

Detection and Quantification of Adulteration in Krill Oil with Raman and Infrared Spectroscopic Methods


Abstract
1. Introduction
- (1)
- Identify the presence of adulterants (palm oil (PO); ω-3 concentrates in ethyl ester (O3C) and fish oil (FO)) in krill oil,
- (2)
- Quantify the amount of adulterant present and
- (3)
- Identify the most promising spectroscopic technique or combination thereof for the quantification of adulterant(s) in krill oil.
2. Results and Discussion
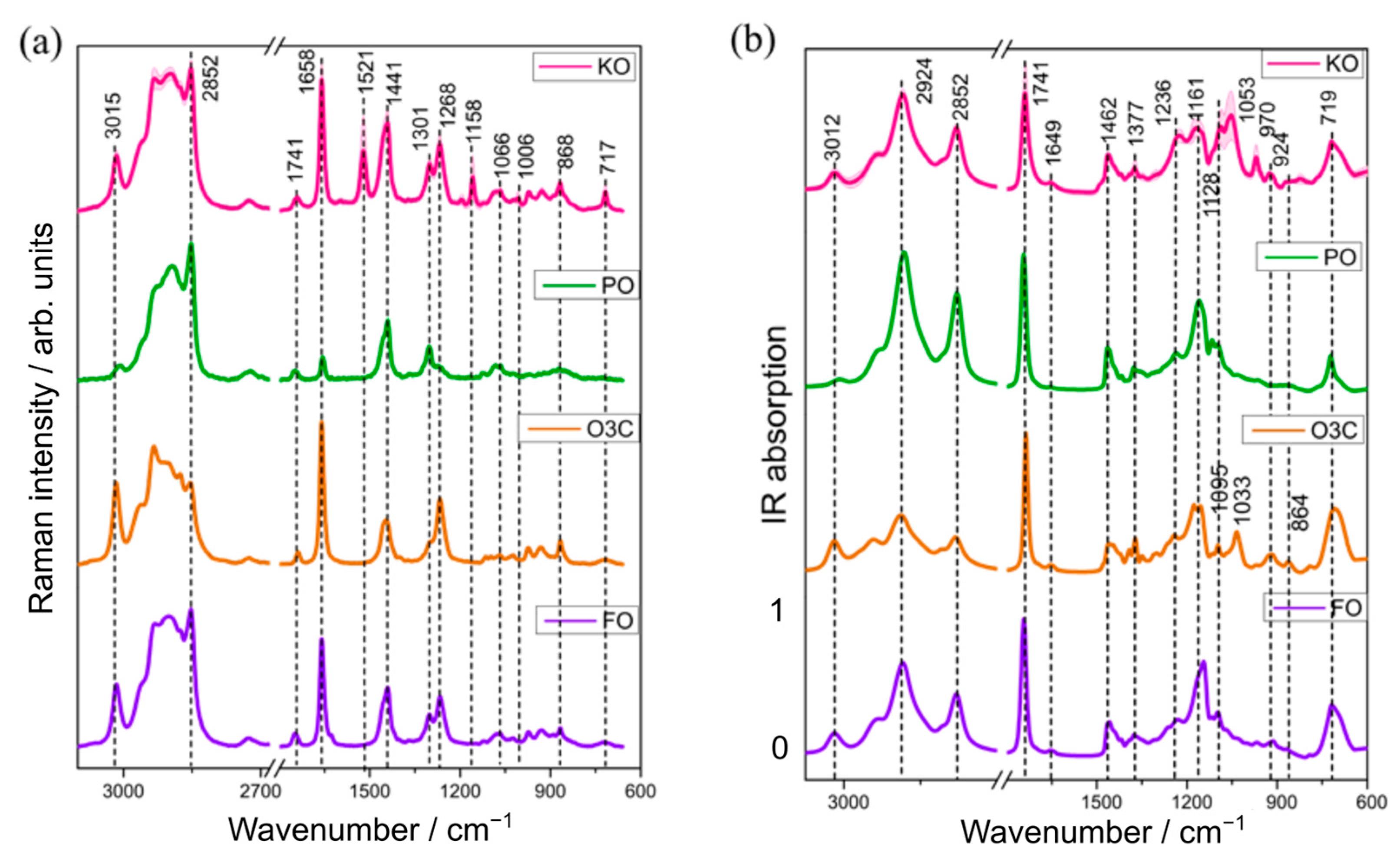
2.1. Raman and Infrared Spectral Features of the Oil Samples
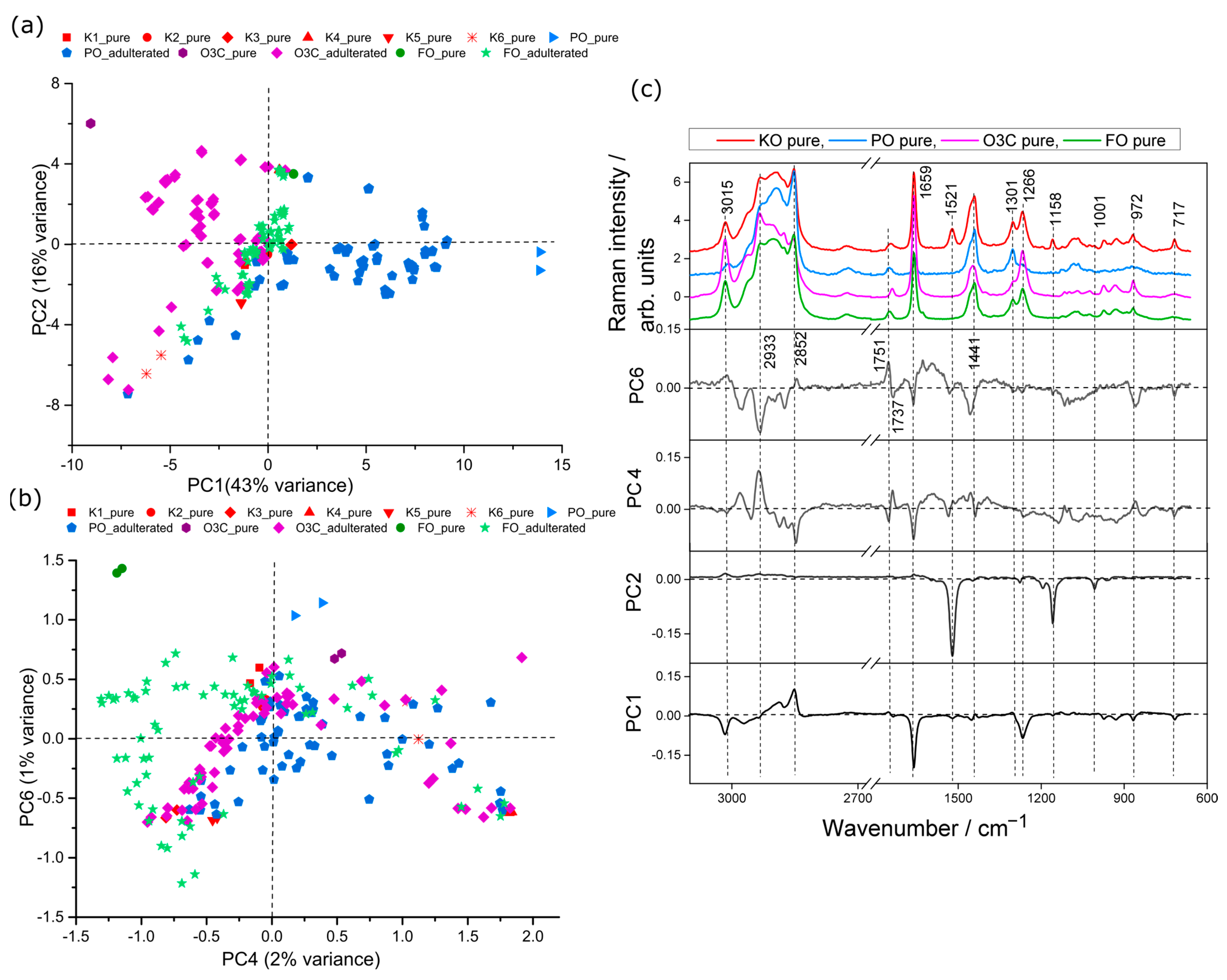
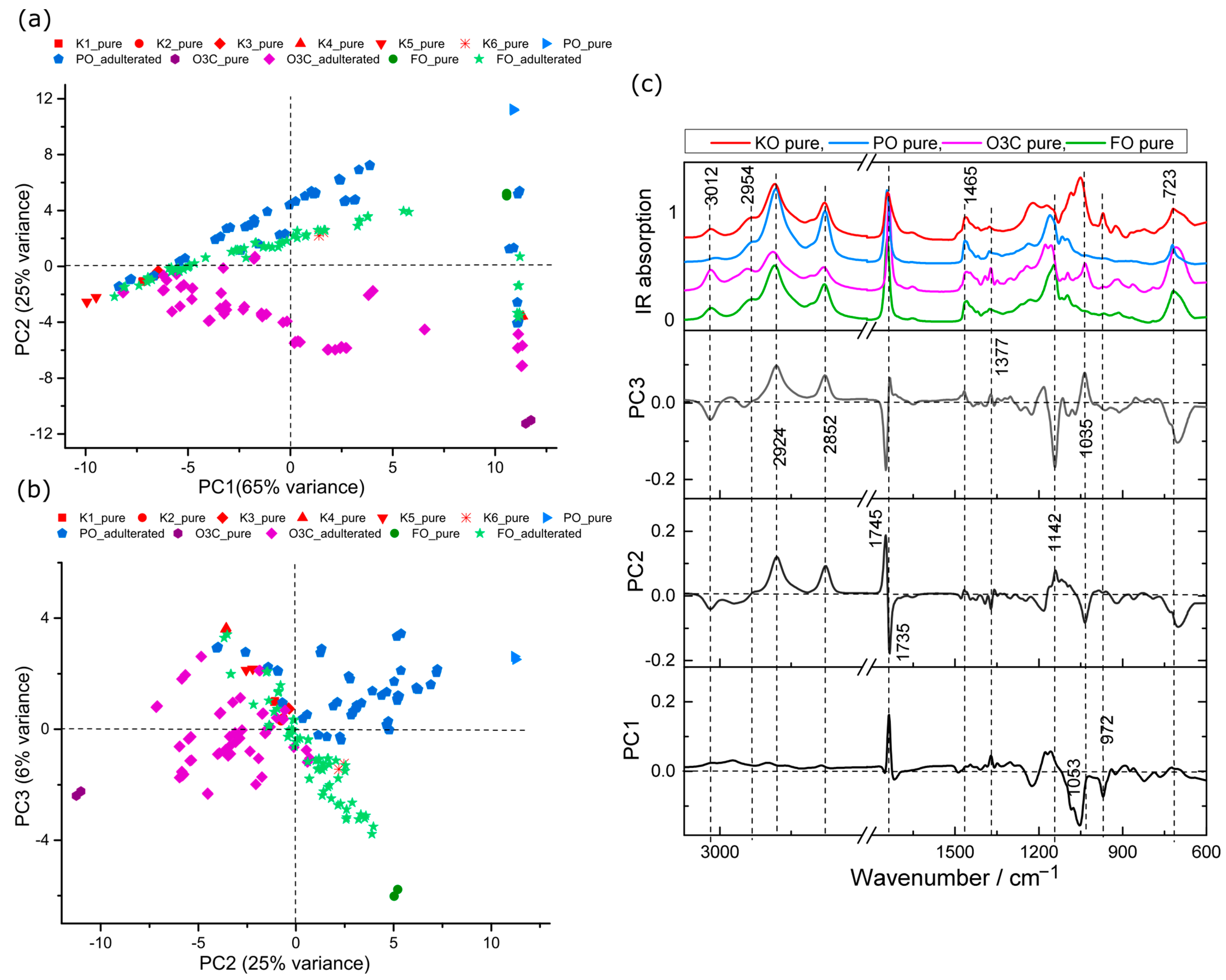
2.2. Principal Component Analysis (PCA)
2.3. SVM Classification of Raman and IR Data
