
A Two-Step Synthesis of Unprotected 3-Aminoindoles via Post Functionalization with Nitrostyrene

 , , , , and
, , , , and 
Abstract
1. Introduction
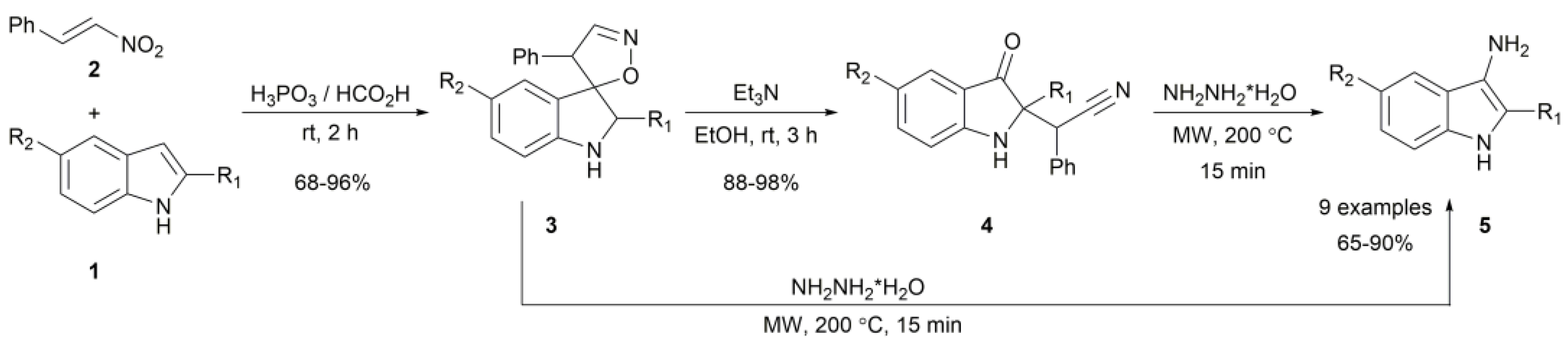
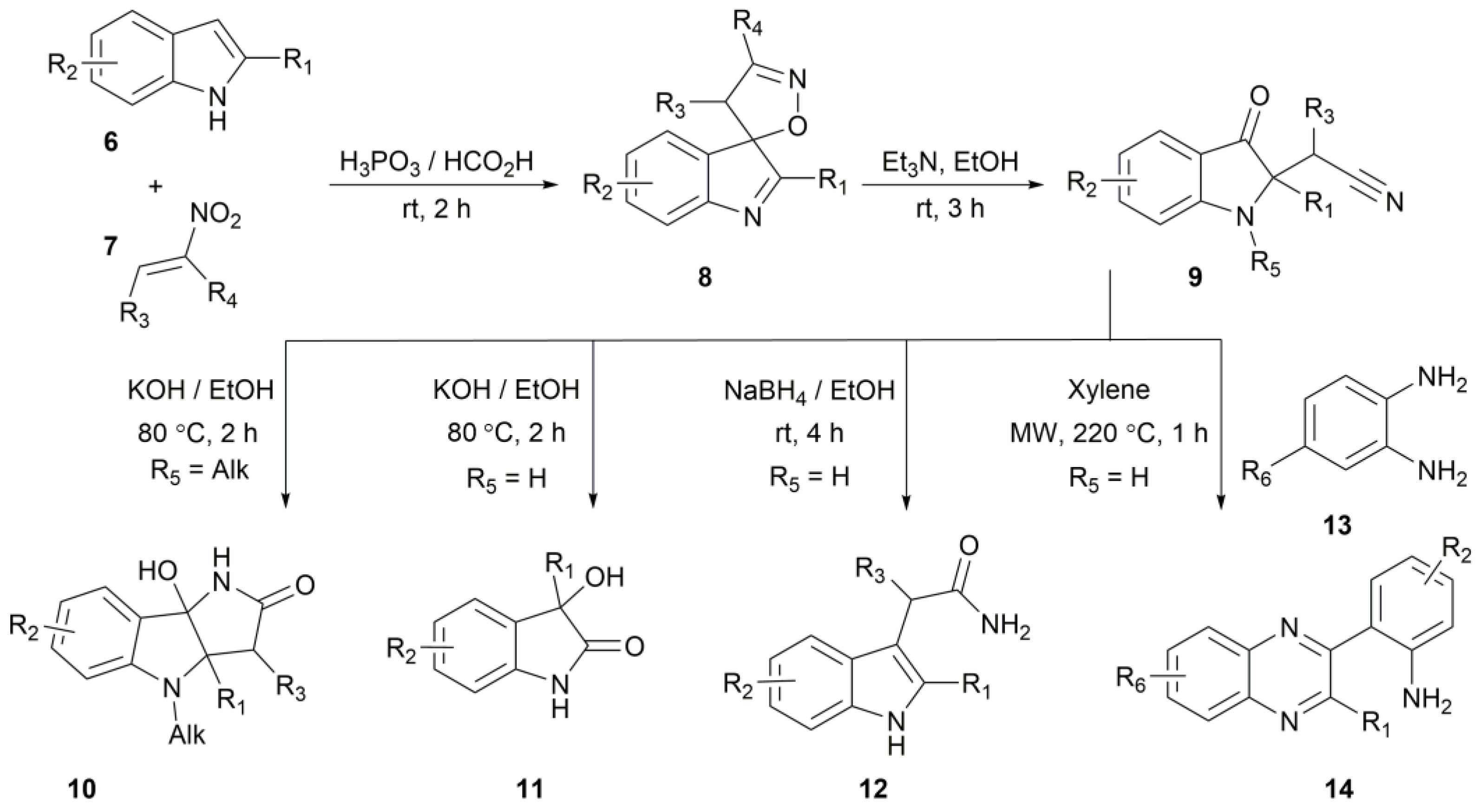
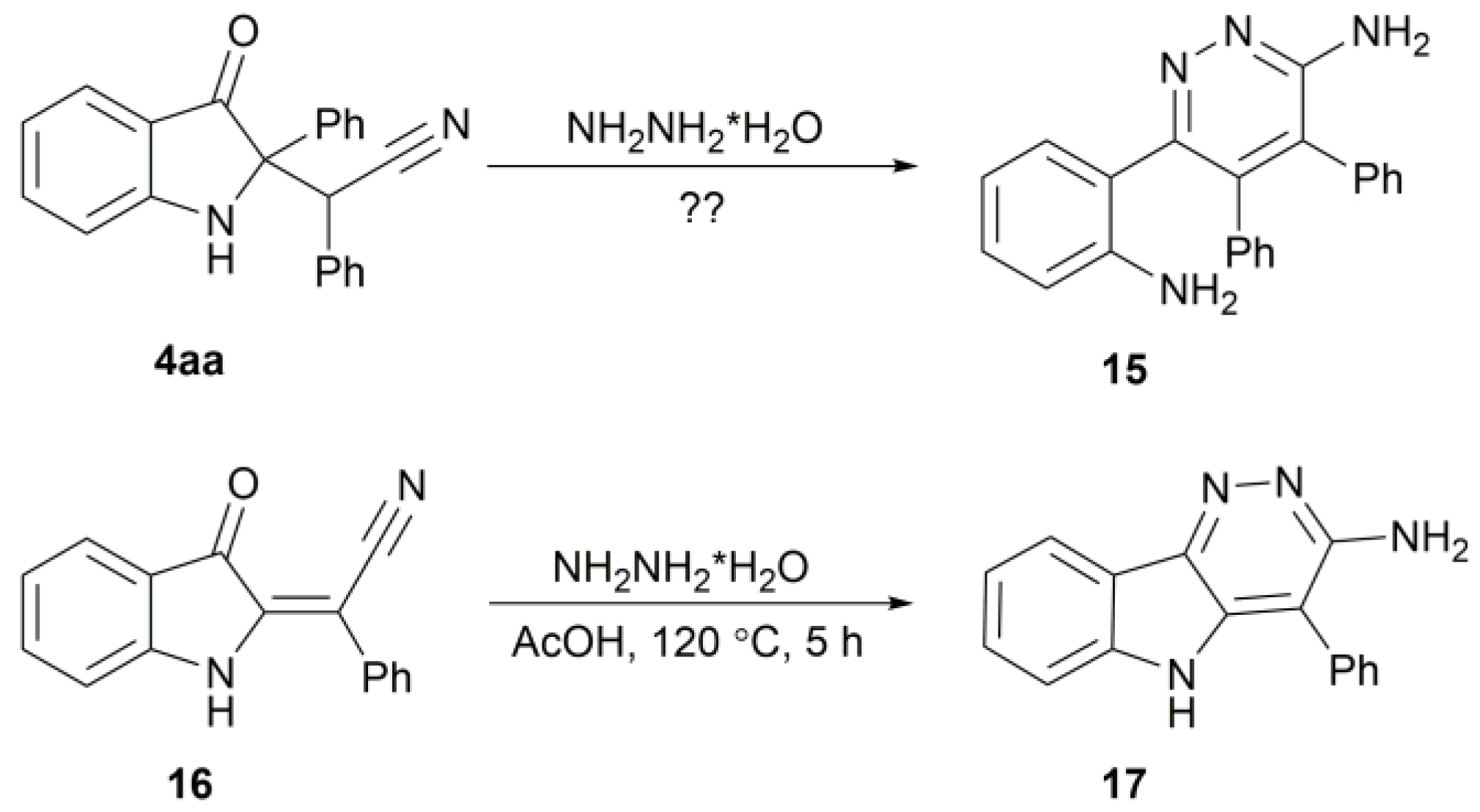
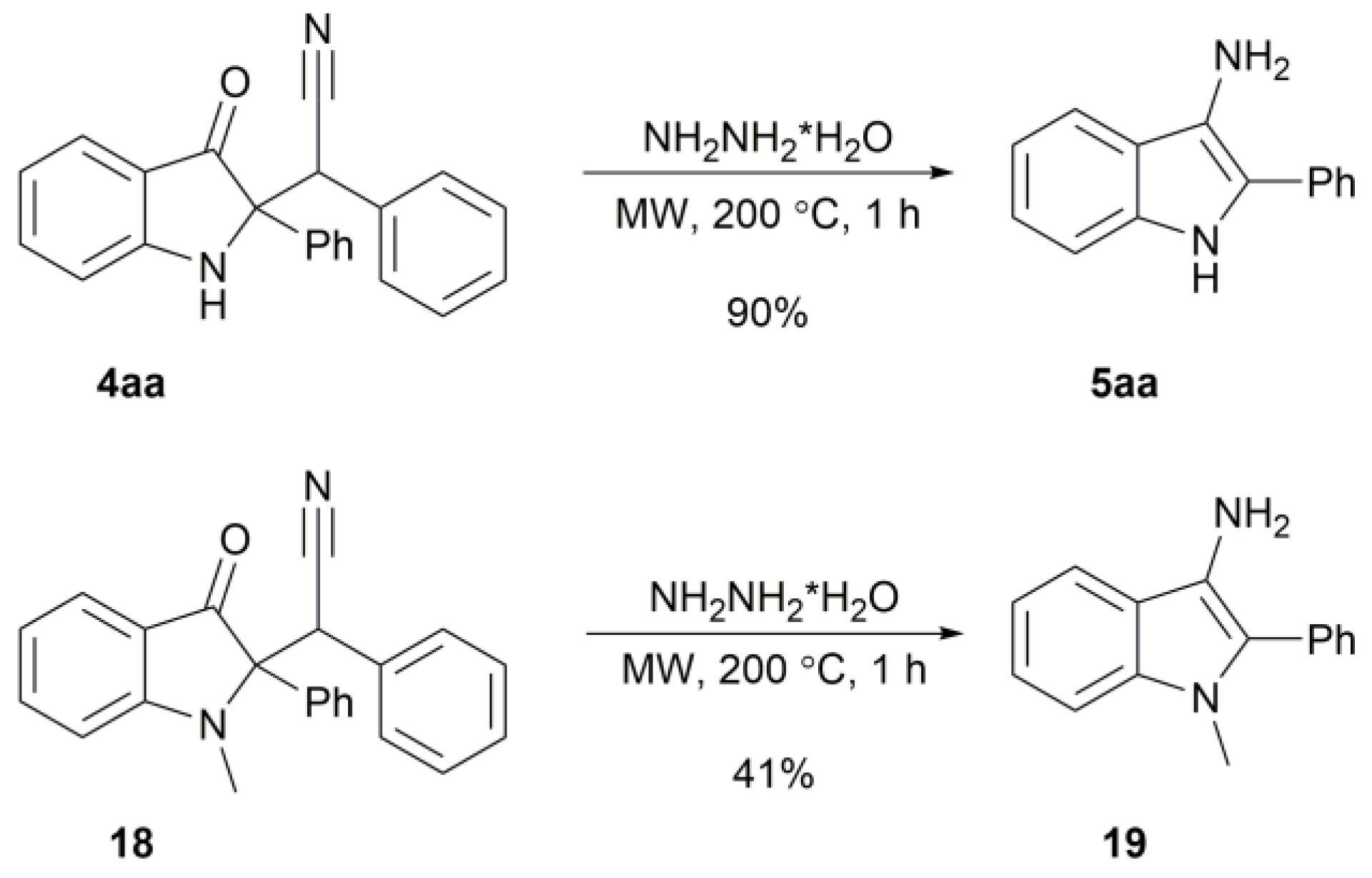
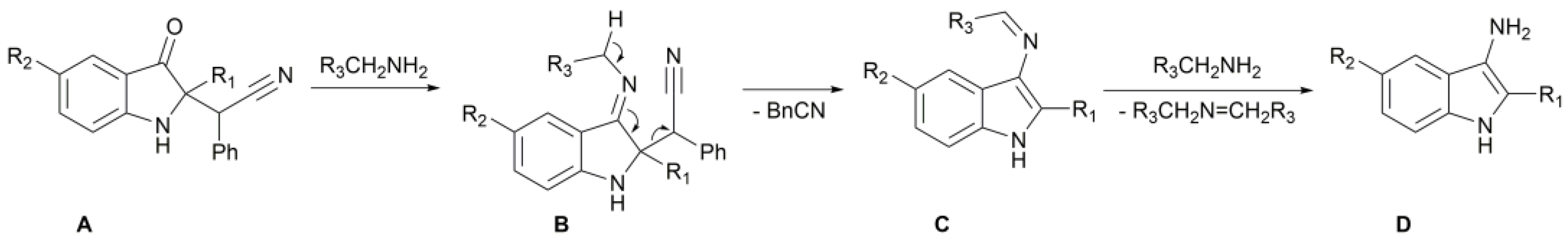
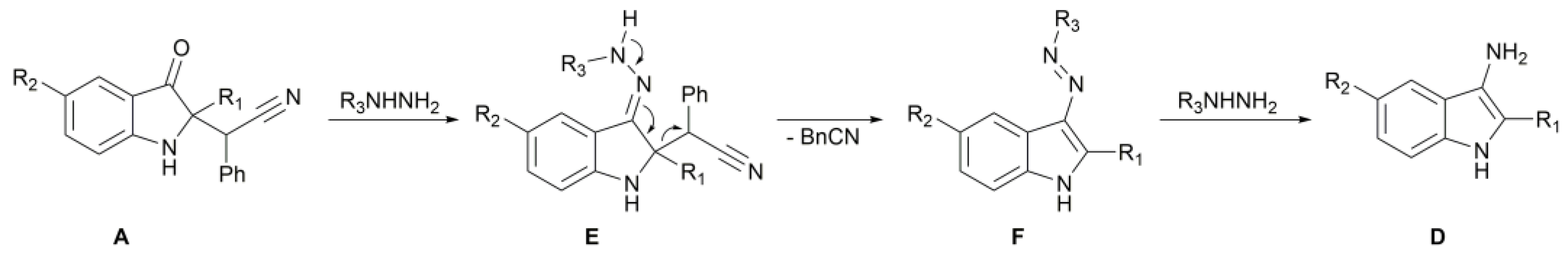
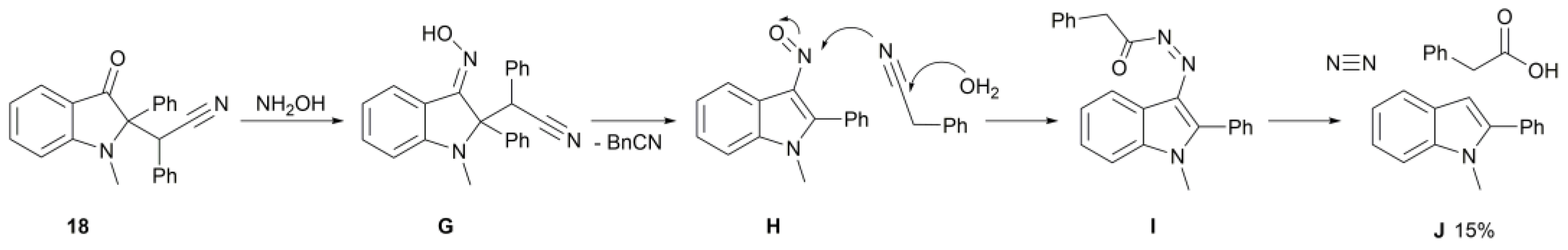
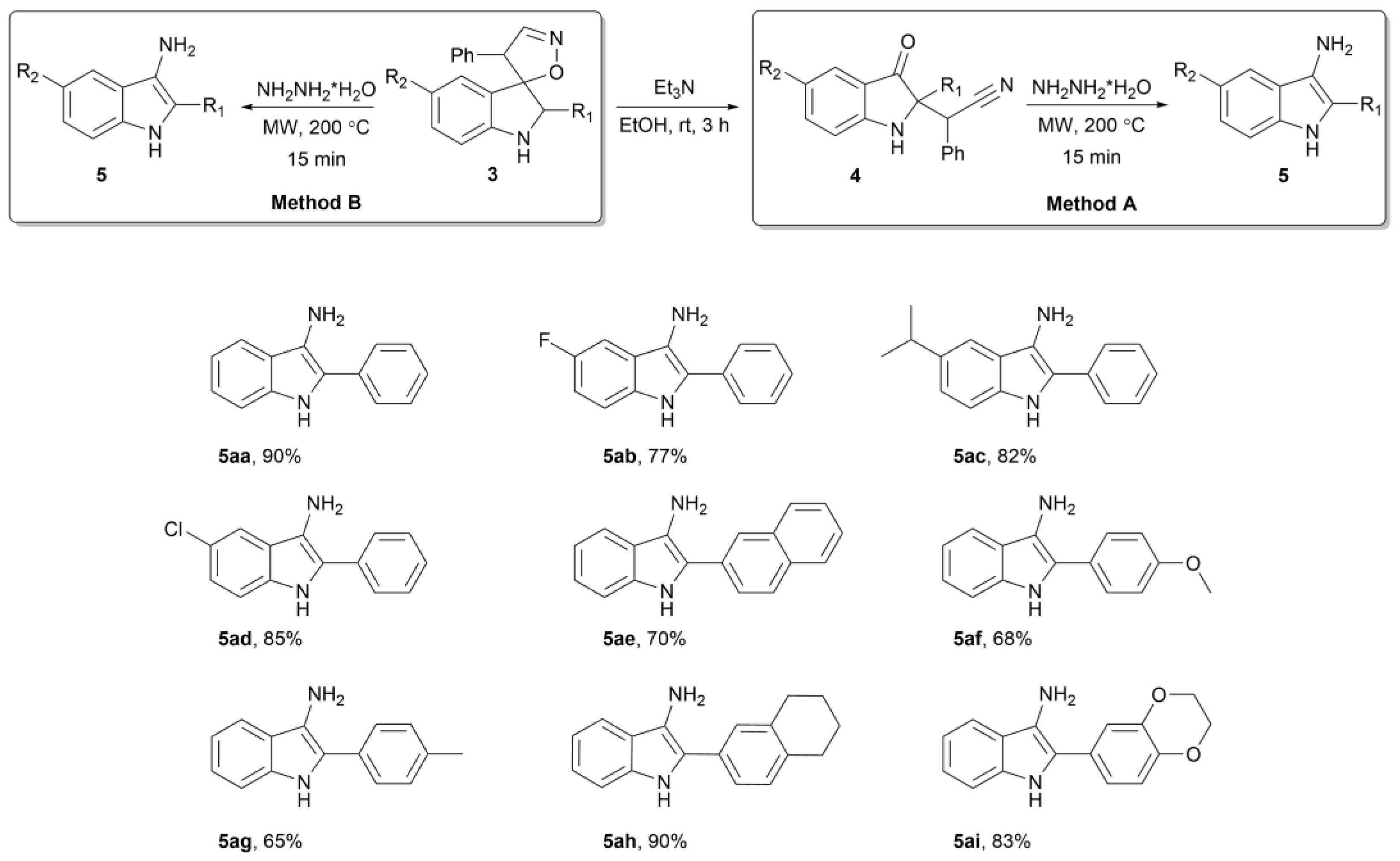
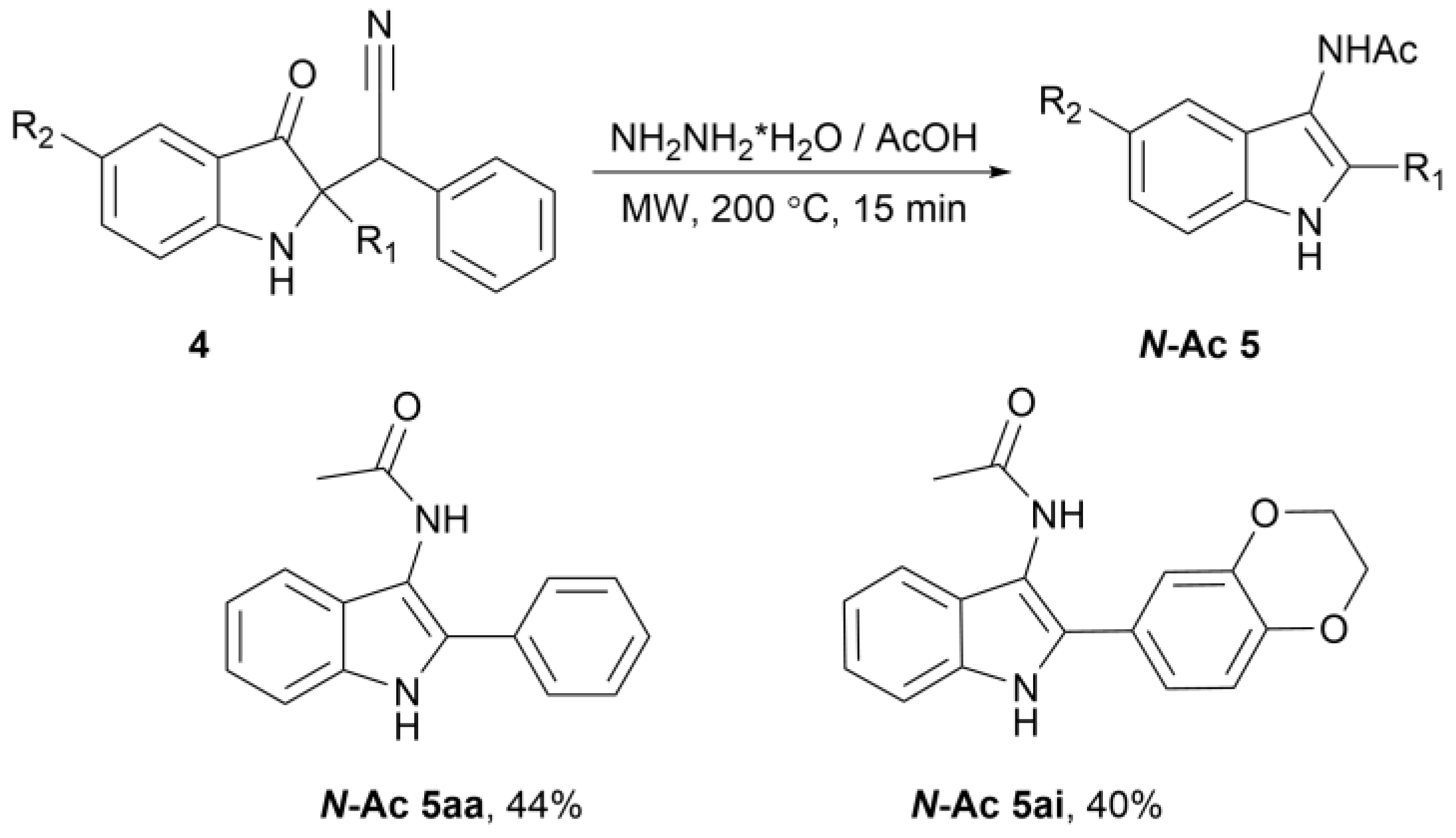
2. Results
3. Materials and Methods
3.1. General Information
3.2. Preparation of 2-(5-Halide-3-Oxo-2-Phenylindolin-2-Yl)-2-Phenylacetonitrile 4ab, 4ad (General Procedure)
3.3. Preparation of 2-Aryl-1H-Indol-3-Amine 5aa–ai (General Procedure)
3.4. Preparation of N-(2-Aryl-1H-Indol-3-yl)acetamide N-Ac 5aa and N-Ac 5ai (General Procedure)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Arzel, E.; Rocca, P.; Grellier, P.; Labaeïd, M.; Frappier, F.; Guéritte, F.; Gaspard, C.; Marsais, F.; Godard, A.; Quéguiner, G. New Synthesis of Benzo-δ-carbolines, Cryptolepines, and Their Salts: In Vitro Cytotoxic, Antiplasmodial, and Antitrypanosomal Activities of δ-Carbolines, Benzo-δ-carbolines, and Cryptolepines. J. Med. Chem. 2001, 44, 949–960. [Google Scholar] [CrossRef]
- Bahekar, R.H.; Jain, M.R.; Goel, A.; Patel, D.N.; Prajapati, V.M.; Gupta, A.A.; Jadav, P.A.; Patel, P.R. Design, synthesis, and biological evaluation of substituted-N-(thieno[2,3-b]pyridin-3-yl)-guanidines, N-(1H-pyrrolo[2,3-b]pyridin-3-yl)-guanidines, and N-(1H-indol-3-yl)-guanidines. Bioorg. Med. Chem. 2007, 15, 3248–3265. [Google Scholar] [CrossRef] [PubMed]
