2.2. Catalysts Based on LSA as a Support Matrix for the Immobilization of Transition Metal Ions (LSA-Mn+)
The adequate acid–base properties shown by
LSA allowed its entrance to the catalysis field in a fruitful manner. From this point, the need to expand their catalytic performance toward other chemical transformations became notably attractive in offering a new class of biobased catalysts produced from this highly abundant industrial waste. However, depending only on acid–base properties is not enough to reach catalytic success in different chemical reactions, and from this situation, the inclusion of well-known catalytic transition metals emerged as a suitable strategy to expand the utility of these catalysts by searching for synergistic effects between the
LSA and catalytic metal ions. In addition, another advantage of this strategy is the reutilization of the material in consecutive reaction cycles due to its insolubility in the reaction media, avoiding the loss of these usually expensive transition metals but also preventing their leakage into the environment. Regarding the above, the first good example of this strategy was achieved in 2013 by Sun and collaborators [
33], where they successfully achieved the immobilization of two different metal triflate derivates, Sc(OTf)
3 and Cu(OTf)
2, on
LSA-Na+ (
Figure 3). The authors took advantage of the charged character of
LSA-Na+ and separately by a simple ion exchange process which achieved the coordination of both triflate species. Typically, the impregnation process was carried out by dispersing a certain amount of
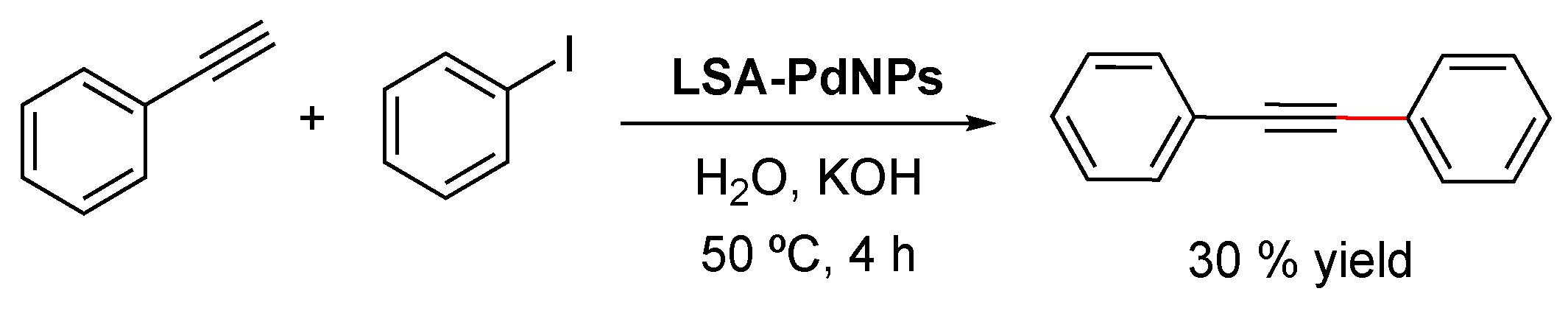
LSA-Na+ into an ethanolic solution of the metal precursor, after which isolation of the hybrid catalyst was easily achieved by filtration, followed by several washing steps, and ending with a final drying stage. It must be mentioned that, in this case, the authors performed the impregnation method at 80 °C and 36 h; however, these parameters should be carefully selected depending on the nature of the metal precursor. Additionally, the authors corroborated the importance of deprotonated sulfonic groups present in the support by observing a direct relation between the metal load and the amount of SO
3− species. After obtaining the catalyst, the most important characterization corresponds to the quantification of the metal loading by means of inductively coupled plasma mass spectrometry (ICP-MS). In this work, the authors reached loading values of 0.022 and 0.044 mmol/g for
LSA-Sc3+ and 0.80 mmol/g for
LSA-Cu2+, which were employed with good results in the catalyzed formation of C–C bonds.