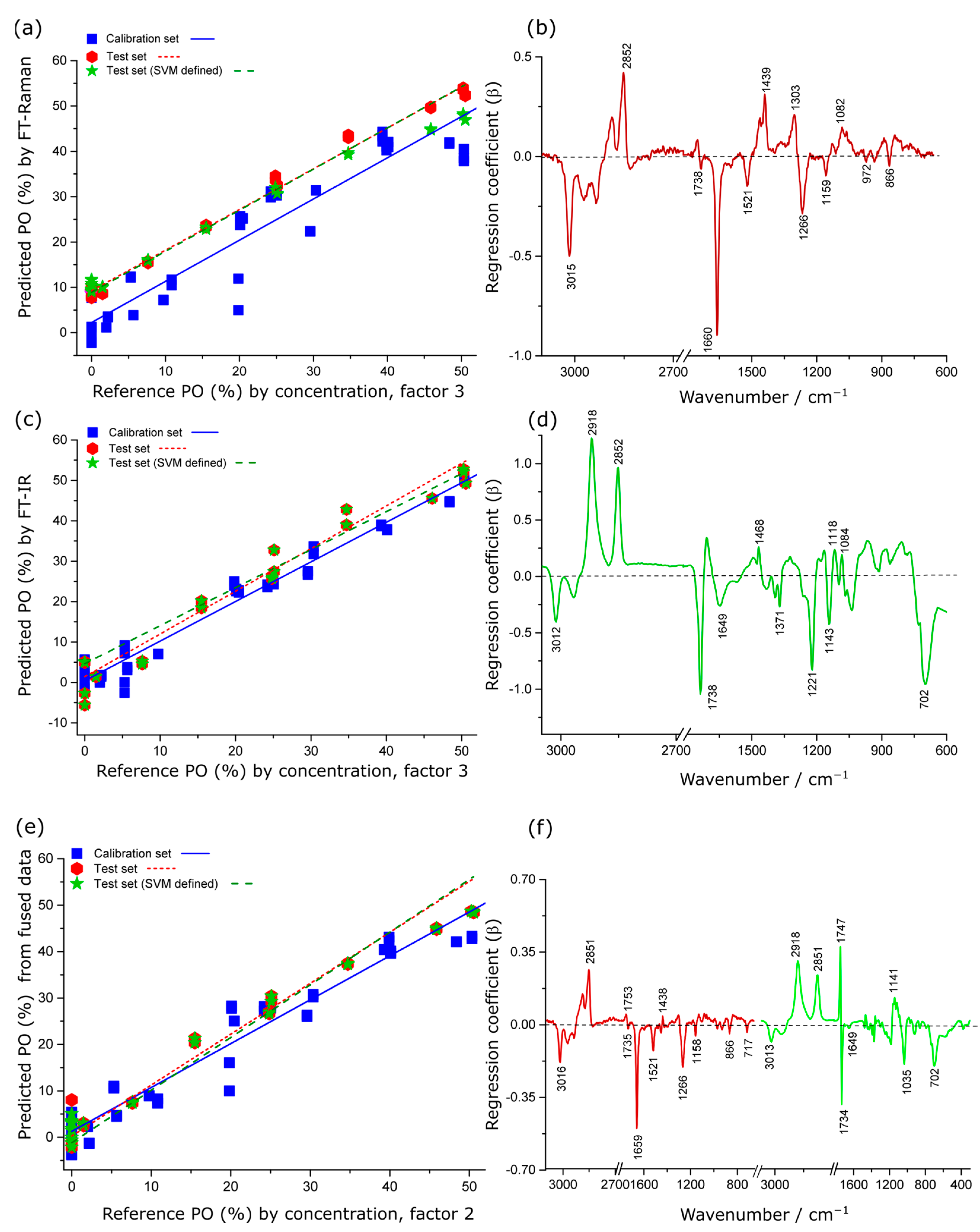
2.4. Quantitative Measurements of Krill Oil Adulteration
- (1)
- Running the samples through the appropriate model based on prior knowledge of the adulterant used. This has the limitation that it does not reflect how a truly unknown sample will perform if we have no prior knowledge of the adulterant present.
