Abstract
Phenols are important readily available synthetic building blocks and starting materials for organic synthetic transformations, which are widely found in agrochemicals, pharmaceuticals, and functional materials. The C–H functionalization of free phenols has proven to be an extremely useful tool in organic synthesis, which provides efficient increases in phenol molecular complexity. Therefore, approaches to functionalizing existing C–H bonds of free phenols have continuously attracted the attention of organic chemists. In this review, we summarize the current knowledge and recent advances in ortho-, meta-, and para-selective C–H functionalization of free phenols in the last five years.
1. Introduction
Phenols are readily available and have emerged as ubiquitous building blocks in natural products, pharmaceuticals, and functional materials [1,2,3,4,5,6]. For example, many natural products such as hormones, antibiotics, vitamins, and neurotransmitters are derived from or contain phenols; Approximately, more than 10% of the top 200 selling pharmaceuticals bear at least one phenol or employ phenols as synthetic intermediates; phenols are also key components of functional materials such as polymer resins (Scheme 1). Consequently, straightforward and highly efficient site-selective C–H bond functionalization of free phenols has continuously attracted the attention of organic chemists [7,8,9,10].
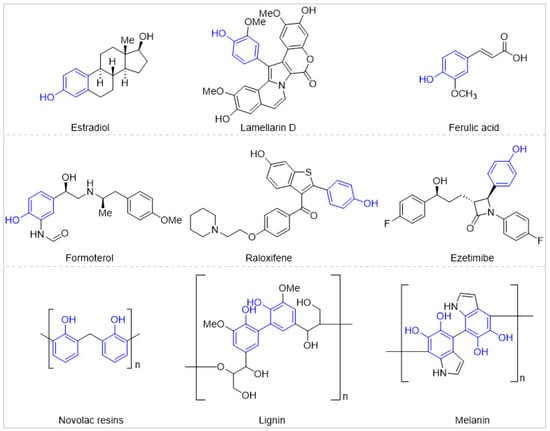
Scheme 1.
Phenol-containing molecules.
With developments made in organic synthesis, the direct C–H functionalization of phenols has been explored extensively as an efficient and powerful tool to synthesize complex phenol molecules. In the previous selective conversion of phenol, most of the directing groups installed on the phenolic hydroxyl group were used to realize the indirect strategy of activation of ortho C–H bond functionalization. However, as the most straightforward and atom-economical strategy, directly using free phenolic hydroxyl groups as directing groups to synthesize highly substituted phenolic derivatives, there are many challenges for chemo- and regioselective C–H functionalization of unprotected free phenols. The major barrier stems from several aspects: (1) the phenolic hydroxyl group has strong acidity and nucleophilicity, so that the reaction site mostly occurs on the phenolic hydroxyl group instead of the benzene ring; (2) the ortho and para positions of the phenolic hydroxyl group in the benzene ring are highly active, which reduces the regioselectivity of the C–H bond functionalization reaction; (3) it is often necessary to add a stoichiometric oxidant in the C–H functionalization reaction, and the phenols are typical electron-rich aromatic hydrocarbons, which are easily oxidatively decomposed during the reaction. Thus, more economical and green synthetic approaches for the direct regioselective C–H bond functionalization of free phenols are a significant challenge of great interest.
Some reviews on the transition-metal-catalyzed functionalization of C–H bonds in phenols have been reported so far. Lumb and co-workers [11] wrote a review on phenol-directed C–H functionalization in 2018. Luo [12] summarized the progress of the transition metal-catalyzed directing-group-assisted C–H activation of phenols until 2019. Immediately afterward, Mamari [13] outlined a comprehensive advance in regard to metal-catalyzed C–H bond functionalization of phenol derivatives in 2020. Later, Youn and co-workers [14] reported a comprehensive overview of transition-metal-catalyzed ortho-selective C–H functionalization reactions of free phenols until 2021. Recently, Zhai [15] published an excellent review on recent advances in catalytic oxidative reactions of phenols and naphthalenols. We want to focus on the recent development on the C–H functionalization of different positions on the free phenol aromatic ring. Thus, we will summarize recent advances since 2018 in regioselective C–H bond functionalization of free phenols in this review. This review is classified into three sections according to the position of functionalization, treating ortho-, meta-, and para-positions of free phenol successively, and only some selected examples are described schematically (Figure 1).
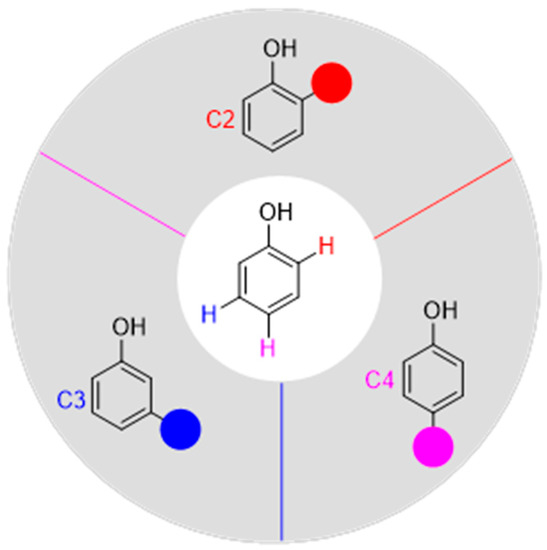
Figure 1.
The regioselective C–H bond functionalization of free phenols.
2. C2-Functionalization
2.1. Metal-Catalyzed C–H Bond Functionalization of Free Phenol
Over the past decades, there have been tremendous advances in the field of catalytic C–H bond functionalization [16,17,18], among which metal-catalyzed C–H bond functionalization of free phenol has proven to be an extremely useful tool in the synthesis of complex phenol molecules.