Interestingly, the authors extended this impregnation protocol and successfully tested the immobilization of catalytic nonmetallic species, that is, an amine-functionalized imidazole-based ionic liquid (
LSA-IL@NH2). Although there are no metal ions involved in this case, it constitutes an isolated example described in the same publication [
33]. Therefore, we have decided to mention it here rather than creating a separate section. Briefly, the catalyst was achieved after performing the ionic exchange process between ammonium lignosulfonate (
LSA-NH4+) and a suitable ionic liquid (IL) derivative [
33], allowing the preparation of a catalytic system with an IL loading of 0.48 mmol/g (calculated from EA). Even when the above work stood out by the wide variety of reactions tested, a very limited catalyst characterization was performed, being mostly based on the quantification of metal loadings. Notwithstanding the above, seven years later (2020), some members of the same group involved in the above work contributed a new and improved methodology for the impregnation of Cu
2+ on
LSA, along with its catalytic assessment [
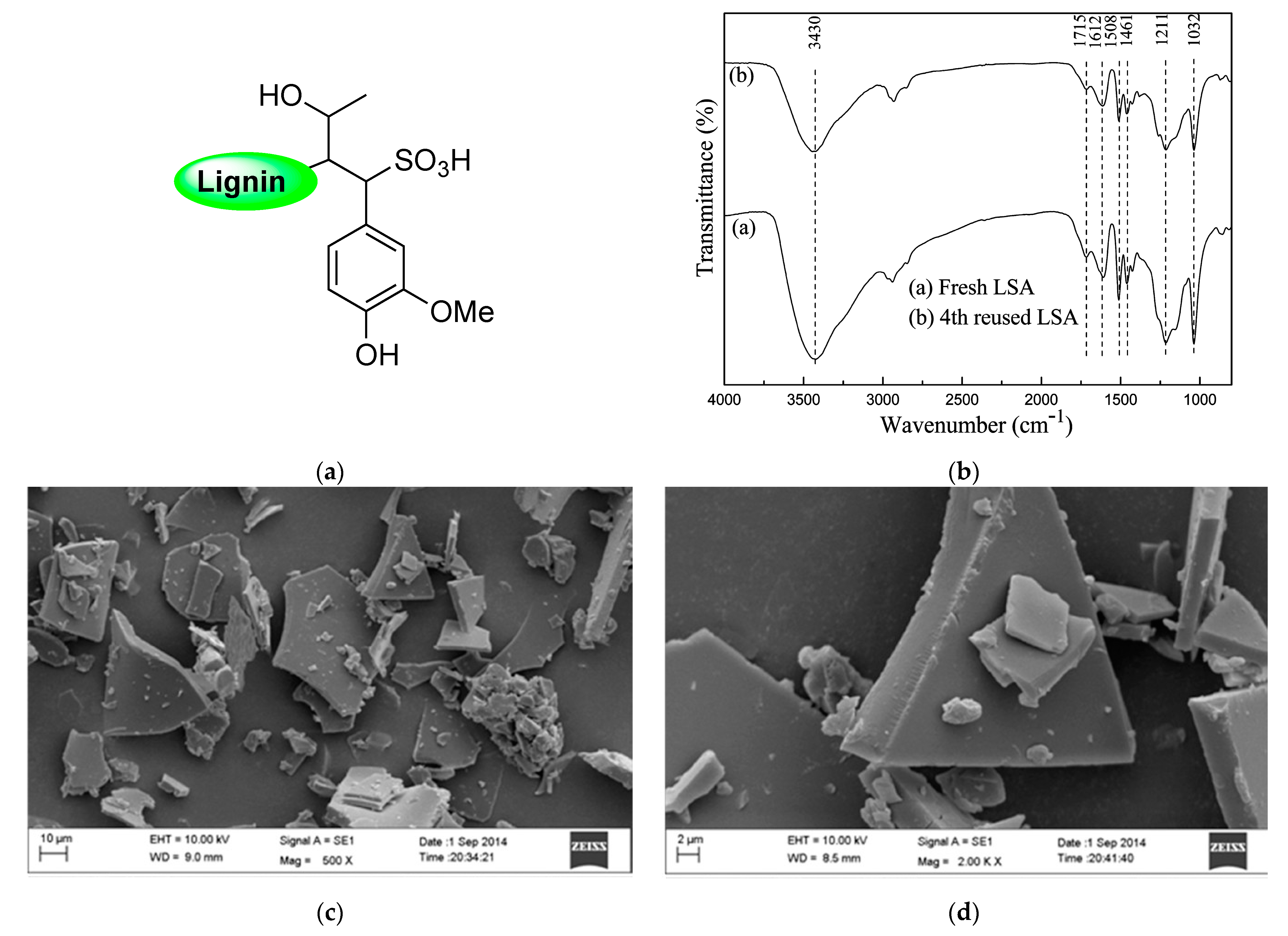
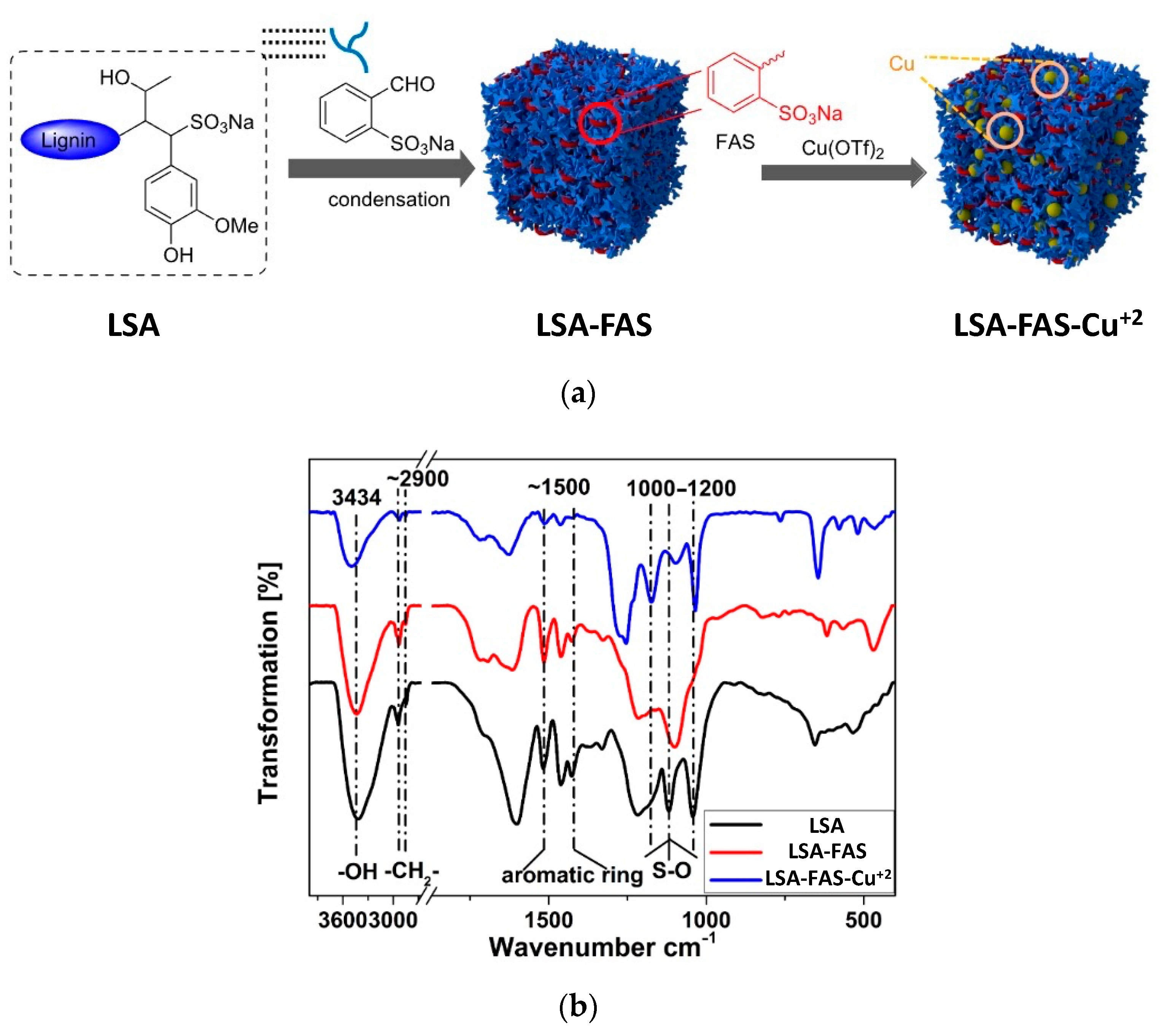
34]. Additionally, in this opportunity, a more in-depth characterization of the catalyst was performed. The new strategy relied on the increment of sulfonic groups by chemically modifying
LSA with 2-formylbenzenesulfonic acid sodium (
FAS) (
Figure 4a). To achieve this, the authors carried out condensation reactions between the phenol residues of
LSA and the aldehyde group of
FAS entities. The process was performed using water as the solvent and concentrated HCl as the catalyst while stirring at 90 °C and 8 h of reaction, after which the obtained solid (
LSA-FAS) was isolated by filtration, abundantly washed, and finally dried at 110 °C. The increase of sulfonic groups was constated by EA, from which an increment of approximately 4% was measured for S% regarding unmodified
LSA. The authors devised the increment of SO
3H entities as an efficient way to introduce more metal-coordinating sites enhancing the metal fixation on the material. In this regard, they achieved the impregnation of Cu(OTf)
2 species by dispersing
LSA-FAS in refluxing ethanol containing the copper precursor. After purifying the
LSA-FAS-Cu2+ catalyst, ICP analysis revealed a Cu
2+ loading of 0.92 mmol/g, which was considerably higher than the values of 0.45 and 0.56 mmol/g reached with two other reference materials, thereby confirming the success of the strategy [
34]. The chemical structure of the material was first analyzed by FTIR spectroscopy, allowing the identification of typical signals ascribed to the lignin backbone, especially those accusing the presence of SO
3− and SO
3H structures, [
35,
36] (
Figure 4b).
