(PPh3)2]: A New Rhodacarborane with Enhanced Activity in the Hydrogenation of 1-Alkenes. Angew. Chem. Int. Ed. 1996, 35, 2251–2253. [Google Scholar] [CrossRef]
- Ferlekidis, A.; Goblet-Stachow, M.; Liégeois, J.F.; Pirotte, B.; Delarge, J.; Demonceau, A.; Fontaine, M.; Noels, A.F.; Chizhevsky, I.T.; Zinevich, T.V.; et al. Ligand effects in the hydrogenation of methacycline to doxycycline and epi-doxycycline catalysed by rhodium complexes molecular structure of the key catalyst [closo-3,3-(η2,3-C7H7CH2)-3,1,2-RhC2B9H11]. J. Organomet. Chem. 1997, 536–537, 405–412. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Half- and Mixed-sandwich Metallacarboranes in Catalysis. In Boron Chemistry in Organometallics, Catalysis, Materials and Medicine; Hosmane, N.S., Eagling, R., Eds.; World Scientific: Singapore, 2018; pp. 27–80. [Google Scholar]
- Fisher, S.P.; Tomich, A.W.; Lovera, S.O.; Kleinsasser, J.F.; Guo, J.; Asay, M.J.; Nelson, H.M.; Lavallo, V. Nonclassical Applications of closo-Carborane Anions: From Main Group Chemistry and Catalysis to Energy Storage. Chem. Rev. 2019, 119, 8262–8290. [Google Scholar] [CrossRef]
- Gunther, S.O.; Lai, Q.; Senecal, T.; Huacuja, R.; Bremer, S.; Pearson, D.M.; DeMott, J.C.; Bhuvanesh, N.; Ozerov, O.V.; Klosin, J. Highly Efficient Carborane-Based Activators for Molecular Olefin Polymerization Catalysts. ACS Catal. 2021, 11, 3335–3342. [Google Scholar] [CrossRef]
- Grishin, I.V.; Zimina, A.M.; Anufriev, S.A.; Knyazeva, N.A.; Piskunov, A.V.; Dolgushin, F.M.; Sivaev, I.B. Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2−Dicarbollide Ligands. Catalysts 2021, 11, 1409. [Google Scholar] [CrossRef]
- Cheng, R.; Zhang, J.; Zhang, H.; Qiu, Z.; Xie, Z. Ir-catalyzed enantioselective B–H alkenylation for asymmetric synthesis of chiral-at-cage o-carboranes. Nat. Commun. 2021, 12, 7146. [Google Scholar] [CrossRef]
- Vaillant, J.F.; Guenther, K.J.; King, A.S.; Morel, P.; Schaffer, P.; Sogbein, O.O.; Stephenson, K. The medicinal chemistry of carboranes. Coord. Chem. Rev. 2002, 232, 173–230. [Google Scholar] [CrossRef]
- Armstrong, A.F.; Vaillant, J.F. The bioinorganic and medicinal chemistry of carboranes: From new drug discovery to molecular imaging and therapy. Dalton Trans. 2007, 38, 4240–4251. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as Pharmacophores: Properties, Synthesis, and Application Strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sarosi, M.-B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef]
- Gozzi, M.; Schwarze, B.; Hey-Hawkins, E. Preparing (Metalla)carboranes for Nanomedicine. ChemMedChem 2021, 16, 1533–1565. [Google Scholar] [CrossRef]
- Marfavi, A.; Kavianpour, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef]
- Chen, Y.; Du, F.; Tang, L.; Xu, J.; Zhao, Y.; Wu, X.; Li, M.; Shen, J.; Wen, Q.; Cho, C.H.; et al. Carboranes as unique pharmacophores in antitumor medicinal chemistry. Mol. Ther. Oncolytics 2022, 24, 400–416. [Google Scholar] [CrossRef]