2.1.1. Cu-Catalyzed C–H Bond Functionalization
In 2018, Jain’s group [19] synthesized a GO-Cu7S4NPs from Cu2S to achieve an highly efficient ortho-selective C–H aminomethylation of free phenol derivatives 1 with N,N-dimethylbenzylamines 2 using tert-Butyl hydroperoxide (TBHP) as an oxidant under solvent-free conditions. The nano-copper catalyst could be reused for Csp2-Csp3 cross-dehydrogenative coupling, with no requirement for pre-functionalization of substrates. A variety of C–C coupled products 3 are obtained with 68–89% yields. Furthermore, more challenging substrates like β-naphthols and substituted halohydroxypyridine derivatives were successfully used in the aminomethylation reaction to afford the corresponding aminomethylated products in 70–80% yields. Control experiments suggested that this reaction may undergo the nucleophilic attack by ortho-phenol carbon 4 on iminium ion 5, which was formed by the loss of hydrogen atom via a single electron transfer under the action of the nano-copper catalyst and TBHP, to generate the Cu-coordinated ketone 7 (Scheme 2).
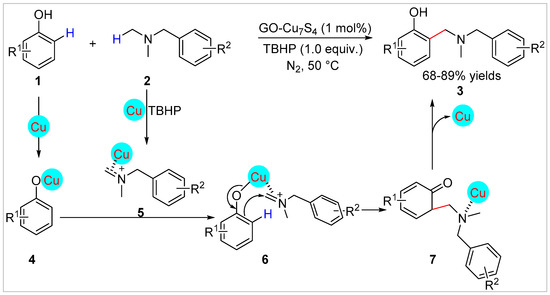
Scheme 2.
GO-Cu7S4 NPs-Catalyzed ortho-aminomethylation of phenols with N,N-dimethylbenzylamines.
Simultaneously, Patureau’s group [20] reported a highly selective Cu(II)-catalyzed cross-dehydrogenative ortho-aminomethylation of free phenols 8 with aniline derivatives 9 using Di-tert-butyl peroxide (DTBP) as an oxidant. As illustrated in Scheme 3, a series of aminomethylated products 10 were obtained with 32–82% yields and based on controlled experiments; the authors proposed that this reaction involved the aminomethyl radical 13 by a Cu-centered six membered transition state 15. However, this method required an excess of amines (6.3–9.5 equiv.) and only tertiary N-methyl aromatic amines were compatible with the reaction system, which limited its synthetic utility.
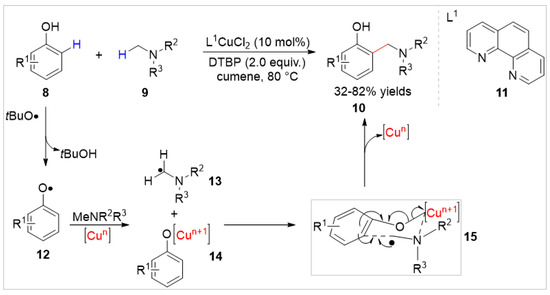
Scheme 3.
Cu-catalyzed ortho-aminomethylation of phenols with aniline derivatives.
In 2021, a copper(II)-modified nitrogen-rich covalent organic polymer (Cu/N-COP-1) was prepared and employed to achieve the cross-dehydrogenative ortho-aminomethylation of free phenols 16 with N,N-dimethylanilines 17 under mild conditions by Xie et al. [21]. Various aminomethylated products 18 were obtained in 42–59% yields (Scheme 4). In addition, the catalyst could be easily reused for at least five consecutive runs without significant loss of catalytic activity, which is attributed to the interaction between the nitrogen-containing group of the covalent organic polymer and copper chloride. Having the di-substituted products 19 and moderate yields (mostly 42–59%) of the desired products were the drawbacks of this method.
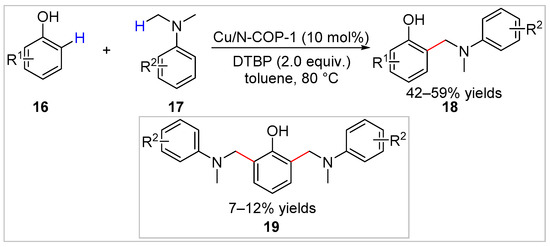
Scheme 4.
Cu/N-COP-1-catalyzed ortho-aminomethylation of phenols with N,N-dimethylanilines.
In 2020, Ma et al. [22] reported the first copper-catalyzed highly efficient ortho-C–H bond functionalization rather than O–H insertion of free phenols 20 with a-aryl-a-diazoesters 21 (Scheme 5). A range of alkylation products 22 were obtained in 76–98% yields under mild reaction conditions. A preliminary mechanistic study indicated that the hydroxyl group was important not only for site-selectivity but also for the reactivity of the C–H bond functionalization reaction, and the ortho-selectivity could be improved via the interaction between the hydroxyl group and the copper catalyst. Herein, the authors proposed two possible reaction mechanisms for this transformation, which involved the Cu-carbene 27 and the carbocation intermediate 28 formed by coordination between the CuCl2 and the nitrogen of a-aryl-a-diazoesters, respectively.
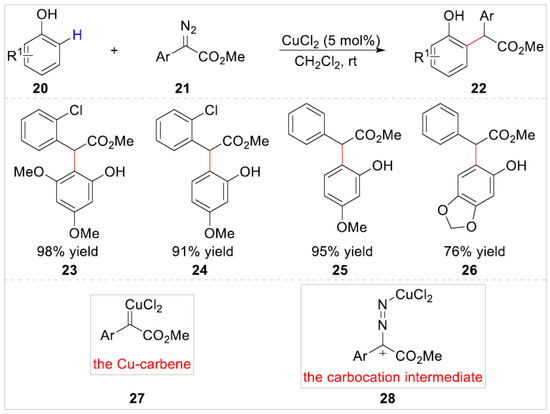
Scheme 5.
Cu-catalyzed ortho-aminomethylation of phenols with a-aryl-a-diazoesters.
One year later, Xie’s group [23] developed a copper-catalyzed tandem cross-coupling/annulation of free phenols 30 with ketoximes 29 via dual C–H functionalization. A variety of oximes, including 1-naphthyl-bearing oxime and heteroaryl-bearing oximes, were compatible for this reaction system and afforded the corresponding products 31 in good yields. Control experiments disclosed that the benzoquinone 33 might be the vital intermediate of the dual C–H functionalization reaction, as the para-dihydroxyl was crucial in the process of reaction (Scheme 6).