Moreover, FTIR spectra of
LSA,
LSA-FAS, and
LSA-FAS-Cu2+ also revealed important pattern changes when compared to each other, where authors attributed this variation first to the incorporation of
FAS units, and then by the complexation of Cu
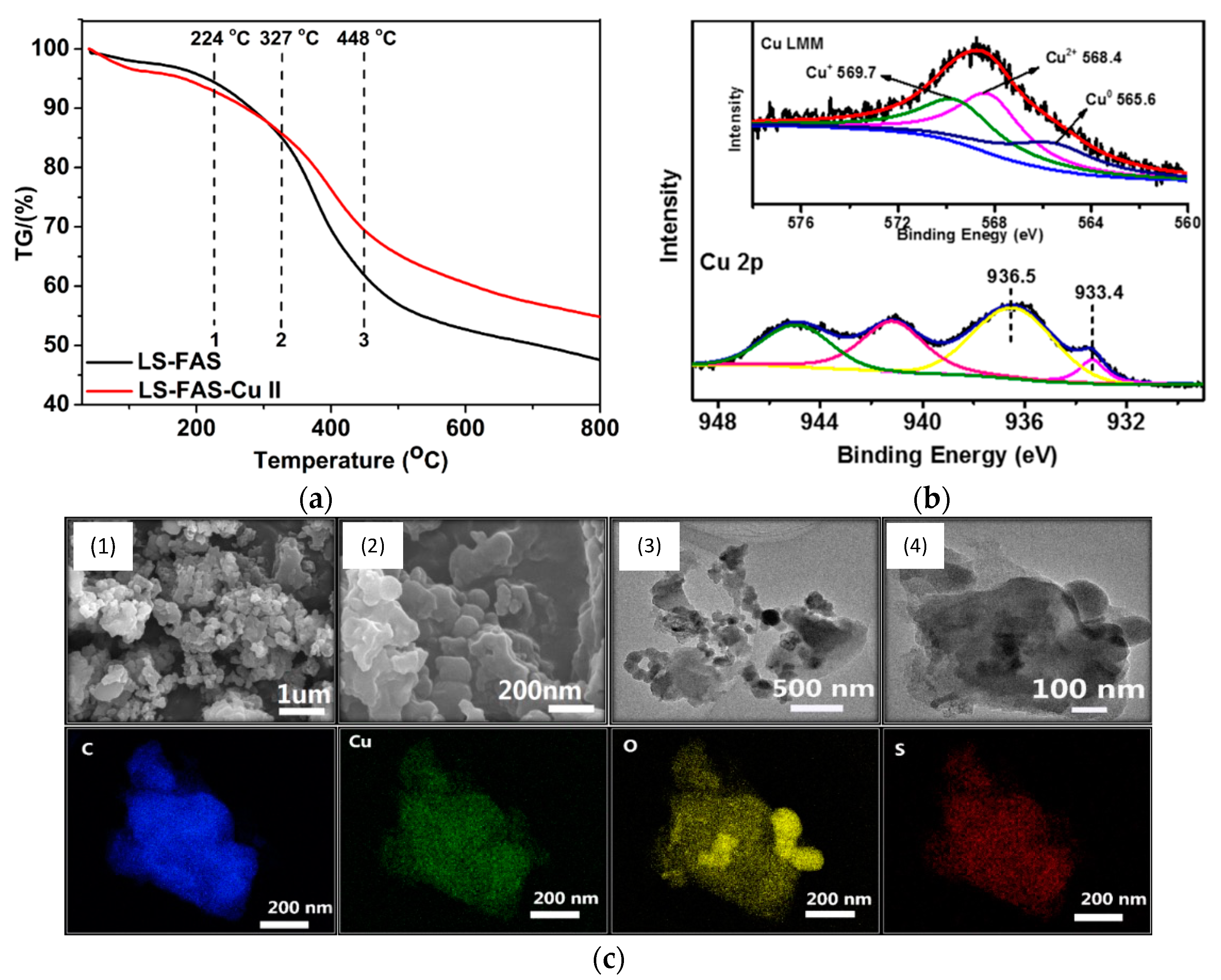
2+ species. Furthermore, the thermal stability of these materials was evaluated by thermogravimetric analysis (TGA), showing that
LSA-FAS and
LSA-FAS-Cu2+, despite subtle differences, exhibited outstanding thermal resistance when considering the temperature conditions usually employed in catalyzed organic transformations (
Figure 5a). Complementing the above, the authors also assigned the different weight losses observed in the thermograms to certain degradation processes, such as moisture loss, elimination of SO
3− species, and finally the volatilization of organic fragments resulting from the main degradation of the polymeric material [
37,
38,
39]. Then, the authors studied the catalyst through X-ray photoelectron spectroscopy (XPS), identifying C, O, F, S, and Cu as the main elements present in the chemical composition of the material’s surface, as was already expected considering the elements present in
LSA-FAS and Cu(OTf)
2. Interestingly, thanks to the high-resolution spectrum of Cu 2p accompanied by the analysis of the Auger Cu LMM spectrum, the coexistence of Cu
2+, Cu
+, and Cu
0 species was confirmed, demonstrating that part of the Cu
2+ precursor was partially reduced during catalyst preparation [
40,
41,
42] (
Figure 5b). Additionally, through field emission scanning electron microscopy (FESEM) and field emission transmission electron microscopy (FETEM), it was possible to observe the surface morphology of
LSA-FAS-Cu2+, showing specimens with a granular/blocky morphology with sizes ranging from 100 to 400 nm. Finally, elemental mapping images showed a uniform distribution of C, O, S, and Cu in
LSA-FAS-Cu2+ (
Figure 5c).
