

One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates

 , and
, and
Abstract
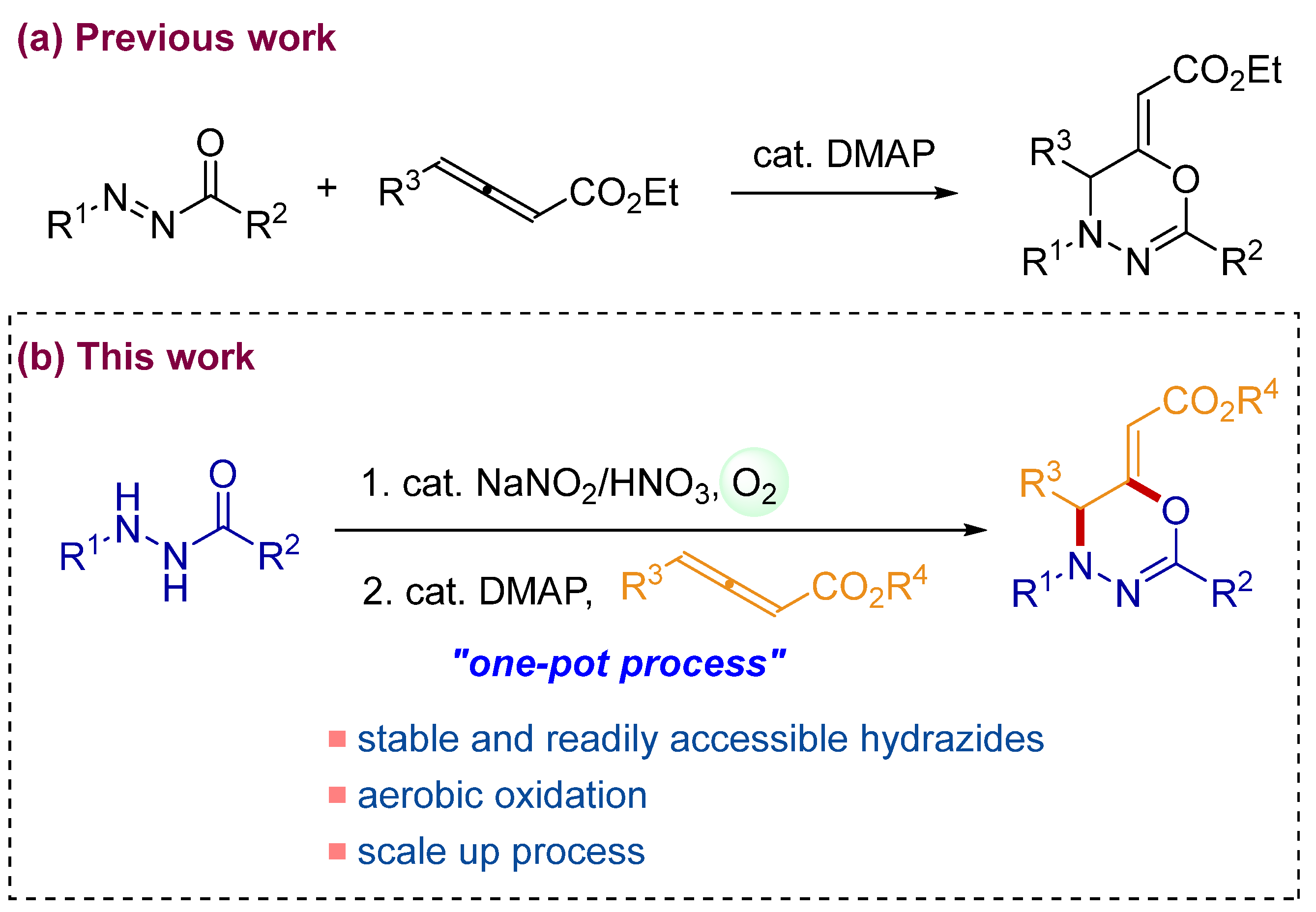
1. Introduction
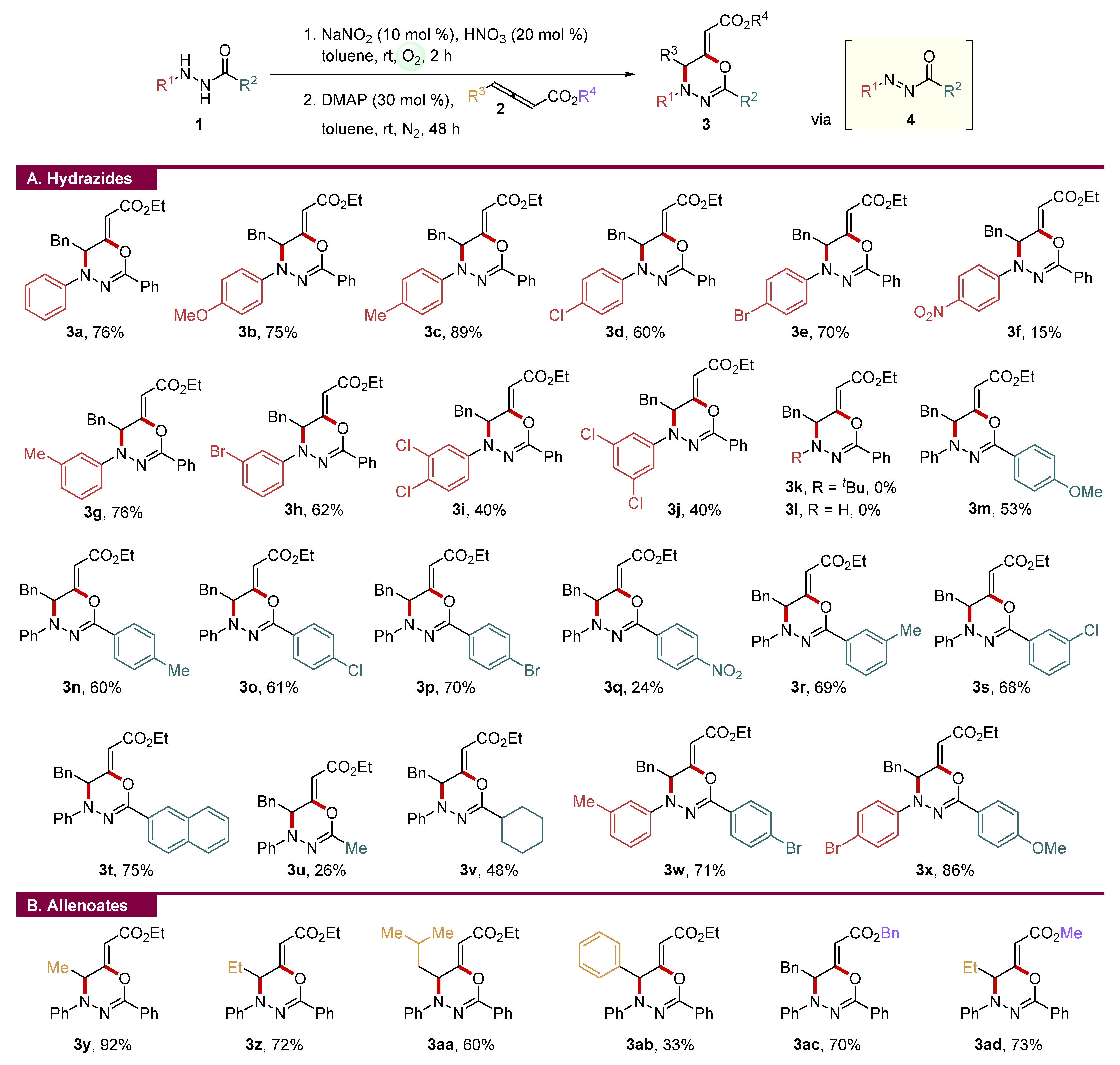
2. Results
3. Materials and Methods
3.1. General Information
3.2. Preparation of Acylhydrazides and Allenoates
3.2.1. Preparation of Acylhydrazides (1a–1j, 1l–1u, and 1w–1x) [49]
3.2.2. Preparation of N′-(tert-butyl)benzohydrazide (1k) [51]
3.2.3. Preparation of N′-phenylcyclohexanecarbohydrazide (1v)
3.2.4. Preparation of Allenoates [52]
3.3. General Procedure for One-Pot Synthesis of 1,3,4-Oxadiazines
3.4. Characterizations of the Newly Synthesized 1,3,4-Oxadiazines
3.5. 1H and 13C NMR Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hunger, K. Industrial Dyes: Chemistry, Properties, Applications; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Merino, E. Synthesis of azobenzenes: The coloured pieces of molecular materials. Chem. Soc. Rev. 2011, 40, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Moroz, E.; Castagner, B.; Leroux, J.-C. Activatable Cell Penetrating Peptide−Peptide Nucleic Acid Conjugate via Reduction of Azobenzene PEG Chains. J. Am. Chem. Soc. 2014, 136, 12868–12871. [Google Scholar] [CrossRef]
- Feringa, B.L.; van Delden, R.A.; Koumura, N.; Geertsema, E.M. Chiroptical Molecular Switches. Chem. Rev. 2000, 100, 1789–1816. [Google Scholar] [CrossRef] [PubMed]
- Bandara, H.M.D.; Burdette, S.C. Photoisomerization in different classes of azobenzene. Chem. Soc. Rev. 2012, 41, 1809–1825. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M.; Mukaiyama, T. Preparation of Esters of Phosphoric Acid by the Reaction of Trivalent Phosphorus Compounds with Diethyl Azodicarboxylate in the Presence of Alcohols. Bull. Chem. Soc. Jpn. 1967, 40, 935–939. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of Esters of Carboxylic and Phosphoric Acid via Quaternary Phosphonium Salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- But, T.Y.S.; Toy, P.H. The Mitsunobu Reaction: Origin, Mechanism, Improvements, and Applications. Chem. Asian J. 2007, 2, 1340–1355. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, F.; Suzuki, K.; Nitta, Y. A New Hydrogen-Abstracting Reaction with Diethyl Azodicarboxylate. J. Am. Chem. Soc. 1966, 88, 2328–2329. [Google Scholar] [CrossRef]
- Yoneda, F.; Suzuki, K.; Nitta, Y. A New Hydrogen-Abstracting Reaction with Diethyl Azodicarboxylate. J. Org. Chem. 1967, 32, 727–729. [Google Scholar] [CrossRef]
- Cao, H.T.; Grée, R. DEAD-(cat) ZnBr2 an efficient system for the oxidation of alcohols to carbonyl compounds. Tetrahedron Lett. 2009, 50, 1493–1494. [Google Scholar] [CrossRef]
- Stone, M.T. An Improved Larock Synthesis of Quinolines via a Heck Reaction of 2-Bromoanilines and Allylic Alcohols. Org. Lett. 2011, 13, 2326–2329. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Mourya, M.; Guin, D.; Joshi, Y.C.; Dobhal, M.P.; Basak, A.K. Diisopropyl azodicarboxylate mediated selective dehydrogenation of 2-amino-3-cyano 4H-chromenes. Tetrahedron Lett. 2017, 58, 1727–1732. [Google Scholar] [CrossRef]
- Bang, S.B.; Kim, J. Efficient dehydrogenation of 1,2,3,4-tetrahydroquinolines mediated by dialkyl azodicarboxylates. Synth. Commun. 2018, 48, 1291–1298. [Google Scholar] [CrossRef]
- Xu, X.; Li, X. Copper/Diethyl Azodicarboxylate Mediated Regioselective Alkynylation of Unactivated Aliphatic Tertiary Methylamine with Terminal Alkyne. Org. Lett. 2009, 11, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.N.; Singh, P.; Kaur, A.; Singh, P. C-1 Alkynylation of N-Methyltetrahydroisoquinolines through CDC: A Direct Access to Phenethylisoquinoline Alkaloids. Synlett 2012, 23, 760–764. [Google Scholar] [CrossRef]
- Huang, W.; Ni, C.; Zhao, Y.; Hu, J. DIAD-mediated metal-free cross dehydrogenative coupling between tertiary amines and α-fluorinated sulfones. New J. Chem. 2013, 37, 1684–1687. [Google Scholar] [CrossRef]