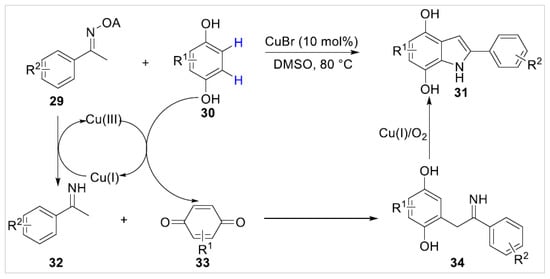
Scheme 6.
Copper-catalyzed annulation of phenols with ketoximes.
In 2022, Zhao et al. [24] reported a copper-catalyzed C2-site selective amination of free p-aminophenol derivatives 35 with arylamines 36 under mild conditions, using air as a terminal oxidant, and a variety of C2-site functional aminophenol derivatives 37 were obtained with 39–78% yields (Scheme 7). Interestingly, the para-benzomorpholine substituted phenol was also compatible with the reaction system for transformation into the desired C2-site selective aminated product 37a in 58% yield with oxidation of the benzomorpholine group to 4H-benzo[b][1,4]oxazine. Based on controlled experiments, the authors proposed that the amination reaction may undergo a process of radical-radical cross-coupling between the phenoxyl radical 39 and the N-radical 40.
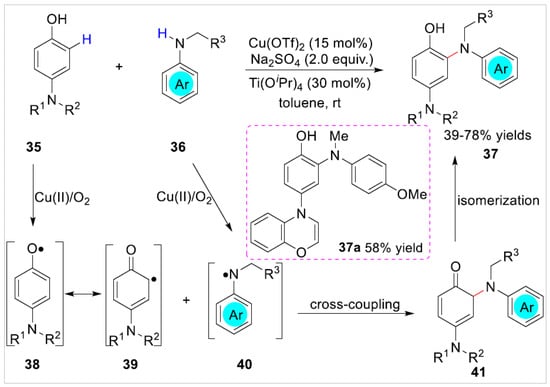
Scheme 7.
Copper-catalyzed C2-site functionalization of p-aminophenols with arylamines.
2.1.2. Co-Catalyzed C–H Bond Functionalization
In 2018, Khakyzadeh, Zolfigol, and co-workers [25] disclosed a Co(II)-catalyzed regioselective synthesis of 2-(aryl/alkylthio)phenols 44 via formal C–H bond functionalization of free phenols 42 with aryl/alkyl thiols 43 using acetic anhydride as the directing group under mild conditions. A variety of 2-(aryl/alkylthio)phenols 44 were obtained with moderate to excellent yields (Scheme 8). In addition, 2-naphthol and 1-naphthol were also compatible with this reaction system. Later, this reaction was further developed through Fe3O4@SiO2-UT@Co(II), using pivalic anhydride as the directing group, by Khaef et al. [26]. In fact, the two reactions were stepwise processes. By introducing directing groups to protect the hydroxyl group, the ortho-C–H functionalization of phenol was achieved. Finally, 2-(aryl/alkylthio)phenols were obtained by an acidic hydrolysis strategy. Depending on the control experiment, both thiolation reactions may undergo a process of free radicals 45.
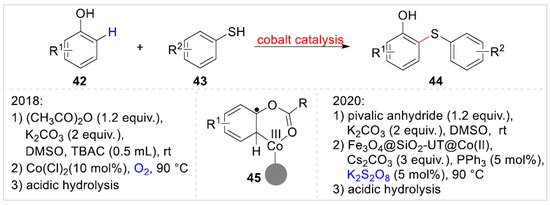
Scheme 8.
Co(II)-catalyzed ortho-C–H functionalization of phenols with aryl/alkyl thiols.
In 2019, a Cobalt(II)[salen]-catalyzed selective aerobic oxidative cross-coupling of electron-rich free phenols 46 with 2-naphthols 47 was developed by the Pappo group [27] in a recyclable 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) solvent. A variety of nonsymmetric biphenols 48 were obtained with 40–92% yields under mild waste-free conditions by a selectivity-driven catalyst design approach. However, the nonsymmetric biphenols 50, 51 and 52 were obtained by the para-C–H bond functionalization rather than ortho-positions of free phenols with 2-naphthols (Scheme 9). Besides, a strong electron-withdrawing group such as ester was not compatible with the current system, failing to afford the corresponding biaryl coupling product. Detailed control experiments and kinetic studies suggested that a liberated phenoxyl radical 53 and a ligated 2-naphthoxyl radical 54 were involved in the reaction pathway.
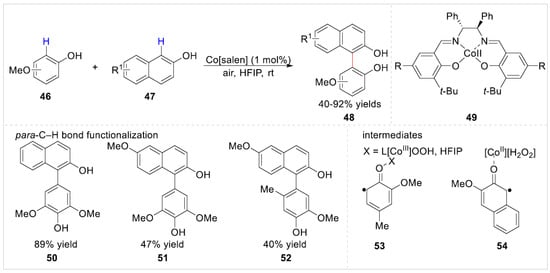
Scheme 9.
Cobalt(II)[salen]-catalyzed ortho-C–H functionalization of phenols with 2-naphthols.
2.2. Electrochemical/Photochemical C–H Bond Functionalization
Electrochemistry/photochemistry has been a powerful synthetic tool for organic chemistry [28,29,30,31,32,33]. Compared with the chemical methods of the C–H functionalization of free phenols, the electrochemistry/photochemistry method uses electrons/light as a renewable and environmentally friendly reagent to replace the metal catalysts and the stoichiometric amounts of oxidant in the C–H functionalization reactions. Undoubtedly, the electrochemical/photochemical C–H bond functionalization of free phenols has attracted increasing attention from organic chemists.