- Waddington, M.A.; Zheng, X.; Stauber, J.M.; Moully, E.H.; Montgomery, H.R.; Saleh, L.M.A.; Král, P.; Spokoyny, A.M. An Organometallic Strategy for Cysteine borylation. J. Am. Chem. Soc. 2021, 143, 8661–8668. [Google Scholar] [CrossRef]
- Gazvoda, M.; Dhanjee, H.H.; Rodriguez, J.; Brown, J.S.; Farquhar, C.E.; Truex, N.L.; Loas, A.; Buchwald, S.L.; Pentelute, B.L. Palladium-Mediated Incorporation of Carboranes into Small Molecules, Peptides, and Proteins. J. Am. Chem. Soc. 2022, 144, 7852–7860. [Google Scholar] [CrossRef]
- Ma, Y.-N.; Gao, Y.; Ma, Y.; Wang, Y.; Ren, H.; Chen, X. Palladium-Catalyzed Regioselective B(9)-Amination of o-Carboranes and m-Carboranes in HFIP with Broad Nitrogen sources. J. Am. Chem. Soc. 2022, 144, 8371–8378. [Google Scholar] [CrossRef]
- Ma, Y.-N.; Ren, H.; Wu, Y.; Li, N.; Chen, F.; Chen, X. B(9)-OH-o-Carboranes: Synthesis, Mechanism, and Property exploration. J. Am. Chem. Soc. 2023, 145, 7331–7342. [Google Scholar] [CrossRef]
- Ren, H.; Zhang, P.; Xu, J.; Ma, W.; Tu, D.; Lu, C.; Yan, H. Direct B–H Functionalization of icosahedral Carboranes via Hydrogen Atom Transfer. J. Am. Chem. Soc. 2023, 145, 7638–7647. [Google Scholar] [CrossRef]
- Bregadze, V. Dicarba-closo-dodecaboranes C2B10H12 and their derivatives. Chem. Rev. 1992, 92, 209–223. [Google Scholar] [CrossRef]
- Junk, P.C.; Cole, M.L. Alkali-metal bis(aryl)formamidinates: A study of coordinative versatility. Chem. Commun. 2007, 16, 1579–1590. [Google Scholar] [CrossRef]
- Edelmann, F.T. Chapter 3—Advances in the Coordination Chemistry of Amidinate and Guanidinate Ligands. Adv. Organomet. Chem. 2008, 57, 183–352. [Google Scholar]
- Edelmann, F.T. Lanthanide amidinates and guanidinates in catalysis and materials science: A continuing success story. Chem. Soc. Rev. 2012, 41, 7657–7672. [Google Scholar] [CrossRef]
- Deacon, G.B.; Hossain, M.E.; Junk, P.C.; Salehisaki, M. Rare-earth N,N′-diarylformamdinate complexes. Coord. Chem. Rev. 2017, 340, 247–265. [Google Scholar] [CrossRef]
- Sengupta, D.; Gómez-Torres, A.; Fortier, S. Guanidinate, Amidinate, and Formamidinate Ligands. In Comprehensive Coordination Chemistry; University of Texas at El Paso: El Paso, TX, USA, 2021; Volume 3, pp. 366–405. [Google Scholar]
- Edelmann, F.T. Carboranylamidinates. Z. Anorg. Allg. Chem. 2013, 639, 655–667. [Google Scholar] [CrossRef]
- Yao, Z.-J.; Jin, G.-X. Transition metal complexes based on carboranyl ligands containing N, P, and S donors: Synthesis, reactivity and applications. Coord. Chem. Rev. 2013, 257, 2522–2535. [Google Scholar] [CrossRef]
- Rädisch, T.; Harmgarth, N.; Liebing, P.; Beltrán-Leiva, M.J.; Páez-Hernández, D.; Arratia-Pérez, R.; Engelhardt, F.; Hilfert, L.; Oehler, F.; Busse, S.; et al. Three new types of transition metal carboranylamidinate complexes. Dalton Trans. 2018, 47, 6666–6671. [Google Scholar] [CrossRef]
- Liebing, P.; Harmgarth, N.; Zörner, F.; Engelhardt, F.; Hilfert, L.; Busse, S.; Edelmann, F.T. Synthesis and Structural Characterization of Two New Main Group Element Carboranylamidinates. Inorganics 2019, 7, 41. [Google Scholar] [CrossRef]