- Singh, K.N.; Kessar, S.V.; Singh, P.; Singh, P.; Kaur, M.; Batra, A. Transition-Metal-Free Arylation of N-Alkyl-tetrahydroisoquinolines under Oxidative Conditions: A Convenient Synthesis of C1-Arylated Tetrahydroisoquinoline Alkaloids. Synthesis 2014, 46, 2644–2650. [Google Scholar] [CrossRef]
- Suga, T.; Iizuka, S.; Akiyama, T. Versatile and highly efficient oxidative C(sp3)–H bond functionalization of tetrahydroisoquinoline promoted by bifunctional diethyl azodicarboxylate (DEAD): Scope and mechanistic insights. Org. Chem. Front. 2016, 3, 1259–1264. [Google Scholar] [CrossRef]
- Kim, Y.H.; Gil, M.G.; Kim, D.Y. Diethyl Azodicarboxylate-promoted Oxidative Coupling Reaction of N-Phenyl Tetrahydroisoquinoline with β-Keto Acids. Bull. Korean Chem. Soc. 2017, 38, 1499–1502. [Google Scholar] [CrossRef]
- Sharma, S.; Han, S.H.; Han, S.; Ji, W.; Oh, J.; Lee, S.-Y.; Oh, J.S.; Jung, Y.H.; Kim, I.S. Rh(III)-Catalyzed Direct Coupling of Azobenzenes with α-Diazo Esters: Facile Synthesis of Cinnolin-3(2H)-ones. Org. Lett. 2015, 17, 2852–2855. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Scheidt, K.A. Direct Amination of Homoenolates Catalyzed by N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2008, 130, 2740–2741. [Google Scholar] [CrossRef]
- Huang, X.-L.; He, L.; Shao, P.-L.; Ye, S. [4+2] Cycloaddition of Ketenes with N-Benzoyldiazenes Catalyzed by N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. 2009, 48, 192–195. [Google Scholar] [CrossRef]
- Morrill, L.C.; Lebl, T.; Slawin, A.M.Z.; Smith, A.D. Catalytic asymmetric α-amination of carboxylic acids using isothioureas. Chem. Sci. 2012, 3, 2088–2093. [Google Scholar] [CrossRef]
- Taylor, J.E.; Daniels, D.S.B.; Smith, A.D. Asymmetric NHC-Catalyzed Redox α-Amination of α-Aroyloxyaldehydes. Org. Lett. 2013, 15, 6058–6061. [Google Scholar] [CrossRef]