2.3. Catalysts Based on LSA as a Support Matrix for the Immobilization of Metal Nanoparticles (LSA-MNPs)
The good results achieved by
LSA-Mn+ materials in the catalytic formation of C–C bonds served as motivation to continue developing new lignin-based systems for this purpose. In this sense, it was a matter of time for nanotechnology to reach this topic, supported by the outstanding performance that metal nanoparticles (MNPs), of diverse nature, have been shown in a large variety of catalytic transformations [
43,
44,
45]. It seems that, unintentionally, the development of
LSA-Mn+ catalysts paved the way to prepare lignin-based nanocomposites containing MNPs due to the easy access to this type of entity from the already impregnated ionic metal species in
LSA-Mn+. Indeed, by subjecting
LSA-Mn+ specimens to a reducing step,
LSA-MNP nanocomposites could be achieved. Notwithstanding the above, another option for the preparation of these materials is by directly incorporating previously synthesized nanoparticles into the lignin matrix, a method that is usually referred to as “wet impregnation” [
46,
47]. Regarding the reducing step, there are a large variety of suitable reducing agents that have been successfully tested in the formation of metal nanoparticles [
48]; however, a high interest has emerged in employing protocols where external reducing agents can be replaced by temperature, avoiding the excessive implication of chemical substances, and thereby offering a greener methodology [
49]. On the other hand, the lignin matrix plays a significant role during the synthesis of MNPs, not just serving as mere support but also being implicated in the stabilization of these nanostructures, controlling both nucleation and growth mechanisms aiming to ideally prevent the appearance of agglomerates or obtain structures exceeding the nanometric dimension. Surprisingly, even though the first report about using lignin as an MNP support and its catalytic assessment for the construction of C–C bonds was more than a decade ago, to date, the literature related to this is still scarce. Indeed, the first report came out in 2009 by Guillén and coworkers [
50]; however, it must be mentioned that in this case, lignin was used as an activated carbon precursor and not in its pristine form. The authors first carried out the impregnation of lignin with H
3PO
4 followed by its activation under an N
2 atmosphere using a conventional tubular furnace at 500 °C for 2 h. After being cooled under an inert atmosphere, the sample was thoroughly rinsed with warm water (60 °C) until neutral pH and there was a lack of phosphate traces in the washing waters. The obtained material was dried at 100 °C, yielding an activated carbon material labeled
LAC. Then, a portion of
LAC was submitted to an additional thermal program consisting of heating the sample up to 900 °C, also under an N
2 atmosphere, achieving the preparation of a second carbonaceous material named
LACT. Both materials were subsequently used as support matrices of palladium nanoparticles (PdNPs), which were incorporated through wet impregnation. Regarding the above,
LAC and
LACT were introduced into an aqueous solution of PdCl
2 with a concentration fixed to reach a metal loading of approximately 0.5 wt%. Finally, the impregnated palladium ions on both supports were reduced at 400 °C using a mixed N
2/H
2 gaseous atmosphere (ratio 3:1), affording
LAC-Pd and
LACT-Pd catalysts. The textural properties of both catalysts were studied by N
2 adsorption–desorption isotherms performed at low temperatures (−196 °C) and with CO
2 at 0 °C, from which a very complete and comprehensive characterization of the porosity of samples was achieved. In this sense, parameters such as the apparent surface area, the external surface area (associated with the nonmicroporous structure), the micropore volume, and the specific surface area were calculated employing well-known fitting models [
51,
52]. From the above results, the authors stated that both materials presented a heterogeneous porous structure characterized by a wide microporous structure but also a well-developed mesoporosity. Moreover, through Brunauer–Emmett–Teller (BET) analysis, surface areas for all obtained materials (with and without PdNPs) were calculated. Interestingly, this last parameter seems to be highly affected in those samples pyrolyzed at 900 °C, where lower values were measured for
LACT compared to
LAC. The authors explained this based on a possible thermal-triggered reorganization experienced by these materials at high temperatures, promoting the shrinking of their porous structure [
53]. On the other hand, even lower surface areas were determined for samples decorated with PdNPs, ascribed to the volume occupation that these entities exert after being generated within the cavities of the material. Therefore, the employment of phosphoric acid as an activating agent is a good strategy to afford an adequate porosity, favoring the crosslinking process between different portions of the polymer through the formation of polyphosphate bridges that are eventually removed during the washing step [
54,
55]. The authors also characterized both carbonaceous matrices and their corresponding Pd-containing catalysts by XPS. Thanks to this technique, they were able to corroborate the expected surface elemental composition of these materials, evidencing the presence of C, O, P, and N in
LAC and
LACT, while Pd was also detected in
LAC-Pd and
LACT-Pd catalysts. The mass percentage of Pd was very close to the targeted nominal value of 0.43% and 0.50% for
LAC-Pd and
LACT-Pd, respectively. The authors explained the presence of the unexpected nitrogen content as part of the initial composition for the Kraft lignin used as a precursor, while the phosphorous content was related to traces that were not removed after the washing step. A more in-depth XPS analysis allowed, in the case of P, verification of its pentavalent tetracoordinated nature, accusing the presence of species such as phosphates and polyphosphates that after the thermal treatment become part of the carbonaceous material [
56,
57]. More specifically, the deconvolution analysis for the P band allowed the identification of phosphorus-containing fragments with different chemical surroundings, for instance, C–O–PO
3, C–PO
3, and C
3P [
58,
59,
60]. Surprisingly, the
LACT sample seems to have a richer composition in C–PO
3 and C
3P species than
LAC, suggesting that thermal treatment has a high impact on this result. Similarly, the authors carried out a study on the Pd content and found that both Pd
0 and Pd
2+ species were present in samples
LAC-Pd and
LACT-Pd. However, the Pd
0 content was always higher (71.7% vs. 28.3% and 59.8% vs. 40.2% for
LAC-Pd and
LACT-Pd, respectively). Finally, by TEM analysis, the authors were able to visualize PdNPs exhibiting sizes of approximately 5 nm together with a proper distribution along the catalyst surface.