- Wang, H. Recent advances on carborane-based ligands in low-valent group 13 and group 14 elements chemistry. Chin. Chem. Lett. 2022, 33, 3672–3680. [Google Scholar] [CrossRef]
- Harmgarth, N.; Hrib, C.G.; Lorenz, V.; Hilfert, L.; Edelmann, F.T. Unprecedented formation of polycyclic diazadiborepine derivatives through cage deboronation of m-carborane. Chem. Commun. 2014, 50, 13239–13242. [Google Scholar] [CrossRef]
- Stalke, D.; Wedler, M.; Edelmann, F.T. Dimere Alkalimetallbenzamidinate: Einfluß des Metallions auf die Struktur. J. Organomet. Chem. 1992, 431, C1–C5. [Google Scholar] [CrossRef]
- Schmidt, J.A.R.; Arnold, J. Synthesis and characterization of a series of sterically-hindered amidines and their lithium and magnesium complexes. Dalton Trans. 2002, 14, 2890–2899. [Google Scholar] [CrossRef]
- Baker, R.J.; Jones, C. Synthesis and characterisation of sterically bulky lithium amidinate and bis-amidinate complexes. J. Organomet. Chem. 2006, 691, 65–71. [Google Scholar] [CrossRef]
- Kahl, S.B.; Kasar, R.A. Simple, High-Yield Synthesis of Polyhedral Carborane Amino Acids. J. Am. Chem. Soc. 1996, 118, 1223–1224. [Google Scholar] [CrossRef]
- Harmgarth, N.; Gräsing, D.; Dröse, P.; Hrib, C.G.; Jones, P.G.; Lorenz, V.; Hilfert, S.; Busse, S.; Edelmann, F.T. Novel inorganic heterocycles from dimetalated carboranylamidinates. Dalton Trans. 2014, 43, 5001–5013. [Google Scholar] [CrossRef]
- Stoe & Cie. X-Area and X-Red; Stoe & Cie: Darmstadt, Germany, 2002. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Farha, O.K.; Spokoyny, A.M.; Mulfort, K.L.; Hawthorne, M.F.; Mirkin, C.A.; Hupp, J.T. Synthesis and Hydrogen Sorption Properties of Carborane Based Metal-Organic Framework Materials. J. Am. Chem. Soc. 2007, 129, 12680–12681. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Maguire, J.A.; Hosmane, N.S. Polyhedral boron clusters in materials science. New J. Chem. 2011, 35, 19551972. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harmgarth, N.; Liebing, P.; Lorenz, V.; Engelhardt, F.; Hilfert, L.; Busse, S.; Goldhahn, R.; Edelmann, F.T. Synthesis and Structural Characterization of p-Carboranylamidine Derivatives. Molecules 2023, 28, 3837. https://doi.org/10.3390/molecules28093837
Harmgarth N, Liebing P, Lorenz V, Engelhardt F, Hilfert L, Busse S, Goldhahn R, Edelmann FT. Synthesis and Structural Characterization of p-Carboranylamidine Derivatives. Molecules. 2023; 28(9):3837. https://doi.org/10.3390/molecules28093837
Chicago/Turabian StyleHarmgarth, Nicole, Phil Liebing, Volker Lorenz, Felix Engelhardt, Liane Hilfert, Sabine Busse, Rüdiger Goldhahn, and Frank T. Edelmann. 2023. "Synthesis and Structural Characterization of p-Carboranylamidine Derivatives" Molecules 28, no. 9: 3837. https://doi.org/10.3390/molecules28093837
APA StyleHarmgarth, N., Liebing, P., Lorenz, V., Engelhardt, F., Hilfert, L., Busse, S., Goldhahn, R., & Edelmann, F. T. (2023). Synthesis and Structural Characterization of p-Carboranylamidine Derivatives. Molecules, 28(9), 3837. https://doi.org/10.3390/molecules28093837