- (2)
- Run the test set data through the SVM classification model to evaluate the adulterant present, then feed those data through the appropriate PLSR quantitative model (see Figure S2 for the workflow). This has the advantage of no user input in the selection of the PLSR model used and will reflect a likely workflow in a truly unknown sample.
2.4.1. Spectroscopic Estimation of % PO in Krill Oil
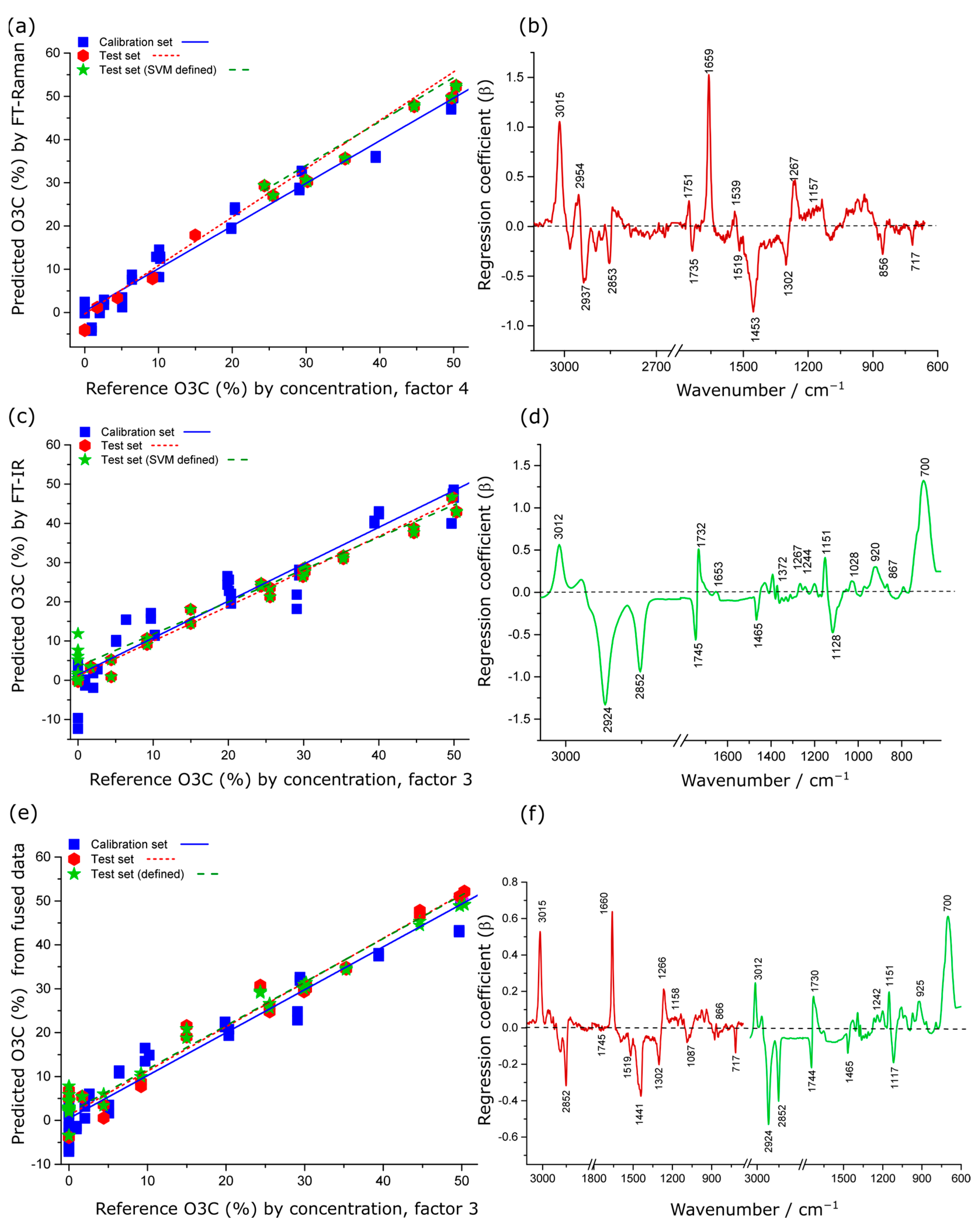
2.4.2. Spectroscopic Estimation of % O3C in Krill Oil
2.4.3. Spectroscopic Estimation of % FO in Krill Oil
3. Materials and Methods
3.1. Spectroscopic Methods
3.2. Spectral Pre-Processing and Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- He, Y.; Bai, X.; Xiao, Q.; Liu, F.; Zhou, L.; Zhang, C. Detection of adulteration in food based on nondestructive analysis techniques: A review. Crit. Rev. Food Sci. Nutr. 2021, 61, 2351–2371. [Google Scholar] [CrossRef]
- Fuller, I.D.; Cumming, A.H.; Card, A.; Burgess, E.J.; Barrow, C.J.; Perry, N.B.; Killeen, D.P. Free Fatty Acids in Commercial Krill Oils: Concentrations, Compositions, and Implications for Oxidative Stability. J. Am. Oil Chem. Soc. 2020, 97, 889–900. [Google Scholar] [CrossRef]
- Ahn, S.H.; Lim, S.J.; Ryu, Y.M.; Park, H.R.; Suh, H.J.; Han, S.H. Absorption rate of krill oil and fish oil in blood and brain of rats. Lipids Health Dis. 2018, 17, 162. [Google Scholar] [CrossRef]
- Berge, K.; Musa-Veloso, K.; Harwood, M.; Hoem, N.; Burri, L. Krill oil supplementation lowers serum triglycerides without increasing low-density lipoprotein cholesterol in adults with borderline high or high triglyceride levels. Nutr. Res. 2014, 34, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.S.; Nielsen, N.S.; Baron, C.P.; Jacobsen, C. Marine phospholipids: The current understanding of their oxidation mechanisms and potential uses for food fortification. Crit. Rev. Food Sci. Nutr. 2017, 57, 2057–2070. [Google Scholar] [CrossRef]
- Zhao, J.; Jiang, K.; Chen, Y.; Chen, J.; Zheng, Y.; Yu, H.; Zhu, J. Preparation and Characterization of Microemulsions Based on Antarctic Krill Oil. Mar. Drugs 2020, 18, 392. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Gong, M.; Wei, W.; Jin, J.; Wang, X.; Wang, X.; Jin, Q. Antarctic Krill (Euphausia superba) Oil: A Comprehensive Review of Chemical Composition, Extraction Technologies, Health Benefits, and Current Applications. Compr. Rev. Food Sci. Food Saf. 2019, 18, 514–534. [Google Scholar] [CrossRef]
- Koçakoğlu, Ş.; Akpınar, E. Apparent Hyperpigmented Skin Blemish That Has Been Incidentally Treated with Oral Krill Oil: A Case Report. J. Med.-Clin. Res. Rev. 2020, 4, 1–3. [Google Scholar] [CrossRef]