- Morrill, L.C.; Smith, S.M.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Mediated Asymmetric Functionalization of 3-Alkenoic Acids. J. Org. Chem. 2014, 79, 1640–1655. [Google Scholar] [CrossRef]
- Yang, L.; Wang, F.; Lee, R.; Lv, Y.; Huang, K.-W.; Zhong, G. Asymmetric NHC-Catalyzed Aza-Diels–Alder Reactions: Highly Enantioselective Route to α-Amino Acid Derivatives and DFT Calculations. Org. Lett. 2014, 16, 3872–3875. [Google Scholar] [CrossRef]
- Savva, A.C.; Mirallai, S.I.; Zissimou, G.A.; Berezin, A.A.; Demetriades, M.; Kourtellaris, A.; Constantinides, C.P.; Nicolaides, C.; Trypiniotis, T.; Koutentis, P.A. Preparation of Blatter Radicals via Aza-Wittig Chemistry: The Reaction of N-Aryliminophosphoranes with 1-(Het)aroyl-2-aryldiazenes. J. Org. Chem. 2017, 82, 7564–7575. [Google Scholar] [CrossRef]
- Bigotto, A.; Forchiassin, M.; Fisaliti, A.; Russo, C. Reactions of cyclohexanone enamines with asymmeric diimides. Tetrahedron Lett. 1979, 20, 4761–4764. [Google Scholar] [CrossRef]
- Forchiassin, M.; Risaliti, A.; Russo, C. 1,3,4-Oxadiazine derivatives from cyclohexanone enamines and asymmetric diimides: Possibility of ring-chain tautomerism in such heterocyclic system. Tetrahedron 1981, 37, 2921–2928. [Google Scholar] [CrossRef]
- Zhou, R.; Han, L.; Zhang, H.; Liu, R.; Li, R. A Deoxygenative [4+1] Annulation Involving N-Acyldiazenes for an Efficient Synthesis of 2,2,5-Trisubstituted 1,3,4-Oxadiazole Derivatives. Adv. Synth. Catal. 2017, 359, 3977–3982. [Google Scholar] [CrossRef]
- Ma, C.; Zhou, J.-Y.; Zhang, Y.-Z.; Mei, G.-J.; Shi, F. Catalytic Asymmetric [2+3] Cyclizations of Azlactones with Azonaphthalenes. Angew. Chem. Int. Ed. 2018, 57, 5398–5402. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Meng, L.-G.; Peng, T.; Zhu, L.; Wang, L. 4-Dimethylaminopyridine-Catalyzed Regioselective [3+2] Cycloaddition of Isatin-Derived Morita−Baylis−Hillman Adducts with Azo Esters: A Simple Protocol to Access 3-Spiropyrazole-2-oxindoles. Adv. Synth. Catal. 2018, 360, 3176–3180. [Google Scholar] [CrossRef]