A year later, the same research group extended this protocol for preparing mesoporous-activated carbons using lignin as the carbon source [
61]. However, unlike their previous work, in this opportunity, the authors tested and compared the catalytic activity of samples in which Pd impregnation was carried out before or after thermal treatment at 900 °C. For clarity, the catalyst obtained after the H
3PO
4 activation stage at 500 °C was labeled
L, and after palladium impregnation/reduction, it was labeled
L-Pd. On the other hand, starting from
L, two different types of samples were achieved: (1) those named
LT-Pd resulting from, first, the heating of
L at 900 °C followed by the impregnation/reducing step of Pd precursors and (2) those labeled
L-PdT in which the impregnation/reducing step of Pd was carried out prior to the pyrolysis process. All materials were subjected to the same type of characterization performed in their first work, observing similar trends. For instance, gas adsorption–desorption isotherms revealed that those samples exposed at 900 °C showed, as expected, a decrease in their surface areas, which was even lower in the presence of palladium. However, a strong point of this report relies on the study of the Pd chemical state dependence when metal impregnation was carried out prior to or after the calcination step. In this sense, through XPS analysis, Pd 3d
5/2 and Pd 3d
3/2 signals indicated the coexistence of both Pd
0 and Pd
2+ species in all catalysts [
62,
63,
64]. Moreover, the deconvolution of the Pd 3d region in the obtained spectra allowed calculating the Pd
0/Pd
2+ ratio and how this value varies depending on the thermal treatment applied to the samples. Surprisingly, the
L-Pd and
LT-Pd catalysts showed a Pd content richer in zero-valent species with percentages of approximately 71.7% and 59.8%, respectively. Conversely,
L-PdT samples exhibited a remarkably higher proportion of Pd
2+ species (77.1%), showing that, apparently, during the pyrolysis stage, an important fraction of the metallic Pd is oxidized. The authors argued that a feasible explanation for the above could be the oxidation of Pd
0 triggered by the reduction of C–O–PO
3 groups to C–PO
3 and C
3PO entities. Additionally, from TGA measurements, it was evidenced that at 900 °C, most of the lignin precursor is decomposed, and due to its chemical structure, it is highly possible to expect CO
2 evolution as a degradation product. Therefore, the authors also proposed that CO
2 could be dissociated on Pd nanoparticle surfaces, generating highly oxidative species that would promote the transformation of Pd
0 into Pd
2+ [
65,
66]. Finally, palladium particles present in the
L-Pd,
LT-Pd, and
L-PdT catalysts were visualized by TEM and analyzed in terms of their size and size distributions.
L-Pd showed a narrower size distribution with entities between 3 and 6 nm and an average value of 4.5 nm. Moreover, this sample stood out by the high dispersion exhibited by PdNPs across the material, ascribed to the high acidity of the carbon support due to the H
3PO
4 activation process that lowers the hydrophobicity of the system [
59,
67]. In contrast, both samples subjected to the thermal treatment showed broader size distributions and larger average particle sizes (11.7 nm and 9.7 nm for
L-PdT and
LT-Pd, respectively), ascribed to sintering processes that metal nanoparticles could experience under these conditions [
68]. Lastly, the authors also confirmed the oxidant resistance of these catalysts by exposing them to thermogravimetric analysis under air conditions. This was explored motivated by the already detected P content in the samples, since it has been well-reported that phosphorus can act as an inhibitor of the oxidation of carbon materials [
69,
70]. As a result, all catalysts showed high oxidation resistance by showing no significant decomposition below 450 °C, allowing us to expand the experimental conditions in which these materials can be used as catalysts.
In 2013, Coccia and coworkers prepared PdNPs using water-soluble lignin as a reducing and stabilizing agent [
71]. This can be considered one of the first reports on using lignin in its pristine form for the elaboration of catalysts for C–C bond formation and not as a mere source of active carbonaceous systems. The method stood out by its green experimental conditions in terms of moderate temperatures, short reaction times, and the use of aerated water as the reaction medium. They employed two types of lignin, the first corresponding to Kraft lignin, while the second was a lignosulfonated sample in the form of an ammonium derivative (
LSA-NH4+). This last lignin was obtained from red pine andwas reacted in a calcium hydrogen sulfite solution. Lignins were codissolved with PdCl
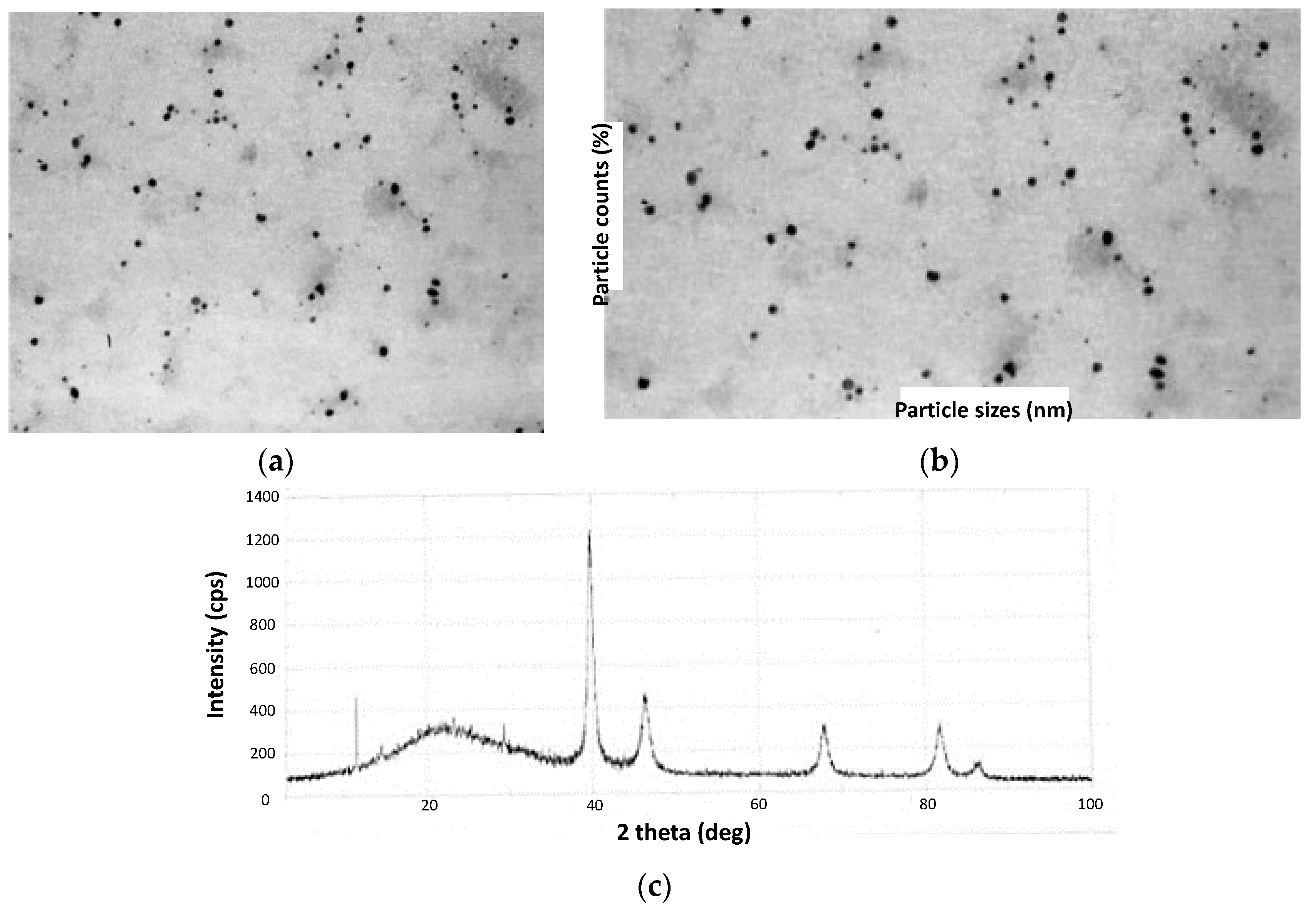
2 in water and heated at 80 °C for 3 h, after which a color change from brown to black was observed. The authors followed the reaction course by ultraviolet-visible (UV-Vis) spectroscopy, determining subtle changes in the visible region of the spectra accusing the formation of PdNPs, which were finally visualized through TEM analysis (
Figure 6a). Spherical nanoparticles were obtained with both lignin materials, with a slight tendency toward agglomeration. However, UV-Vis analysis over time demonstrated good stability by not detecting relevant changes in the spectra even after a month of storage under aerobic conditions and room temperature (RT). Interestingly, PdNP sizes were highly influenced by the type of lignin used. In this regard, when using
LSA-NH4+, values between 16 and 20 nm were achieved, while the use of Kraft lignin allowed us to reach values in the range of 8–14 nm (
Figure 6b). On the other hand, the obtained lignin/PdNP catalysts in powder form were also characterized by X-ray diffraction (XRD), revealing a clear crystalline structure with peaks centered at 39.9°, 46.3°, 67.4°, 82.5°, and 86.9° attributed to PdNPs (
Figure 6c) [
72,
73]. The authors also evidenced a thinner structure for these peaks in the sample prepared from Kraft lignin, as was expected due to the lower particle size. After confirming the presence of PdNPs, the authors carried out proton nuclear magnetic resonance (
1H-NMR) and FTIR experiments aiming to investigate possible changes suffered by lignin after the reducing step. Surprisingly,
1H-NMR spectra signals ascribed to small amounts of acetic acid and formic acid, as well as methanol, were found that, based on the author’s arguments, could be assigned to oxidation products coming from the polymer backbone. On the other hand, FTIR spectra revealed that most of the vibrational bands ascribed to pristine lignin remained unchanged, with the exception of a new weak signal centered at 1730 cm
−1 that could be related to carbonyl species that would emerge from, for example, the oxidation of alcoholic residues that participate during Pd
+2 reduction [
74].