- Köhler, A.; Sarkkinen, E.; Tapola, N.; Niskanen, T.; Bruheim, I. Bioavailability of fatty acids from krill oil, krill meal and fish oil in healthy subjects–a randomized, single-dose, cross-over trial. Lipids Health Dis. 2015, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, J.P.; Schneider, I.; Meyer, H.; Neubronner, J.; von Schacky, C.; Hahn, A. Incorporation of EPA and DHA into plasma phospholipids in response to different omega-3 fatty acid formulations-a comparative bioavailability study of fish oil vs. krill oil. Lipids Health Dis. 2011, 10, 145. [Google Scholar] [CrossRef]
- Lee, D.-S.; Noh, B.-S.; Bae, S.-Y.; Kim, K. Characterization of fatty acids composition in vegetable oils by gas chromatography and chemometrics. Anal. Chim. Acta 1998, 358, 163–175. [Google Scholar] [CrossRef]
- Murota, K.; Takagi, M.; Watanabe, Y.; Tokumura, A.; Ohkubo, T. Roe-derived phospholipid administration enhances lymphatic docosahexaenoic acid-containing phospholipid absorption in unanesthetized rats. Prostaglandins Leukot. Essent. Fat. Acids 2018, 139, 40–48. [Google Scholar] [CrossRef]
- Wen, M.; Zhao, Y.; Shi, H.; Wang, C.; Zhang, T.; Wang, Y.; Xue, C. Short-term supplementation of DHA as phospholipids rather than triglycerides improve cognitive deficits induced by maternal omega-3 PUFA deficiency during the late postnatal stage. Food Funct. 2021, 12, 564–572. [Google Scholar] [CrossRef]
- Ahmmed, M.K.; Ahmmed, F.; Tian, H.S.; Carne, A.; Bekhit, A.E. Marine omega-3 (n-3) phospholipids: A comprehensive review of their properties, sources, bioavailability, and relation to brain health. Compr. Rev. Food Sci. Food Saf. 2020, 19, 64–123. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Jin, J.; Sun, J.; Liang, L.; Wang, X.; Zhang, W.; Wang, X.; Jin, Q. Comparison of solvents for extraction of krill oil from krill meal: Lipid yield, phospholipids content, fatty acids composition and minor components. Food Chem. 2017, 233, 434–441. [Google Scholar] [CrossRef]
- Bengtson Nash, S.M.; Schlabach, M.; Nichols, P.D. A nutritional-toxicological assessment of Antarctic krill oil versus fish oil dietary supplements. Nutrients 2014, 6, 3382–3402. [Google Scholar] [CrossRef]
- Parry, L. The adulateration of cod-liver oil. Lancet 1904, 163, 757. [Google Scholar] [CrossRef]
- Araujo, P.; Tilahun, E.; Zeng, Y. A novel strategy for discriminating marine oils by using the positional distribution (sn-1, sn-2, sn-3) of omega-3 polyunsaturated fatty acids in triacylglycerols. Talanta 2018, 182, 32–37. [Google Scholar] [CrossRef] [PubMed]
- The National Library of Medicine Case Number 1015. Adulteration and Misbranding of cod Liver Oil. U. S. v. The Swiftide Co. Plea of Nolo Contendere. Fine, $100. Available online: https://fdanj.nlm.nih.gov/catalog/ddnj01015 (accessed on 27 March 2023).