The highly efficient arylation of benzothiophenes 55 with free phenols 56 was realized by the Waldvogel group [34] under exogenous oxidant-free and metal catalyst-free electrochemical oxidation conditions in 2018. The 2- or 3-(hydroxyphenyl)benzo[b]thiophenes 57 were regioselective, obtained with 38–88% yields in an undivided cell equipped with a BDD anode and a BDD cathode at a constant current of 5.2 mA/cm2, using Bu3NMeO3SOMe as the electrolyte at RT or 40 °C (Scheme 10). Later, Yue et al. [35] developed an electrochemical oxidative C–H/C–H coupling of free phenols 58 with 3-phenylbenzothiophenes 59, which delivered highly tunable benzothiophene derivatives 60 with 33–89% yields in an undivided cell equipped with a RVC anode and a Pt plate cathode at a constant current of 10 mA, using n-Bu4NPF6 as the electrolyte at 25 °C under the protection of nitrogen (Scheme 10). Both monofunctional and bifunctional groups could be achieved in this electrochemical oxidation reaction under external oxidant- and catalyst-free conditions. However, the thiophene and benzothiophene were not compatible with the reaction system. Control experiments suggested that a cross-coupling process between the p-methoxylphenol radical 62 and the 3-phenylbenzothiophene radical cation 63 might be involved in this electrochemical transformation.
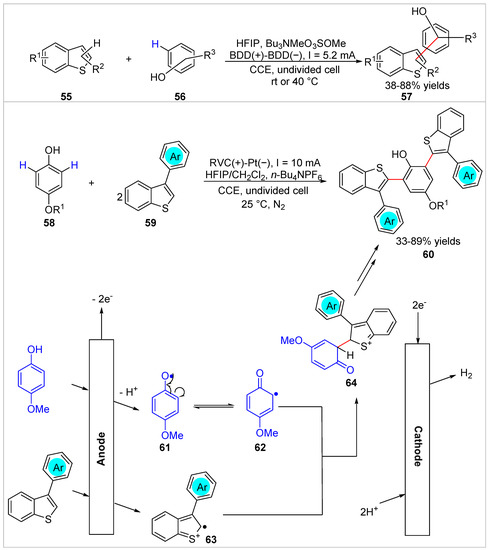
Scheme 10.
Electrochemical C–H functionalization of phenols with benzothiophenes.
In 2018, the Lei group [36] reported an electrochemical oxidative C–H amination of free phenols 65 with phenothiazine derivatives 66 under metal catalyst- and chemical oxidant-free conditions. A host of N-aryl phenothiazines 67 were obtained with 17–93% yields in an undivided cell equipped with a graphite rod anode and a nickel plate cathode at a constant current of 7 mA, using n-Bu4NBF4 as the electrolyte at room temperature under nitrogen protection (Scheme 11). Notably, 2-naphthol and 1-naphthol were also compatible with this reaction system, the products of the ortho-C–H bond functionalization of 2-naphthol and the para-C–H bond functionalization of 1-naphthol were obtained in 89% yield and 88% yield, respectively. Besides, the electro-oxidative double C–H amination of ortho-substituted phenols was also observed in this reaction. Based on cyclic voltammetry (CV) experiments, the authors proposed that the electrocatalytic amination reaction involved the oxidation of amine substrate to generate the radical cation 70.
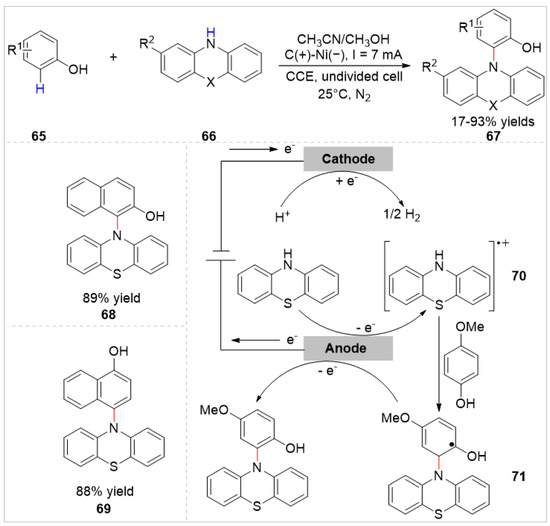
Scheme 11.
Electrochemical ortho-C–H functionalization of phenols with phenothiazine derivatives.
Subsequently, Feng et al. [37] reported an electro-oxidative and regioselective C–H azolation of free phenols 72 under external oxidant-free conditions with H2 evolution. The electrosynthesis proceeds in a simple undivided cell equipped with two platinum electrodes at a constant potential of 2.5 V, using n-Bu4NPF6 as an electrolyte at room temperature under the protection of nitrogen. Moreover, the para-methoxyl aniline derivatives were successfully employed as the reaction substrate under standard conditions. Detailed experiments and cyclic voltammetry studies suggested that a radical coupling process between the carbon radical 76 and the nitrogen-centered radical 77 might be involved in this electrochemical transformation (Scheme 12).
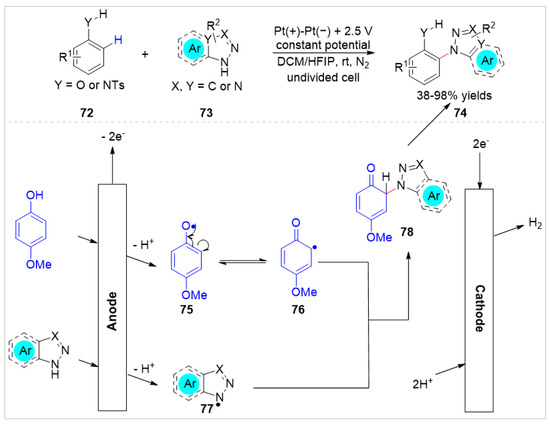
Scheme 12.
Electrochemical regioselective C–H functionalization of phenols with aniline derivatives.
In 2019, Sun, Zeng, and co-workers [38] reported a selective electro-oxidative cross-coupling of different free phenols 79 and naphthols 80 using tri(p-bromophenyl)amine(TBPA) 82 as a redox mediator. A variety of non-symmetric biphenols 81 were obtained with 27–83% yields in an H-type divided cell equipped with a Pt plate anode and a graphite plate cathode at 0.8 V vs. Ag/AgNO3, using LiClO4 as the electrolyte (Scheme 13). When 2,6-dimethoxyphenol 84 was used as a substrate, the para-C–H bond functionalization of 2,6-dimethoxyphenol was observed. Based on the CV analyses, the authors proposed that this cross-coupling reaction started from an electrochemical oxidation of TBPA 82 at the surface of anode to generate its cation radical TBPA+• 83. Then, homogeneous electron transfer of TBPA+• with 2,6-dimethoxyphenol 84 formed the cation radical 85, which underwent nucleophilic addition with 2-naphthol.