Three years later, in 2016, another report dealing with the deposition of PdNPs on lignin (
LSA-PdNPs) and its catalytic evaluation in Mizoroki–Heck couplings was performed by Marulasiddeshwara and Kumar [
61]. Here, the authors synthesized PdNPs from PdCl
2 aqueous solutions in the presence of lignin. To achieve this, two strategies were tested: (1) mixing PdCl
2 and lignin in water at an ambient temperature, followed by the addition of hydrazine monohydrate as a reducing agent and (2) mixing PdCl
2 and lignin in water followed by boiling. In the latter case, lignin serves both as a reducing and stabilizing agent. Nevertheless, both strategies showed similar qualitative behavior, in which red solutions ascribed to the presence of the PdCl
2 complex turned dark gray due to the formation of PdNPs. Indeed, a good option for monitoring the reaction was through UV-Vis analysis, from which the band’s disappearance centered at 430 nm indicates the consumption of PdCl
2 [
75,
76,
77]. After completing the reaction time according to each strategy, catalysts were isolated by removing the solvent under reduced pressure, washing the gray residue, and ending with a drying step in a vacuum oven. The obtained specimens turned out to be insoluble in conventional organic solvents, promoting their use as heterogeneous catalysts. However, a series of advantages were found for strategy two, such as better stability against aggregation phenomena, since a more intimate interaction between PdNPs and lignin is achieved by the dual function that this biopolymer exerts in this case. Therefore, initially, the coordination of palladium cations on the polymer backbone is expected, followed by their reduction by the lignin’s functional groups. Additionally, by high-resolution ICP-AES analysis, it was found that the amount of loaded Pd was higher using this strategy, being approximately 0.0467 mmol/g. Based on the above arguments, the authors centered the catalytic evaluation using the sample achieved by strategy two. TEM images allowed the visualization of PdNPs impregnated on the lignin matrix, from which spherical entities were identified with sizes between 1 and 5 nm. As a complement, SEM allowed the authors to detect a proper dispersion of these nanoentities over the lignin surface, also finding a good correlation with the size measured by TEM. Furthermore, energy dispersive X-ray (EDX) analysis confirmed the elemental composition of the observed nanostructures. The catalyst powder was subjected to XRD analysis, revealing the presence of sharp peaks that corroborated the high crystallinity of the obtained PdNPs characterized by an fcc structure [
78,
79]. Finally, some insights into the role of lignin as a reducing agent during nanoparticle synthesis were identified through FTIR analysis. In this regard, the FTIR spectra of pure lignin and the catalyst showed a very similar pattern with subtle differences that could be related to the involvement of lignin during the reducing step and the presence of interactions between PdNPs and the lignin structure. Therefore, the participation of this biopolymer in the reduction of Pd
2+ would be supported by the appearance of a new vibrational band centered at 1703 cm
−1, typically observed for carbonyl species formed after the oxidation of, for instance, hydroxyl groups. In addition, lignin/PdNP interactions could be associated with the redshift observed for the O–H band after nanoparticle incorporation.