- Pews-Davtyan, A.; Tillack, A.; Schmöle, A.-C.; Ortinau, S.; Frech, M.J.; Rolfs, A.; Beller, M. A new facile synthesis of 3-amidoindole derivatives and their evaluation as potential GSK-3β inhibitors. Org. Biomol. Chem. 2010, 8, 1149–1153. [Google Scholar] [CrossRef]
- Nirogi, R.V.; Deshpande, A.D.; Kambhampati, R.; Badange, R.K.; Kota, L.; Daulatabad, A.V.; Shinde, A.K.; Ahmad, I.; Kandikere, V.; Jayarajan, P.; et al. Indole-3-piperazinyl derivatives: Novel chemical class of 5-HT6 receptor antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Sarkar, T.; Carrion, M.D.; Lopez-Cara, L.C.; Cruz-Lopez, O.; Preti, D.; Tabrizi, M.A.; Tolomeo, M.; Grimaudo, S.; et al. Synthesis and Biological Evaluation of 1-Methyl-2-(3′,4′,5′-trimethoxybenzoyl)-3-aminoindoles as a New Class of Antimitotic Agents and Tubulin Inhibitors. J. Med. Chem. 2008, 51, 1464–1468. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.; Warnhoff, E.W. New reactions of 2-substituted indoles. Can. J. Chem. 1976, 54, 1020–1028. [Google Scholar] [CrossRef]
- Leijendekker, L.H.; Weweler, J.; Leuther, T.M.; Streuff, J. Catalytic Reductive Synthesis and Direct Derivatization of Unprotected Aminoindoles, Aminopyrroles, and Iminoindolines. Angew. Chem. Int. Ed. 2017, 56, 6103–6106. [Google Scholar] [CrossRef]
- Leijendekker, L.H.; Weweler, J.; Leuther, T.M.; Kratzert, D.; Streuff, J. Development, Scope, and Applications of Titanium(III)-Catalyzed Cyclizations to Aminated N-Heterocycles. Chem.-A Eur. J. 2019, 25, 3382–3390. [Google Scholar] [CrossRef]
- Zhang, G.; Lin, L.; Yang, K.; Wang, S.; Feng, Q.; Zhu, J.; Song, Q. 3-Aminoindole Synthesis from 2-Nitrochalcones and Ammonia or Primary Amines. Adv. Synth. Catal. 2019, 361, 3718–3722. [Google Scholar] [CrossRef]