- Adulteration and Misbranding of Halibut Liver Oil Capsules. U. S. v. 24,000, 75,000, and 90,000 Halibut Liver Oil Capsules. Consent decrees of Condemnation. Product Ordered Released under Bond for Relabeling. Available online: https://fdanj.nlm.nih.gov/catalog/ddnj00354 (accessed on 27 March 2023).
- Albert, B.B.; Derraik, J.G.; Cameron-Smith, D.; Hofman, P.L.; Tumanov, S.; Villas-Boas, S.G.; Garg, M.L.; Cutfield, W.S. Fish oil supplements in New Zealand are highly oxidised and do not meet label content of n-3 PUFA. Sci. Rep. 2015, 5, 7928. [Google Scholar] [CrossRef]
- Killeen, D.P.; Marshall, S.N.; Burgess, E.J.; Gordon, K.C.; Perry, N.B. Raman spectroscopy of fish oil capsules: Polyunsaturated fatty acid quantitation plus detection of ethyl esters and oxidation. J. Agric. Food Chem. 2017, 65, 3551–3558. [Google Scholar] [CrossRef]
- Amorim, T.L.; Fuente, M.A.d.l.; Oliveira, M.A.L.d.; Gómez-Cortés, P. ATR-FTIR and Raman Spectroscopies Associated with Chemometrics for Lipid Form Evaluation of Fish Oil Supplements: A Comparative Study. ACS Food Sci. Technol. 2021, 1, 318–325. [Google Scholar] [CrossRef]
- Rohman, A.; Che Man, Y.B. Application of Fourier transform infrared (FT-IR) spectroscopy combined with chemometrics for authentication of cod-liver oil. Vib. Spectrosc. 2011, 55, 141–145. [Google Scholar] [CrossRef]
- Putri, A.R.; Rohman, A.; Riyanto, S. Authentication of Patin (Pangasius Micronemus) Fish Oil Adulterated with Palm Oil Using Ftir Spectroscopy Combined with Chemometrics. Int. J. Appl. Pharm. 2019, 11, 195–199. [Google Scholar] [CrossRef]
- Ali, H.; Saleem, M.; Anser, M.R.; Khan, S.; Ullah, R.; Bilal, M. Validation of Fluorescence Spectroscopy to Detect Adulteration of Edible Oil in Extra Virgin Olive Oil (EVOO) by Applying Chemometrics. Appl. Spectrosc. 2018, 72, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, S.; Spandana, K.; Mahato, K.; Mazumder, N. Purity Analysis of Adulterated Vegetable Oils by Raman and FTIR Spectroscopy. In Frontiers in Optics; Optical Society of America: washington, DC, USA, 2018. [Google Scholar]
- Neves, M.G.; Poppi, R.J. Authentication and identification of adulterants in virgin coconut oil using ATR/FTIR in tandem with DD-SIMCA one class modeling. Talanta 2020, 219, 121338. [Google Scholar] [CrossRef] [PubMed]
- Lingzhi, X.; Fei, G.; Zengling, Y.; Lujia, H.; Xian, L. Discriminant analysis of terrestrial animal fat and oil adulteration in fish oil by infrared spectroscopy. Int. J. Agric. Biol. Eng. 2016, 9, 179–185. [Google Scholar]
- Ahmmed, F.; Killeen, D.P.; Gordon, K.C.; Fraser-Miller, S.J. Rapid Quantitation of Adulterants in Premium Marine Oils by Raman and IR Spectroscopy: A Data Fusion Approach. Molecules 2022, 27, 4534. [Google Scholar] [CrossRef]
- Kizil, R.; Irudayaraj, J. Spectroscopic Technique: Fourier Transform Raman (FT-Raman) Spectroscopy. In Modern Techniques for Food Authentication; Academic Press: Cambridge, MA, USA, 2018; pp. 193–217. [Google Scholar]
- Afseth, N.K.; Wold, J.P.; Segtnan, V.H. The potential of Raman spectroscopy for characterisation of the fatty acid unsaturation of salmon. Anal. Chim. Acta 2006, 572, 85–92. [Google Scholar] [CrossRef]
- Timlin, J.A.; Collins, A.M.; Beechem, T.A.; Shumskaya, M.; Wurtzel, E.T. Localizing and Quantifying Carotenoids in Intact Cells and Tissues. In Carotenoids; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar]
- Bekhit, M.Y.; Grung, B.; Mjøs, S.A. Determination of omega-3 fatty acids in fish oil supplements using vibrational spectroscopy and chemometric methods. Appl. Spectrosc. 2014, 68, 1190–1200. [Google Scholar] [CrossRef]