Later, in 2020, Marulasiddeshwara and Kumar contributed again to the development of a palladium-containing lignin catalyst, but in this opportunity, the sample was endowed with a magnetic response due to the presence of Fe
3O
4 nanoparticles (Fe
3O
4NPs) [
80]. The synthesis of the catalyst started by preparing Fe
3O
4 nanoparticles through chemical coprecipitation employing FeCl
3 and FeSO
4 in a 2:1 ratio as sources of Fe
3+ and Fe
2+, respectively [
81]. Both salts were dissolved in water and heated to 85 °C under constant stirring and an inert atmosphere. Then, a NaOH aqueous solution was added to achieve the formation of the Fe
3O
4NPs in the form of a black precipitate, which was washed thoroughly with water and dried. Subsequently, the hybrid
LSA-Fe3O4NP system was prepared by first dispersing certain aTmounts of Fe
3O
4NPs through sonication in an ethanol/water mixture (1:1) followed by the addition of lignin. The mixture was maintained for 5 h at 40 °C, and the solid was extracted by magnetic decantation. Finally, the preparation of the catalyst was carried out by adding a PdCl
2 solution dropwise over an aqueous dispersion of
LSA-Fe3O4NPs to then incorporate a NaOH solution. The above mixture was heated under reflux for 6 h, after which the reaction mixture turned black. Catalyst purification was achieved through magnetic decantation, meticulous washing steps, and a final drying stage at 100 °C in a vacuum oven. Similar to their previous work, the consumption of PdCl
2 was confirmed by the vanishing of the band centered at 430 nm in the UV-Vis spectrum, indicating the formation of Pd
0 species. On the other hand, in addition to signals belonging to lignin, FTIR analysis allowed the identification of signals at 574 and 572 cm
−1 attributed to Fe–O vibrations, corroborating the presence of Fe
3O
4NPs [
82,
83,
84]. Then,
LSA-Fe3O4NPs and
LSA-Fe3O4NPs-PdNPs were analyzed by XRD, corroborating the simultaneous presence of both Fe
3O
4NP and PdNP species thanks to the characteristic peaks observed in the diffraction patterns, also demonstrating their crystalline structure (
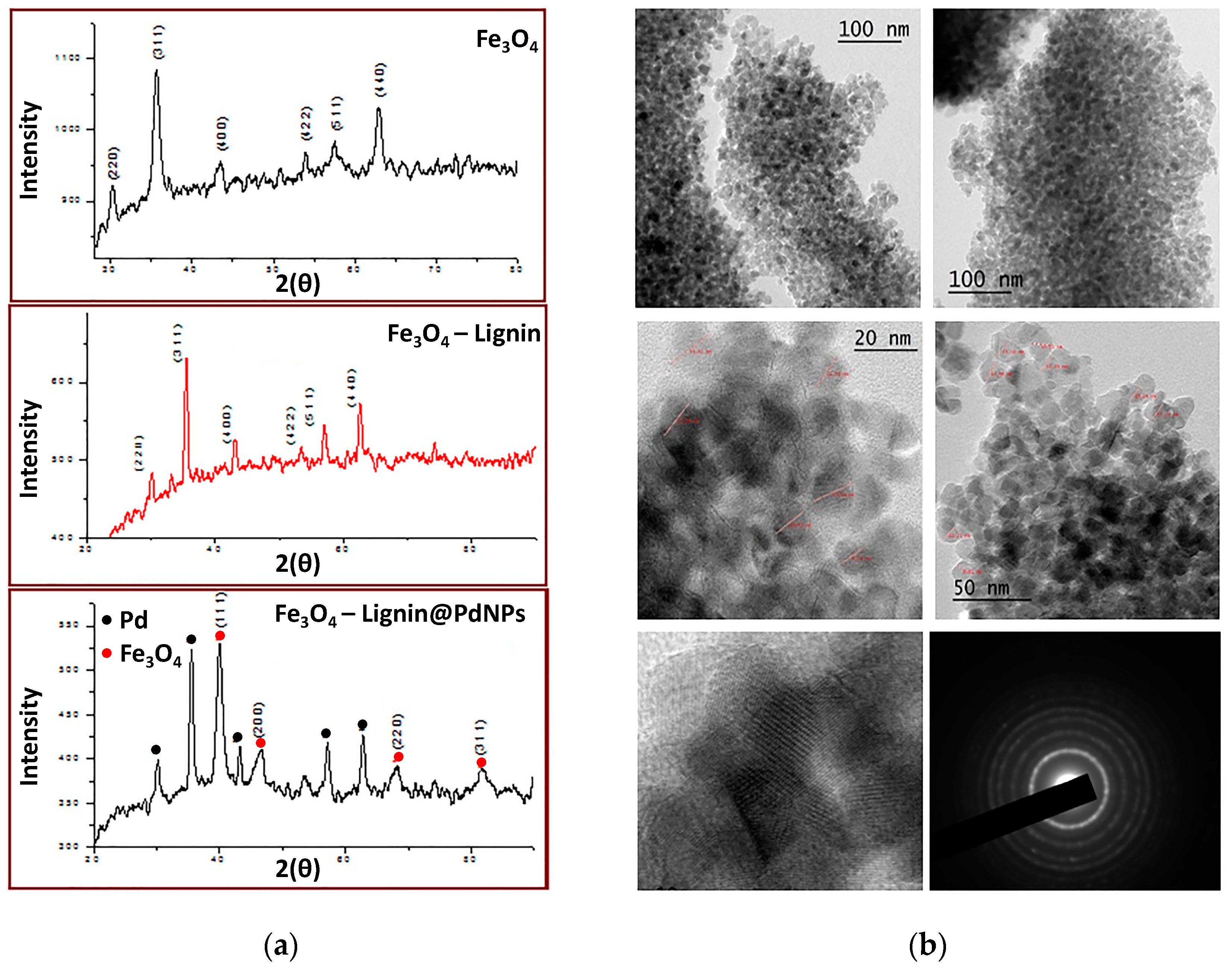
Figure 7a). The authors estimated a nanoparticle size between 5 and 10 nm from the XRD data using the Debye–Scherer equation. On the other hand, they calculated an average size of 20 nm for Fe
3O
4NPs by TEM analysis (
Figure 7b). With the aim of confirming the presence of both types of nanostructures, the authors resorted to EDX measurements through which Pd and Fe were successfully detected, while the quantification of both elements was measured by means of HRICP-AES, reaching values of 11.88 and 10.90 wt%, respectively, per gram of catalyst. The inclusion of nanostructures with a magnetic response, such as Fe
3O
4NPs, could open the possibility of achieving a more facile recovery of the catalytic system, prolonging its useful life.