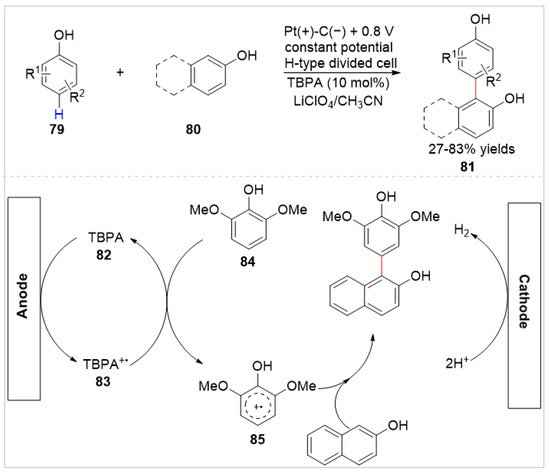
Scheme 13.
Electrochemical C–H functionalization of phenols and naphthols.
Afterwards, the Waldvogel group [39] further developed an electrochemical dehydrogenative coupling reaction of free phenols 86 carrying electron-withdrawing groups using DIPEA as an additive. The homo-coupling reaction was conducted in an undivided cell equipped with BDD electrodes at a constant current of 5 mA/cm2, lacking a supporting electrolyte at room temperature. Besides, this method could be extended to cross-coupling reactions with naphthalenes to form biaryls and precursors for dibenzofurans. Nevertheless, the moderate yield of the products was the drawback of this method. Control experiments suggested that the C–C coupling reaction may involve the coupling of free radicals (Scheme 14).

Scheme 14.
Electrochemical C–H functionalization of phenols with electron-withdrawing groups.
In 2021, Wen, Yang, and co-workers [40] reported an electrochemical in situ oxidative sulfonylation of free phenols 88 with sulfinic acids 89 to give sulfonylated hydroquinones 90 with 32–95% yields in an undivided cell equipped with a graphite rod anode and a platinum plate cathode at a constant current of 10 mA, using LiClO4 as the electrolyte (Scheme 15). When the applicable scope of phenols with different substituents was explored under standard conditions, the regional selectivity of the sulfonylated reaction is slightly poor, due to the steric and electronic effect of the phenols. Besides, the aliphatic sulfinic acids were not compatible with this reaction system. Based on controlled experiments, two postulated reaction pathways were involved in sulfonylated reactions, which were radical-radical cross-coupling between the sulfone radical 98 and the carbon radical 95 or the anodic oxidation of hydroquinone to form benzoquinone 92 followed by Michael addition with sulfinic acids.
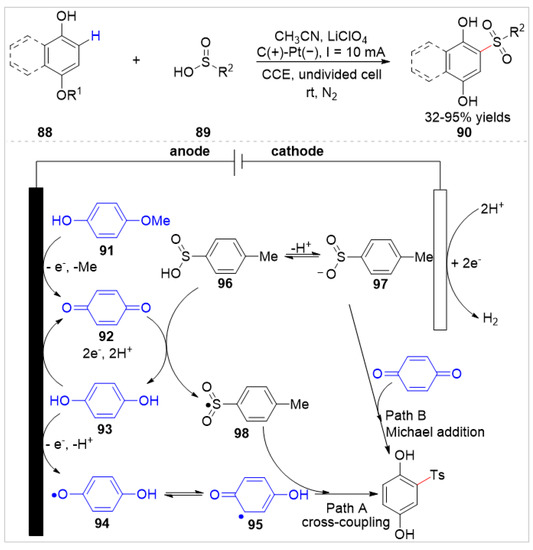
Scheme 15.
Electrochemical C–H functionalization of phenols with sulfinic acids.
In 2021, Li and co-workers [41] reported a catalyst-free visible-light-driven ortho-C(sp2)–H arylation of free phenols 100 with arylbromides 99 under irradiation of a blue LED or natural sunlight. A series of 2-arylated phenols 101, including the electron-donating and electron-withdrawing groups, were obtained with 38–73% yields at room temperature (Scheme 16). However, anisole was not successfully used in the arylation reaction to afford the corresponding product, and heteroaryl halides were also incompatible with this reaction system. Based on controlled experiments, the authors proposed that the reaction proceeded via visible light photoexcitation of an EDA complex 102 between an aryl bromide and a phenolate ion.

Scheme 16.
Photochemical C–H functionalization of free phenols with arylbromides.
Subsequently, a photocatalytic oxidative coupling of free phenols 103 with alkenylphenols was reported, using a recyclable heterogeneous titanium dioxide photocatalyst in air and visible light, by Kozlowski group [42]. When 2,6-dimethylphenol and 2-isopropyl-5-methylphenol were used as substrates in homo-coupling reaction, the para-C–H bond functionalization was observed. The incomplete conversion with moderate yields of the desired products limited the application of the reaction. The bare leak of the phenol hydroxyl group played a key role in the reaction, since anisole was incompatible with this reaction system. Depending on the control experiment, the authors proposed that this reaction may involve the TiO2–phenol complex 105, which was activated through a ligand to metal charge transfer effect (LMCT) (Scheme 17).
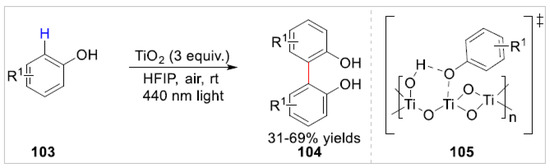
Scheme 17.
Photochemical C–H functionalization of free phenols with alkenylphenols.