- Nettleton, J.A. Omega-3 fatty acids and health. In Omega-3 Fatty Acids and Health; Springer: Berlin/Heidelberg, Germany, 1995; pp. 64–76. [Google Scholar]
- Rohman, A.; Che Man, Y.B. The use of Fourier transform mid infrared (FT-MIR) spectroscopy for detection and quantification of adulteration in virgin coconut oil. Food Chem. 2011, 129, 583–588. [Google Scholar] [CrossRef]
- Ping, B.T.Y.; Yusof, M. Characteristics and properties of fatty acid distillates from palm oil. Oil Palm Bull. 2009, 59, 5–11. [Google Scholar]
- Ahmmed, F.; Fuller, I.D.; Killeen, D.P.; Fraser-Miller, S.J.; Gordon, K.C. Raman and Infrared Spectroscopic Data Fusion Strategies for Rapid, Multicomponent Quantitation of Krill Oil Compositions. ACS Food Sci. Technol. 2021, 1, 570–578. [Google Scholar] [CrossRef]
- Vongsvivut, J.; Miller, M.R.; McNaughton, D.; Heraud, P.; Barrow, C.J. Rapid Discrimination and Determination of Polyunsaturated Fatty Acid Composition in Marine Oils by FTIR Spectroscopy and Multivariate Data Analysis. Food Bioprocess. Technol. 2014, 7, 2410–2422. [Google Scholar] [CrossRef]
- Jamwal, R.; Kumari, S.; Balan, B.; Kelly, S.; Cannavan, A.; Singh, D.K. Rapid and non-destructive approach for the detection of fried mustard oil adulteration in pure mustard oil via ATR-FTIR spectroscopy-chemometrics. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2021, 244, 118822. [Google Scholar] [CrossRef] [PubMed]
- Olsen, E.F.; Rukke, E.-O.; Egelandsdal, B.; Isaksson, T. Determination of omega-6 and omega-3 fatty acids in pork adipose tissue with nondestructive Raman and Fourier transform infrared spectroscopy. Appl. Spectrosc. 2008, 62, 968–974. [Google Scholar] [CrossRef]
- Park, S.E.; Yu, H.Y.; Ahn, S. Development and Validation of a Simple Method to Quantify Contents of Phospholipids in Krill Oil by Fourier-Transform Infrared Spectroscopy. Foods 2021, 11, 41. [Google Scholar] [CrossRef]
- Vongsvivut, J.; Heraud, P.; Zhang, W.; Kralovec, J.A.; McNaughton, D.; Barrow, C.J. Quantitative determination of fatty acid compositions in micro-encapsulated fish-oil supplements using Fourier transform infrared (FTIR) spectroscopy. Food Chem. 2012, 135, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Guillen, M.D.; Cabo, N. Characterization of edible oils and lard by Fourier transform infrared spectroscopy. Relationships between composition and frequency of concrete bands in the fingerprint region. J. Am. Oil Chem. Soc. 1997, 74, 1281–1286. [Google Scholar] [CrossRef]
- Boqué, R.; Rius, F.X. Multivariate detection limits estimators. Chemom. Intell. Lab. Syst. 1996, 32, 11–23. [Google Scholar] [CrossRef]
- Loock, H.-P.; Wentzell, P.D. Detection limits of chemical sensors: Applications and misapplications. Sens. Actuators B Chem. 2012, 173, 157–163. [Google Scholar] [CrossRef]
- Mah, P.T.; Fraser, S.J.; Reish, M.E.; Rades, T.; Gordon, K.C.; Strachan, C.J. Use of low-frequency Raman spectroscopy and chemometrics for the quantification of crystallinity in amorphous griseofulvin tablets. Vib. Spectrosc. 2015, 77, 10–16. [Google Scholar] [CrossRef]
- Strachan, C.J.; Taday, P.F.; Newnham, D.A.; Gordon, K.C.; Zeitler, J.A.; Pepper, M.; Rades, T. Using terahertz pulsed spectroscopy to quantify pharmaceutical polymorphism and crystallinity. J. Pharm. Sci. 2005, 94, 837–846. [Google Scholar] [CrossRef]
- Czamara, K.; Majzner, K.; Pacia, M.Z.; Kochan, K.; Kaczor, A.; Baranska, M. Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 2015, 46, 4–20. [Google Scholar] [CrossRef]