 and
and 
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
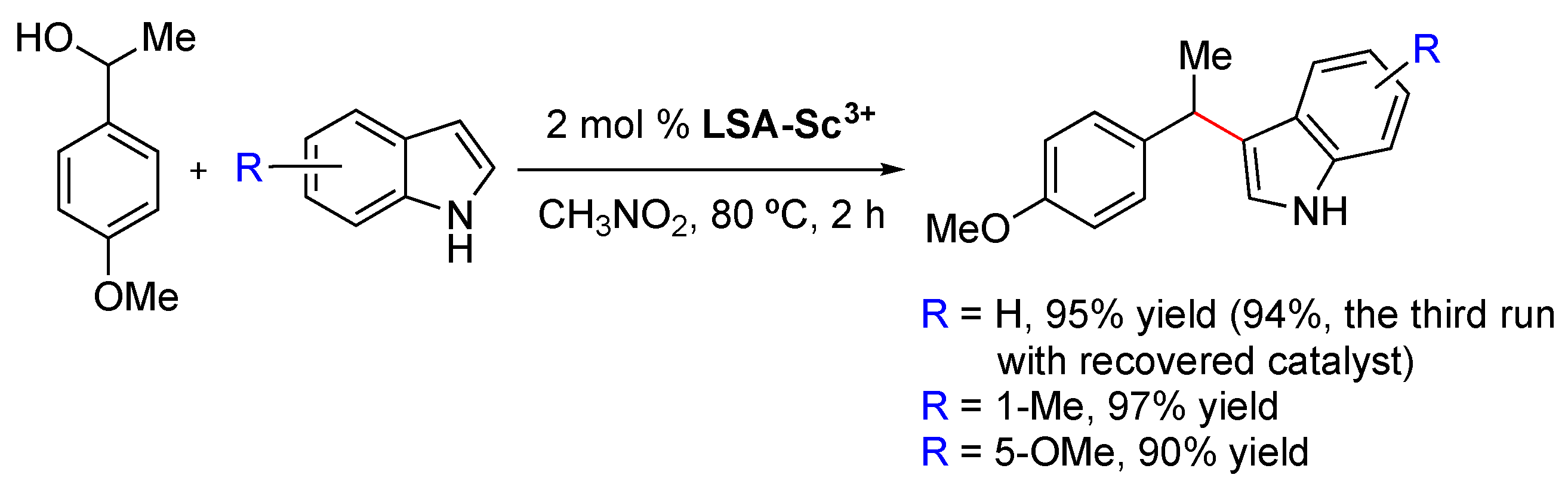{kind=link}
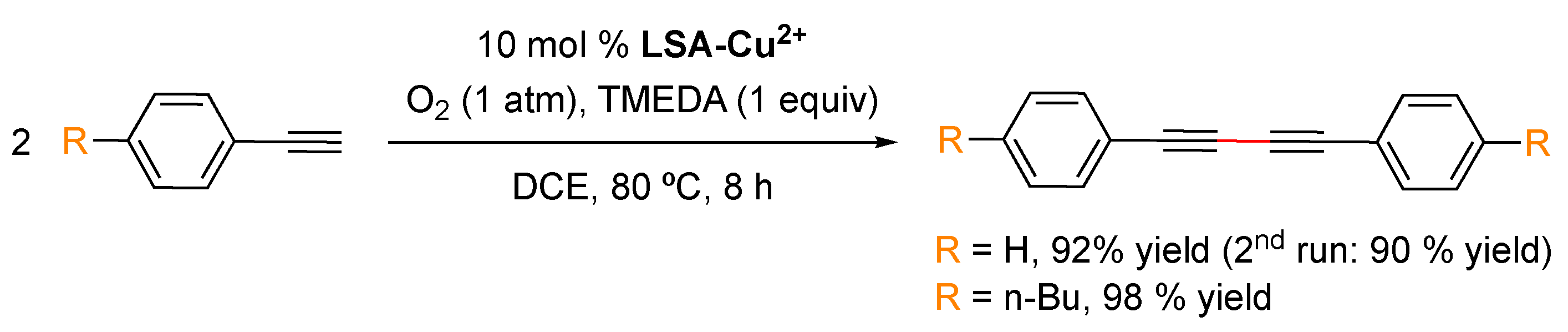{kind=link}
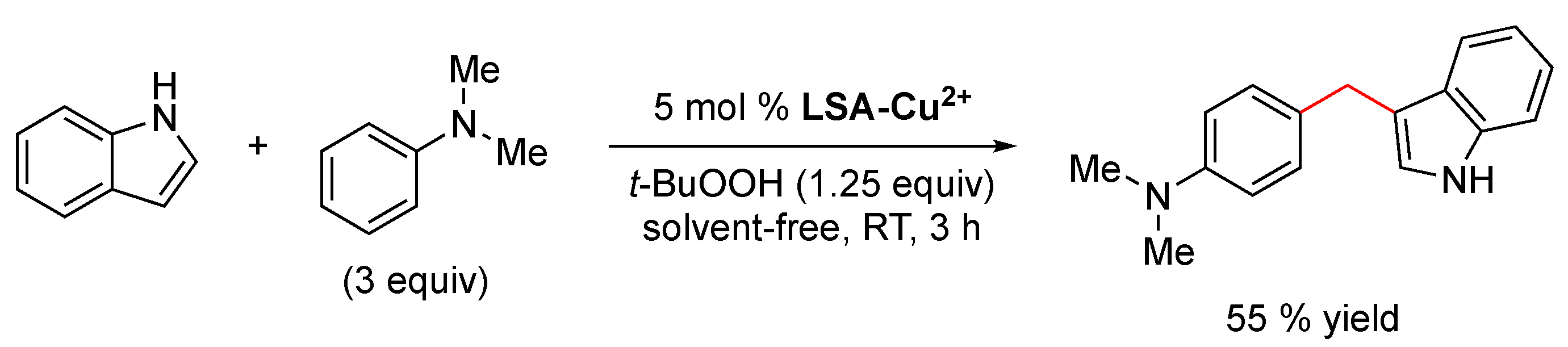{kind=link}
{kind=link}
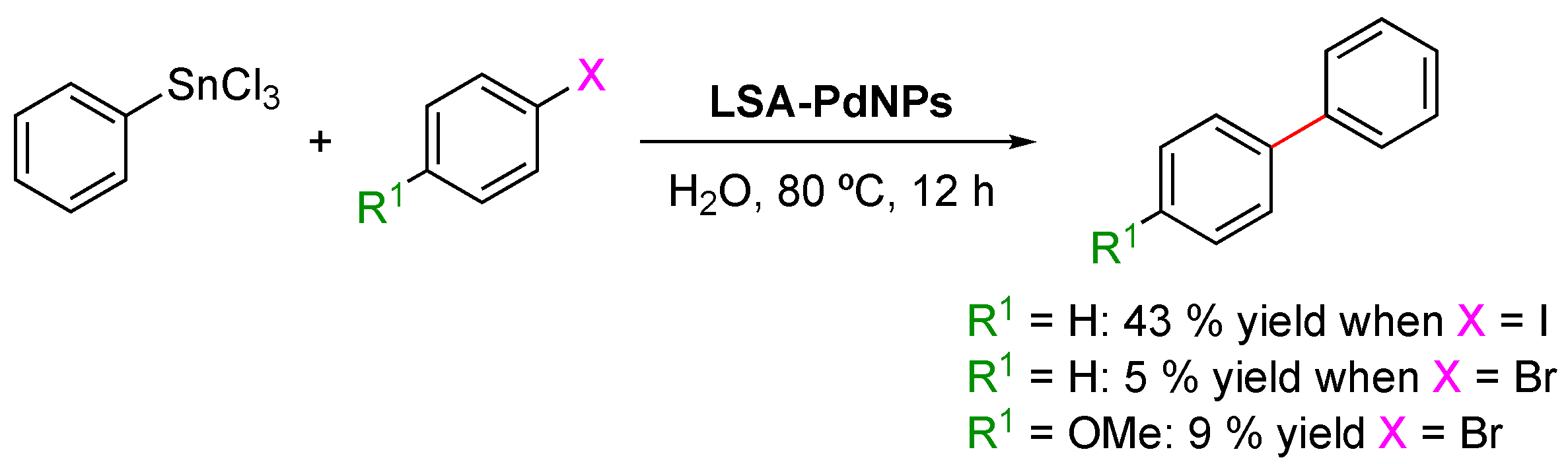{kind=link}


