2.3. Others
In 2018, Li and co-workers [43] reported a B(C6F5)3-catalyzed hydroarylation of free phenols 106 with 1,3-dienes 107 under mild reaction conditions. A series of structurally diverse ortho-allyl phenols 108 were obtained with 32–91% yields. When the phenol was applied as the substrate, a small quantity of para-allylation products was obtained. Moreover, 1,4-dimethoxybenzene was not successfully used in the hydroarylation reaction to afford the corresponding product. A preliminary mechanistic study suggested that the hydroarylation reaction took place via a borane-promoted protonation/Friedel-Crafts pathway, which involved a π-complex of carbocation-anion contact ionpairs 109 (Scheme 18). Subsequently, this method was developed by Zhou et al. to realize a B(C6F5)3 catalyzed hydroarylation of free phenols 110 with terminal alkynes 111 at room temperature (Scheme 18) [44]. As compared with hydroarylation of 1,3-dienes, the difference was that when 2,6-disubstituted free phenol was employed as the substrate, no para-hydroarylation product was observed in the hydroarylation of terminal alkynes with free phenols.
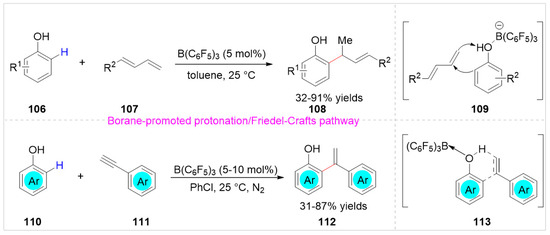
Scheme 18.
B(C6F5)3-catalyzed hydroarylation of free phenols with 1,3-dienes or terminal alkynes.
In 2020, Lei’s group [45] reported a K2S2O8 -induced azolation of electron-rich free phenol derivatives 114 with pyrazoles 115 under catalyst-free conditions. A variety of N-arylazoles 116 bearing the electrondonating groups were obtained with 20–76% yields via oxidant-induced strategy. Detailed experiment studies suggested that a radical coupling process between the N-centered radical 117 and the C-centered radical 118 might be involved in this azolation transformation, which was generated by the oxidation of K2S2O8 (Scheme 19).
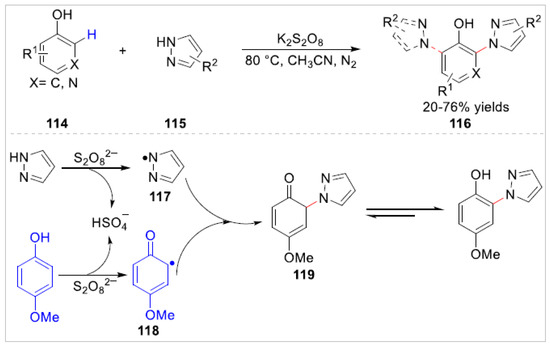
Scheme 19.
K2S2O8-induced azolation of electron-rich free phenols with pyrazoles.
In 2021, Liu and co-workers [46] reported a Brønsted acid catalyzed chemo- and ortho-selective aminomethylation of free phenols 120 with N,O-acetals 121 under mild conditions. Various aminomethylated phenol products 122 were obtained with 42–97% yields. The high ortho-selectivity was attributed to the formation of an intermediate 123 via the interaction of CF3COOH with the free phenol (Scheme 20). Later, Zhou et al. [47] developed an aqueous C–H ortho-aminomethylation of free phenols 124 with trifluoroborates 125 by iodine catalysis, affording various functionalized phenol derivatives 126 in 20–98% yields. Based on controlled experiments, the authors proposed that the aminomethyl radical intermediate 127 was involved in the reaction pathway by oxidation of the hypoiodous acid (Scheme 20).
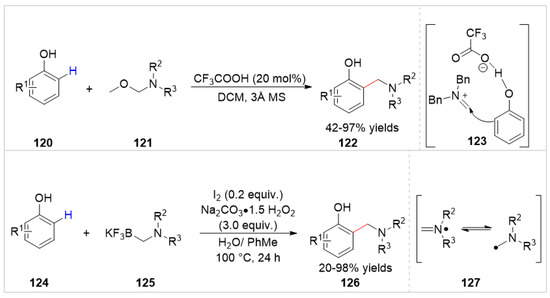
Scheme 20.
ortho-selective aminomethylation of free phenols with N,O-acetals/trifluoroborates.
3. C3-Functionalization
The meta functionalization of free phenols is challenging because the phenolic hydroxyl group can not only activate the aromatic ring for aromatic electrophilic substitution, but also guide its ortho or para-substitution [48,49,50]. Besides, the meta-sites of phenol are not easily activated by metal chelation [51,52]. As a result, the meta-C–H functionalization of free phenols could not be achieved via existing methods based on conventional C–H functionalization, electrophilic aromatic substitution, or oxidative coupling. Recently, Senior et al. [53] reported a meta-selective C–H arylation of polysubstituted free phenols 130 via regiodiversion of electrophilic aromatic substitution. This method achieved the meta-selective C–H arylation of sterically congested phenols through a Bi(V)-mediated electrophilic arylation and a subsequent aryl migration/rearomatization. A variety of the products of the meta-selective C–H arylation of phenols 135, including the electron-donating and electron-withdrawing groups, were obtained in excellent yields. A preliminary mechanistic study suggested that this reaction involved a phenonium ion intermediate 133 (Scheme 21).
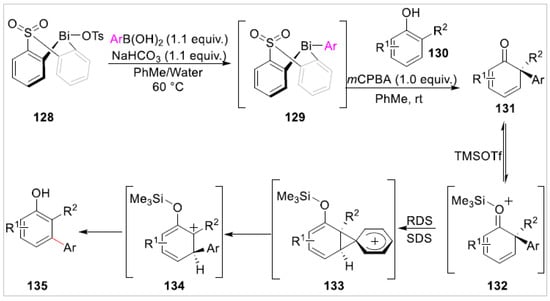
Scheme 21.
meta-selective arylation of phenols.
4. C4-Functionalization
The electron-donating hydroxyl group and the reduced steric of C4 relative to either the C2 or the C6 of free phenols are capable of promoting the para-selective C–H functionalization of free phenols. Here, we introduced the para-selective C–H functionalization of free phenols according to metal catalysis, electrocatalysis, and other methods.