- Li, C.; Xu, D.-N.; Ma, C.; Mei, G.-J.; Shi, F. Diastereo- and Enantioselective Construction of Dihydrobenzo[e]indole Scaffolds via Catalytic Asymmetric [3 + 2] Cycloannulations. J. Org. Chem. 2018, 83, 9190–9200. [Google Scholar] [CrossRef]
- Zhang, Q.; Meng, L.-G.; Zhang, J.; Wang, L. DMAP-Catalyzed [2+4] Cycloadditions of Allenoates with N-Acyldiazenes: Direct Method to 1,3,4-Oxadiazine Derivatives. Org. Lett. 2015, 17, 3272–3275. [Google Scholar] [CrossRef]
- Guo, X.; Chen, X.; Cheng, Y.; Chang, X.; Li, X.; Li, P. Organocatalytic enantioselective [2 + 4]-annulation of γ-substituted allenoates with N-acyldiazenes for the synthesis of optically active 1,3,4-oxadiazines. Org. Biomol. Chem. 2021, 19, 1727–1731. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Aerobic Oxidation of Alkyl 2-Phenylhydrazinecarboxylates Catalyzed by CuCl and DMAP. J. Org. Chem. 2018, 83, 1673–1679. [Google Scholar] [CrossRef]
- Jo, G.; Kim, M.H.; Kim, J. A practical route to azo compounds by metal-free aerobic oxidation of arylhydrazides using an NOx system. Org. Chem. Front. 2020, 7, 834–839. [Google Scholar] [CrossRef]
- Kim, J.; Lee, D.H.; Kim, J. Cu-Catalyzed Aerobic Oxidative Azo-Ene Cyclization. Adv. Synth. Catal. 2021, 363, 4728–4733. [Google Scholar] [CrossRef]
- Jung, D.; Kim, M.H.; Kim, J. Cu-Catalyzed Aerobic Oxidation of Di-tert-butyl Hydrazodicarboxylate to Di-tert-butyl Azodicarboxylate and Its Application on Dehydrogenation of 1,2,3,4-Tetrahydroquinolines under Mild Conditions. Org. Lett. 2016, 18, 6300–6303. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Jang, S.H.; Yim, T.; Kim, J. Oxidation Potential Tunable Organic Molecules and Their Catalytic Application to Aerobic Dehydrogenation of Tetrahydroquinolines. Org. Lett. 2018, 20, 6436–6439. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Ren, L.; Hou, J.; Yu, W.; Chang, J. Annulation Reactions of In-Situ-Generated N-(Het)aroyldiazenes with Isothiocyanates Leading to 2-Imino-1,3,4-oxadiazolines. Org. Lett. 2019, 21, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Q.; Zhao, Q.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-thiadiazoles from Hydrazides and Isothiocyanates via Sequential Oxidation and P(NMe2)3-Mediated Annulation Reactions. Org. Lett. 2020, 22, 4378–4382. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, Q.; Yi, X.; Zhao, Z.; Yu, W.; Chang, J. Synthesis of 2-Imino-1,3,4-Selenadiazoles via Tributylphosphine-Mediated Annulation of N-Aroyldiazenes with Isoselenocyanates. Adv. Synth. Catal. 2021, 363, 4894–4898. [Google Scholar] [CrossRef]
- Kim, S.B.; Baek, S.E.; Lim, J.H.; Kim, J. One-pot synthesis of 2-imino-1,3,4-thiadiazolines from acylhydrazides and isothiocyanates. Bull. Korean Chem. Soc. 2022, 43, 1014–1018. [Google Scholar] [CrossRef]
- Lim, J.H.; Baek, S.E.; Lad, B.; Kim, J. Synthesis of 2-Imino-1,3,4-oxadiazolines from Acylhydrazides and Isothiocyanates via Aerobic Oxidation and DMAP-Mediated Annulation Sequence. ACS Omega 2022, 7, 28148–28159. [Google Scholar] [CrossRef]
- Hirose, D.; Taniguchi, T.; Ishibashi, H. Recyclable Mitsunobu Reagents: Catalytic Mitsunobu Reactions with an Iron Catalyst and Atmospheric Oxygen. Angew. Chem. Int. Ed. 2013, 52, 4613–4617. [Google Scholar] [CrossRef]
- Fang, T.; Tan, O.; Ding, Z.; Liu, B.; Bin Xu, B. Pd-Catalyzed Oxidative Annulation of Hydrazides with Isocyanides: Synthesis of 2-Amino-1,3,4-oxadiazoles. Org. Lett. 2014, 16, 2342–2345. [Google Scholar] [CrossRef]
- Xu, S.; Zhou, L.; Ma, R.; Song, H.; He, Z. Phosphane-Catalyzed [3 + 2] Annulation of Allenoates with Aldehydes: A Simple and Efficient Synthesis of 2-Alkylidenetetrahydrofurans. Chem. Eur. J. 2009, 15, 8698–8702. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