- Nettekoven, M. Combinatorial synthesis of libraries of indole derivatives. Bioorg. Med. Chem. Lett. 2001, 11, 2169–2171. [Google Scholar] [CrossRef]
- Harjani, J.R.; Tang, A.X.; Norton, R.S.; Baell, J.B. A new methodology for the synthesis of 3-amino-1H-indole-2-carboxylates. Tetrahedron 2014, 70, 8047–8055. [Google Scholar] [CrossRef]
- Geng, X.; Wu, X.; Wang, C.; Zhao, P.; Zhou, Y.; Sun, X.; Wang, L.-J.; Guan, W.-J.; Wu, Y.-D.; Wu, A.-X. NaHS·nH2O-induced umpolung: The synthesis of 2-acyl-3-aminoindoles from aryl methyl ketones and 2-aminobenzonitriles. Chem. Commun. 2018, 54, 12730–12733. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Roy, S.; Gribble, G.W. Efficient reductive acylation of 3-nitroindoles. Tetrahedron Lett. 2008, 49, 1531–1533. [Google Scholar] [CrossRef]
- Seong, C.M.; Park, C.M.; Choi, J.; Park, N.S. An efficient base-mediated intramolecular condensation of 2-(disubstituted amino)-benzonitriles to 3-aminoindoles. Tetrahedron Lett. 2009, 50, 1029–1031. [Google Scholar] [CrossRef]
- Gribble, G.; Saulnier, M.; Pelkey, E.; Kishbaugh, T.; Liu, Y.; Jiang, J.; Trujillo, H.; Keavy, D.; Davis, D.; Conway, S.; et al. Novel Indole Chemistry in the Synthesis of Heterocycles. Curr. Org. Chem. 2005, 9, 1493–1519. [Google Scholar] [CrossRef]
- Dang, Q.; Bai, X.; Xu, G.; Zheng, L. Total Synthesis of 4-Azaeudistomin Y1 and Analogues by Inverse-ElectronDemand Diels-Alder Reactions of 3-Aminoindoles with 1,3,5-Triazines. Synthesis 2013, 45, 743–752. [Google Scholar] [CrossRef]
- Gribble, G.W.; Roy, S. Convenient Synthesis of Masked Aminoindoles by Indium Mediated Ont-Pot Reductive Acylation of 3- and 2-Nitroindoles. Heterocycles 2006, 70, 51. [Google Scholar] [CrossRef]
- Prasad, P.K.; Kalshetti, R.G.; Reddi, R.N.; Kamble, S.P.; Sudalai, A. I2-mediated regioselective C-3 azidation of indoles. Org. Biomol. Chem. 2016, 14, 3027–3030. [Google Scholar] [CrossRef]