- Ahmmed, F.; Fraser-Miller, S.J.; Garagoda Arachchige, P.S.; Schallenberg, M.; Novis, P.; Gordon, K.C. Lake snow caused by the invasive diatom Lindavia intermedia can be discriminated from different sites and from other algae using vibrational spectroscopy. J. Raman Spectrosc. 2021, 52, 2597–2608. [Google Scholar] [CrossRef]
- Demšar, J.; Curk, T.; Erjavec, A.; Gorup, Č.; Hočevar, T.; Milutinovič, M.; Možina, M.; Polajnar, M.; Toplak, M.; Starič, A. Orange: Data mining toolbox in Python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Shpachenko, I.G.; Brandt, N.N.; Chikishev, A.Y. Time Variations in the FTIR Spectra of Liquids Measured in the ATR Configuration. Mosc. Univ. Phys. Bull. 2018, 73, 644–650. [Google Scholar] [CrossRef]
- Fraser-Miller, S.J.; Rooney, J.S.; Lau, M.; Gordon, K.C.; Schultz, M. Can Coupling Multiple Complementary Methods Improve the Spectroscopic Based Diagnosis of Gastrointestinal Illnesses? A Proof of Principle Ex Vivo Study Using Celiac Disease as the Model Illness. Anal. Chem. 2021, 93, 6363–6374. [Google Scholar] [CrossRef]
- Robert, C.; Jessep, W.; Sutton, J.J.; Hicks, T.M.; Loeffen, M.; Farouk, M.; Ward, J.F.; Bain, W.E.; Craigie, C.R.; Fraser-Miller, S.J.; et al. Evaluating low- mid- and high-level fusion strategies for combining Raman and infrared spectroscopy for quality assessment of red meat. Food Chem. 2021, 361, 130154. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adulterant | Method | No. Factors | Calibration | Prediction (Test Set) | Prediction (Test Set) SVM | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r2 | RMSEC (%) | r2 | Slope | Offset | RMSEP (%) | r2 | Slope | Offset | RMSEP (%) | |||
| PO% | R | 3 | 0.89 | 5.6 | 0.86 | 0.76 | 10.7 | 7.0 | 0.85 | 0.77 | 10.3 | 7.7 |
| I | 3 | 0.97 | 2.4 | 0.96 | 3.7 | 0.97 | 3.5 | 0.96 | 1.0 | 0.31 | 3.5 | |
| F | 2 | 0.94 | 4.2 | 0.97 | 0.93 | 3.3 | 3.6 | 0.98 | 0.96 | 1.9 | 2.7 | |
| O3C% | R | 4 | 0.98 | 2.4 | 0.98 | 1.1 | −1.5 | 2.4 | 0.95 | 0.96 | 2.8 | 2.3 |
| I | 2 | 0.91 | 5.3 | 0.96 | 0.87 | 1.2 | 3.5 | 0.95 | 0.83 | 2.7 | 4.2 | |
| F | 3 | 0.96 | 3.7 | 0.96 | 1.0 | 1.2 | 3.3 | 0.97 | 0.94 | 2.8 | 3.0 | |
| FO% | R | 4 | 0.95 | 3.4 | 0.90 | 0.92 | 6.3 | 5.8 | 0.31 | 0.59 | 18.8 | 15.6 |
| I | 3 | 0.90 | 5.0 | 0.78 | 0.85 | −3.1 | 8.4 | 0.73 | 0.72 | 0.12 | 9.6 | |
| F | 2 | 0.82 | 6.7 | 0.75 | 0.84 | −3.1 | 9.1 | 0.76 | 0.89 | −5.2 | 9.1 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmmed, F.; Gordon, K.C.; Killeen, D.P.; Fraser-Miller, S.J. Detection and Quantification of Adulteration in Krill Oil with Raman and Infrared Spectroscopic Methods. Molecules 2023, 28, 3695. https://doi.org/10.3390/molecules28093695
Ahmmed F, Gordon KC, Killeen DP, Fraser-Miller SJ. Detection and Quantification of Adulteration in Krill Oil with Raman and Infrared Spectroscopic Methods. Molecules. 2023; 28(9):3695. https://doi.org/10.3390/molecules28093695
Chicago/Turabian StyleAhmmed, Fatema, Keith C. Gordon, Daniel P. Killeen, and Sara J. Fraser-Miller. 2023. "Detection and Quantification of Adulteration in Krill Oil with Raman and Infrared Spectroscopic Methods" Molecules 28, no. 9: 3695. https://doi.org/10.3390/molecules28093695
APA StyleAhmmed, F., Gordon, K. C., Killeen, D. P., & Fraser-Miller, S. J. (2023). Detection and Quantification of Adulteration in Krill Oil with Raman and Infrared Spectroscopic Methods. Molecules, 28(9), 3695. https://doi.org/10.3390/molecules28093695