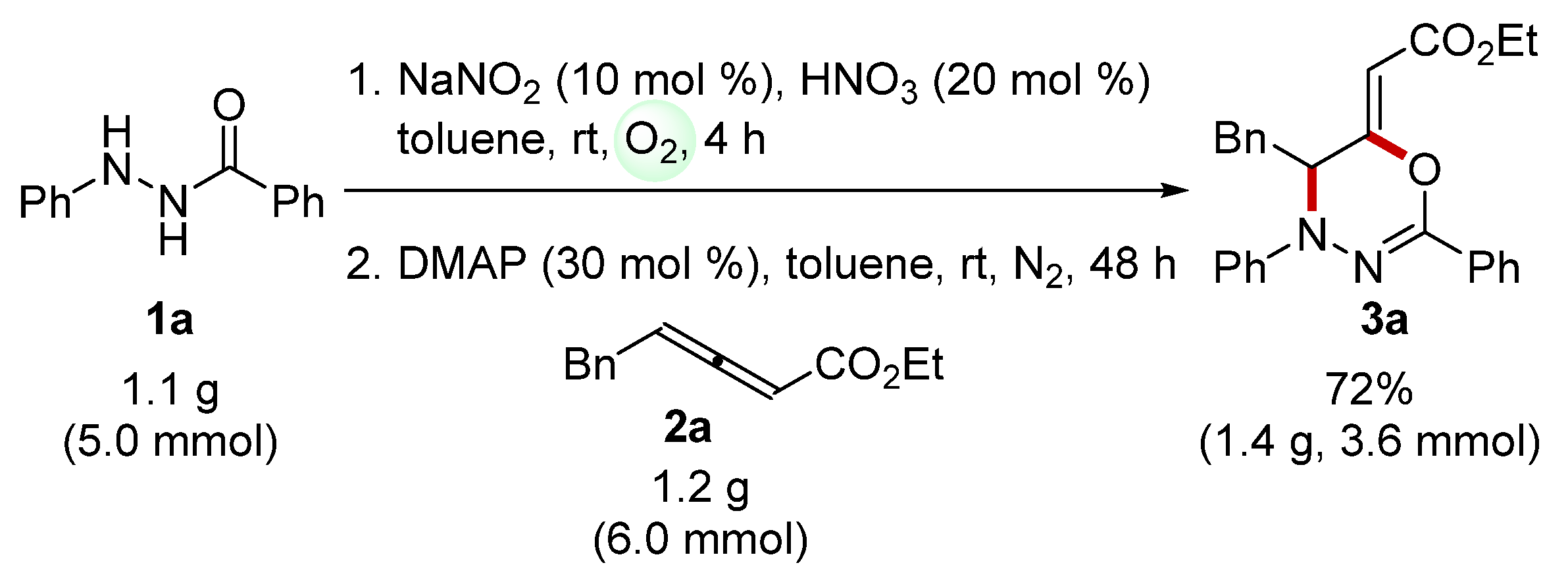{kind=link}
 | ||
|---|---|---|
| Entry | Catalyst (mol %) | Yield (%) b |
| 1 | CuCl (10)/DMAP (60) | 14 |
| 2 | Fe(Pc) (10)/DMAP (40) | 15 |
| 3 | Mn(Pc) (10)/DMAP (40) | 19 |
| 4 | NaNO2 (10)/HNO3 (20)/DMAP (40) | 30 |
 | ||||
|---|---|---|---|---|
| Entry | Catalyst (mol %) | Base | Solvent | Yield (%) b |
| 1 | CuCl (10)/DMAP (20) | DMAP | toluene | 24 |
| 2 | Fe(Pc) (10) | DMAP | toluene | 30 |
| 3 | Mn(Pc) (10) | DMAP | toluene | 13 |
| 4 | NaNO2 (10)/HNO3 (20) | DMAP | toluene | 75 |
| 5 | NaNO2 (10)/HNO3 (20) | pyridine | toluene | 13 |
| 6 | NaNO2 (10)/HNO3 (20) | DBU | toluene | 0 |
| 7 | NaNO2 (10)/HNO3 (20) | DMAP | CH3CN | 20 |
| 8 | NaNO2 (10)/HNO3 (20) | DMAP | CH2Cl2 | 46 |
| 9 | NaNO2 (10)/HNO3 (20) | DMAP | 1,4-dioxane | 45 |
| 10 | NaNO2 (10)/HNO3 (20) | DMAP | EtOH | 25 |
| 11 c | NaNO2 (10)/HNO3 (20) | DMAP | toluene | 76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.B.; Maiti, S.; Park, E.S.; Kim, G.Y.; Choun, Y.; Ahn, S.K.; Kim, J.K.; Kim, J. One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates. Molecules 2023, 28, 3815. https://doi.org/10.3390/molecules28093815
Kim SB, Maiti S, Park ES, Kim GY, Choun Y, Ahn SK, Kim JK, Kim J. One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates. Molecules. 2023; 28(9):3815. https://doi.org/10.3390/molecules28093815
Chicago/Turabian StyleKim, Su Been, Santanu Maiti, Eun Sun Park, Ga Young Kim, Yunji Choun, Soon Kil Ahn, Jae Kwang Kim, and Jinho Kim. 2023. "One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates" Molecules 28, no. 9: 3815. https://doi.org/10.3390/molecules28093815
APA StyleKim, S. B., Maiti, S., Park, E. S., Kim, G. Y., Choun, Y., Ahn, S. K., Kim, J. K., & Kim, J. (2023). One-Pot Synthesis of 1,3,4-Oxadiazines from Acylhydrazides and Allenoates. Molecules, 28(9), 3815. https://doi.org/10.3390/molecules28093815