- Przheval’skii, N.M.; Skvortsova, N.S.; Magedov, I.V. New derivatives of 3-Aminoindole. Synthesis of 2-Aryl(Hetaryl)-3-(3,5-Dimethyl-1-Pyrazolyl)indoles. Chem. Heterocycl. Compd. 2004, 40, 1435–1441. [Google Scholar] [CrossRef]
- Przheval’Skii, N.M.; Skvortsova, N.S.; Magedov, I.V. Fischer Synthesis of 3-(N-Acylamino)-2-phenylindoles. Chem. Heterocycl. Compd. 2002, 38, 1055–1061. [Google Scholar] [CrossRef]
- Chernyak, D.; Chernyak, N.; Gevorgyan, V. Efficient and General Synthesis of 3-Aminoindolines and 3-Aminoindoles via Copper-Catalyzed Three-Component Coupling Reaction. Adv. Synth. Catal. 2010, 352, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Pews-Davtyan, A.; Beller, M. Efficient and simple zinc-mediated synthesis of 3-amidoindoles. Org. Biomol. Chem. 2011, 9, 6331–6334. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Z.; Li, Z. Switchable Synthesis of 2-Methylene-3-aminoindolines and 2-Methyl-3-aminoindoles Using Calcium Carbide as a Solid Alkyne Source. Org. Lett. 2022, 24, 8067–8071. [Google Scholar] [CrossRef] [PubMed]
- Diao, P.-C.; Hu, M.-J.; Yang, H.-K.; You, W.-W.; Zhao, P.-L. Facile one-pot synthesis, antiproliferative evaluation and structure-activity relationships of 3-amino-1H-indoles and 3-amino-1H-7-azaindoles. Bioorg. Chem. 2019, 88, 102914. [Google Scholar] [CrossRef]
- Gribble, G.W. Synthesis and Reactions of Nitroindoles. In Progress in Heterocyclic Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; Volume 31, pp. 83–117. [Google Scholar] [CrossRef]
- Abdel-Rahman, A.A.; El-Latif, M.M.A.; El-Essawy, F.A.; Barakat, Y.A. Synthesis and Antiviral Activity of 3-Aminoindole Nucleosides of 2-Acetamido-2-deoxy-D-glucose. Bull. Korean Chem. Soc. 2012, 33, 3417–3422. [Google Scholar] [CrossRef]