4.1. Metal-Catalyzed C–H Bond Functionalization of Free Phenol
In 2018, Bhanage and co-workers [54] reported a palladium-catalyzed aerobic oxidative carbonylation of a C–H bond of free phenols 136 for the synthesis of p-hydroxybenzoates 137 in 50–76% yields using molecular oxygen as a terminal oxidant. The substrates bearing the electron-withdrawing meta-nitro, meta-trifluoromethyl groups on phenol were not compatible with this reaction system and no corresponding products were observed. Besides, p-cresol with methanol also failed to react under the optimized reaction conditions. Based on controlled experiments, the authors proposed that the carbonylation reaction involved the in-situ production of an intermediate 4-iodophenol 138 that subsequently underwent the nucleophilic attack of alcohol to offered p-hydroxybenzoate 137 via reductive elimination of palladium metal (Scheme 22).
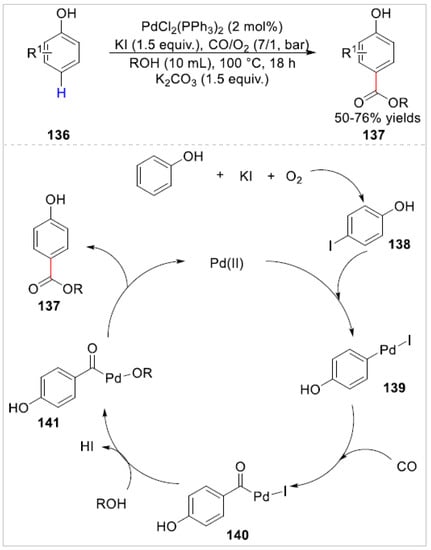
Scheme 22.
Pd(II)-catalyzed para-selective carbonylation of phenols.
Later, a gold-catalyzed highly chemo- and regioselective C–H bond functionalization of free phenols 142 with haloalkynes 143 was developed at room temperature by Hashmi’s group [55]. A series of the products of para-C–H functionalization instead of OH-additions 144 were obtained in good to excellent regio-selectivity and diastereoselectivity, which was attributed to the catalyst and the base-free condition in this reaction. When the preferred position was blocked, the ortho-C–H functionalization of free phenols was feasible. A preliminary mechanistic study suggested that the gold catalyst directly coordinated the chloroalkyne carbon, which formed the more stable π-activated alkyne 145/vinylcation 146. Then, the C-4 position of free phenols attacked the highly electrophilic alkyne instead of O-H insertion to generate β-haloalkenes 144 (Scheme 23).
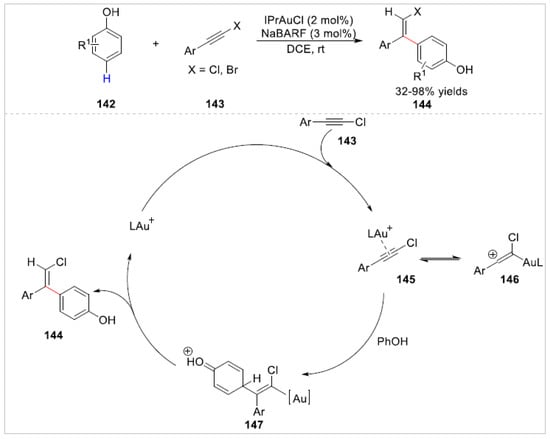
Scheme 23.
Au(I)-catalyzed para-C–H bond functionalization of phenols with haloalkynes.
In 2021, the Liu group [56] reported a regiospecific and site-selective C–H allylation of free phenols 148 with vinyldiazo compounds 149 catalyzed by In(OTf)3. A series of phenol-allylation products instead of OH-additions 151 were obtained with 50–99% yields (Scheme 24). The reactions of aryl substituted vinyldiazoacetates with phenols provided the para-C–H allylation products, but alkyl-substituted vinyldiazoacetates transformed the ortho-selective-C–H allylation products under the standard catalytic conditions. Based on controlled experiments, the authors proposed that this allylation reaction involved a carbene intermediate 150, which was generated during the reaction process by In(OTf)3 releasing of N2.
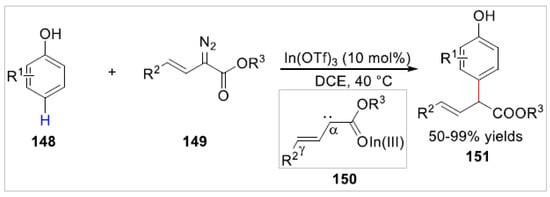
Scheme 24.
In(III)-catalyzed para-C–H allylation of phenols with vinyldiazo compounds.
4.2. Electrochemical C–H Bond Functionalization
Waldvogel’s group [57] reported an efficient electrochemical synthesis by dehydrogenative coupling of free 2,6- or 2,5-substituted phenols under metal catalyst- and oxidant-free conditions in 2019. A variety of the desired 4,4′-biphenols 154 were obtained with 10–77% yields by anodic dehydrogenative cross- and homo-coupling in an undivided beaker-type cell equipped with two boron-doped diamond (BDD) electrodes at a constant current of 5.7 mA/cm2, using Bu3NMeO3SOMe as the electrolyte in 1,1,1,3,3,3-hexafluoropropan-2-ol with 0–6 vol.% water (Scheme 25). A preliminary mechanistic study suggested that this reaction involved a radical intermediate produced by the anodizing of the phenol at the anode, which was subsequently trapped by the phenolic component.
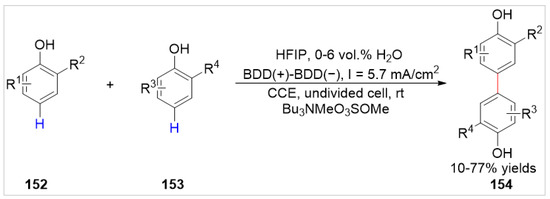
Scheme 25.
Electrochemical C–H functionalization of 2,6- or 2,5-substituted phenols.