- Suvorov, N.N.; Velezheva, V.S.; Yarosh, A.V.; Erofeev, Y.V.; Kozik, T.N. Indole derivatives. Chem. Heterocycl. Compd. 1975, 11, 959–964. [Google Scholar] [CrossRef]
- Hooper, M.W.; Utsunomiya, M.; Hartwig, J.F. Scope and Mechanism of Palladium-Catalyzed Amination of Five-Membered Heterocyclic Halides. J. Org. Chem. 2003, 68, 2861–2873. [Google Scholar] [CrossRef]
- Muralidhar, P.; Kumar, B.S.; Nagaraju, K.; Maddila, S. A novel method for the synthesis of 3-aminoindoles using iodine and Cs2CO3 as catalyst. Chem. Data Collect. 2021, 33, 100731. [Google Scholar] [CrossRef]
- Ortiz, G.X., Jr.; Hemric, B.N.; Wang, Q. Direct and Selective 3-Amidation of Indoles Using Electrophilic N-[(Benzenesulfonyl)oxy]amides. Org. Lett. 2017, 19, 1314–1317. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yamaguchi, E.; Itoh, A. Cross-Dehydrogenative C–H Amination of Indoles under Aerobic Photo-oxidative Conditions. Org. Lett. 2017, 19, 1282–1285. [Google Scholar] [CrossRef]
- Chen, W.-L.; Li, K.; Liao, W.-C.; Liang, W.-F.; Qiu, P.-W.; Liang, C.; Su, G.-F.; Mo, D.-L. An iron(iii)-catalyzed dehydrogenative cross-coupling reaction of indoles with benzylamines to prepare 3-aminoindole derivatives. Green Chem. 2021, 23, 9610–9616. [Google Scholar] [CrossRef]
- Benkovics, T.; Guzei, I.A.; Yoon, T.P. Oxaziridine-Mediated Oxyamination of Indoles: An Approach to 3-Aminoindoles and Enantiomerically Enriched 3-Aminopyrroloindolines. Angew. Chem. Int. Ed. 2010, 49, 9153–9157. [Google Scholar] [CrossRef]
- Sokolovs, I.; Lubriks, D.; Suna, E. Copper-Catalyzed Intermolecular C–H Amination of (Hetero)arenes via Transient Unsymmetrical λ3-Iodanes. J. Am. Chem. Soc. 2014, 136, 6920–6928. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Aksenov, N.A.; Aksenov, D.A.; Khamraev, V.F.; Rubin, M. Nitrostyrenes as 1,4-CCNO-dipoles: Diastereoselective formal [4+1] cycloaddition of indoles. Chem. Commun. 2018, 54, 13260–13263. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, D.A.; Arutiunov, N.A.; Aksenov, N.A.; Aleksandrova, E.V.; Zhao, Z.; Du, L.; Kornienko, A.; Rubin, M. Synthesis of Spiro[indole-3,5′-isoxazoles] with Anticancer Activity via a Formal [4 + 1]-Spirocyclization of Nitroalkenes to Indoles. J. Org. Chem. 2019, 84, 7123–7137. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Aksenov, D.A.; Aksenov, N.A.; Aleksandrova, E.V.; Rubin, M. Preparation of Stereodefined 2-(3-Oxoindolin-2-yl)-2-Arylacetonitriles via One-Pot Reaction of Indoles with Nitroalkenes. J. Org. Chem. 2019, 84, 12420–12429. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Aksenov, N.A.; Aleksandrova, E.V.; Aksenov, D.A.; Grishin, I.Y.; Sorokina, E.A.; Wenger, A.; Rubin, M. Direct Conversion of 3-(2-Nitroethyl)-1H-Indoles into 2-(1H-Indol-2-yl)Acetonitriles. Molecules 2021, 26, 6132. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aleksandrova, E.V.; Aksenov, D.A.; Aksenova, A.A.; Aksenov, N.A.; Nobi, M.A.; Rubin, M. Synthetic Studies toward 1,2,3,3a,4,8b-Hexahydropyrrolo[3,2-b]indole Core. Unusual Fragmentation with 1,2-Aryl Shift. J. Org. Chem. 2022, 87, 1434–1444. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Kirilov, N.K.; Arutiunov, N.A.; Aksenov, D.A.; Kuzminov, I.K.; Aksenov, N.A.; Turner, D.N.; Rogelj, S.; Kornienko, A.; Rubin, M. Reductive Cleavage of 4′H-Spiro[indole-3,5′-isoxazoles] En Route to 2-(1H-Indol-3-yl)acetamides with Anticancer Activities. J. Org. Chem. 2022, 87, 13955–13964. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Arutiunov, N.A.; Aksenov, D.A.; Samovolov, A.V.; Kurenkov, I.A.; Aksenov, N.A.; Aleksandrova, E.A.; Momotova, D.S.; Rubin, M. A Convenient Way to Quinoxaline Derivatives through the Reaction of 2-(3-Oxoindolin-2-yl)-2-phenylacetonitriles with Benzene-1,2-diamines. Int. J. Mol. Sci. 2022, 23, 11120. [Google Scholar] [CrossRef]
- Velezheva, V.S.; Brennan, P.J.; Marshakov, V.Y.; Gusev, D.V.; Lisichkina, I.N.; Peregudov, A.S.; Tchernousova, L.N.; Smirnova, T.G.; Andreevskaya, S.N.; Medvedev, A.E. Novel Pyridazino[4,3-b]indoles with Dual Inhibitory Activity against Mycobacterium tuberculosis and Monoamine Oxidase. J. Med. Chem. 2004, 47, 3455–3461. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.S.; Gowda, S.; Gowda, D.C. Facile Transfer Hydrogenation of Azo Compounds to Hydrazo Compounds and Anilines by Using Raney Nickel and Hydrazinium Monoformate. Synth. Commun. 2004, 34, 1–10. [Google Scholar] [CrossRef]
- Pasha, M.; Nanjundaswamy, H. Reductive fission of azoarenes to aminoarenes by aluminium/hydrazine hydrate. J. Chem. Res. 2004, 2004, 750–752. [Google Scholar] [CrossRef]
- Pasha, M.A.; NanjundaSwamy, H.M. Uncatalyzed Reductive Fission of Azoarenes to Aminoarene(s) by Hydrazine Hydrate. Synth. Commun. 2005, 35, 897–900. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| # | Binucleophile, RXHNH2 | Conditions | Yield 5aa(Isolated), % |
| 1 | Ethylenediamine | 1-butanol, MW 200 °C, 0.5 h | 37 |
| 2 | Propane-1,2-diamine | 1-butanol, MW 200 °C, 0.5 h | 41 |
| 3 | Formamidine acetate | 1-butanol, MW 185 °C, 1 h | decomposed |
| 4 | Thiosemicarbazide | Xylene, MW 180 °C, 0.5 h | 15 |
| 5 | 2-hydrazinylpyridine | Xylene, MW 160 °C, 0.5 h | 20 |
| 6 | Phenylhydrazine | Xylene, MW 200 °C, 0.5 h | 35 |
| 7 | Tosylhydrazine | Xylene, MW 200 °C, 0.5 h | 39 |
| 8 | Benzohydrazide | Xylene, MW 200 °C, 0.5 h | 26 |
| 9 | Hydrazine hydrate | neat, MW 200 °C, 15 min | 90 |
| 10 | Hydroxylamine | Xylene, MW 200 °C, 0.5 h | n.d. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksenov, N.A.; Arutiunov, N.A.; Kurenkov, I.A.; Malyuga, V.V.; Aksenov, D.A.; Momotova, D.S.; Zatsepilina, A.M.; Chukanova, E.A.; Leontiev, A.V.; Aksenov, A.V. A Two-Step Synthesis of Unprotected 3-Aminoindoles via Post Functionalization with Nitrostyrene. Molecules 2023, 28, 3657. https://doi.org/10.3390/molecules28093657
Aksenov NA, Arutiunov NA, Kurenkov IA, Malyuga VV, Aksenov DA, Momotova DS, Zatsepilina AM, Chukanova EA, Leontiev AV, Aksenov AV. A Two-Step Synthesis of Unprotected 3-Aminoindoles via Post Functionalization with Nitrostyrene. Molecules. 2023; 28(9):3657. https://doi.org/10.3390/molecules28093657
Chicago/Turabian StyleAksenov, Nicolai A., Nikolai A. Arutiunov, Igor A. Kurenkov, Vladimir V. Malyuga, Dmitrii A. Aksenov, Daria S. Momotova, Anna M. Zatsepilina, Elizaveta A. Chukanova, Alexander V. Leontiev, and Alexander V. Aksenov. 2023. "A Two-Step Synthesis of Unprotected 3-Aminoindoles via Post Functionalization with Nitrostyrene" Molecules 28, no. 9: 3657. https://doi.org/10.3390/molecules28093657
APA StyleAksenov, N. A., Arutiunov, N. A., Kurenkov, I. A., Malyuga, V. V., Aksenov, D. A., Momotova, D. S., Zatsepilina, A. M., Chukanova, E. A., Leontiev, A. V., & Aksenov, A. V. (2023). A Two-Step Synthesis of Unprotected 3-Aminoindoles via Post Functionalization with Nitrostyrene. Molecules, 28(9), 3657. https://doi.org/10.3390/molecules28093657