In 2022, Banerjee and co-workers [58] developed an efficient electrochemical approach to the Ritter-type reaction at the C(sp2)–H of free phenols 155 in the presence of acetonitrile 156 for the direct synthesis of paracetamol 157 under exogenous oxidant- and catalyst-free conditions. The amination reaction was conducted in an undivided cell equipped with a graphite anode and a nickel cathode at a constant current of 6.0 mA, using n-Bu4NPF6 as the electrolyte at room temperature (Scheme 26). The substrate range of this reaction was mainly free phenol derivatives, and the naphthols (α and β) and benzyl cyanide were not compatible with these reaction conditions. Based on the results of this reaction, the electrochemical C–H amination reaction was proposed to initiate via anodic oxidation of the phenol to generate carbocation 158, which underwent classical Ritter steps in the presence of acetonitrile 156 to deliver the desired product 157.
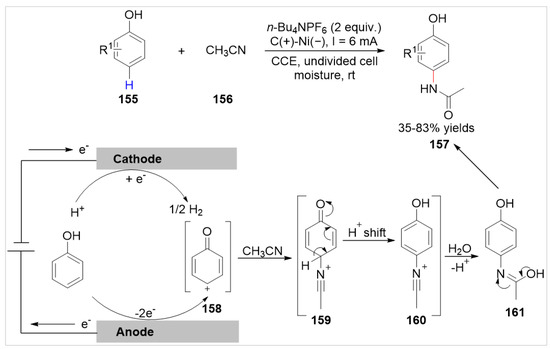
Scheme 26.
Electrochemical Ritter-type C–H amination of phenols with acetonitrile.
Recently, the Banerjee group [59] further developed an electrochemical sulfinylation of free phenols 162 with sulfides 163 under catalyst- and oxidant-free conditions. The aromatic sulfoxides 164 were obtained with 38–65% yields in an undivided cell equipped with a graphite anode and a nickel cathode at a constant current of 15 mA, using n-Bu4NPF6 as the electrolyte at room temperature (Scheme 27). The phenols bearing the electron-withdrawing groups such as nitro, ester, and aldehyde, were unsuccessfully employed as the reaction substrates under standard conditions. Control experiments suggested that the electrochemical sulfonated reaction was proposed to initiate via the anodic oxidation of phenol to generate carbocation, which was then attacked by the nucleophilic sulfide to generate the desired product.
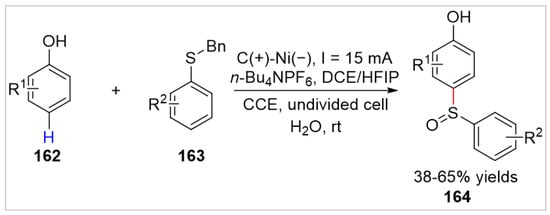
Scheme 27.
Electrochemical sulfinylation of phenols with sulfides.
4.3. Others
In 2020, Procter and co-workers [60] revealed an efficient transition-metal free C–H/C–H-type cross-coupling of free phenols 166 with benzothiophenes 165 to form C2/C3 arylated benzothiophenes 167 with 34–95% yields in trifluoroacetic acid using TFAA as an activator. A variety of substrates, including C2-substituted benzothiophene S-oxides and C3-substituted benzothiophene S-oxides, were successfully used in the arylated reaction to realize the C–H bond functionalization of free phenols. However, when using 4-methoxyphenol as the substrate, the ortho-difunctionalization of 4-methoxyphenol was observed to give the corresponding product in 85% yield. Based on controlled experiments, the authors proposed that this reaction was initiated by reaction of the benzothiophene S-oxides with TFAA to give the activated sulfoxides 168, which could engage directly at the para-position of phenol to generate the corresponding products by a vinylogous Pummerer-type mechanism (Scheme 28).
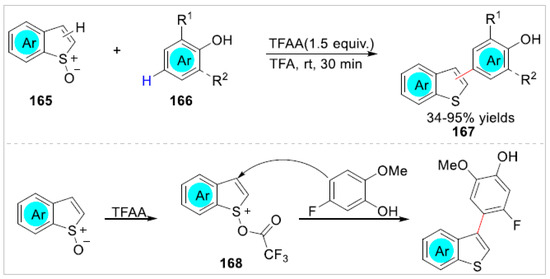
Scheme 28.
TFAA-promoted arylated reaction of phenols with benzothiophenes.
Later, a triflic acid-catalyzed chemo- and site-selective C–H bond functionalization of free phenols 170 with 1,3-dienes 169 was realized by Liu et al. [61] in 2020. A series of para-selective allyl phenols 171 were obtained with 40–88% yields under mild conditions in a site-selective manner. However, the ortho-selective allylic alkylation of phenols with 1,3-dienes were observed under standard conditions in this transformation, and the ortho-C–H bond allylic alkylation product was obtained with 40% yield when the p-cresol was employed as the substrate to react with diene. The authors proposed that this HOTf-catalyzed allylic alkylation involved an ion-pair 172 including allylic cation and triflate anion, which was pushed away by the phenolic hydroxyl, owing to its steric hindrance to giving the para-selective product (Scheme 29).
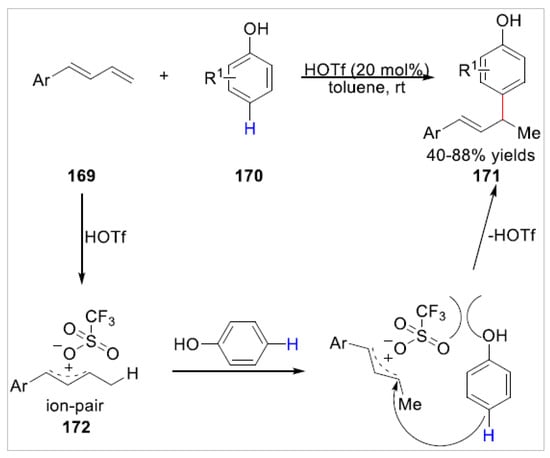
Scheme 29.
Triflic acid-catalyzed para-selective allylic alkylation of phenols with 1,3-dienes.
5. Ortho-Functionalization-Cyclization Process
There were also some examples in which the -OH group took part in further transformations. Due to the 1C,3O-bisnucleophilic reactivity, free phenols have been applied to a variety of [3 + n] cycloaddition reactions with biselectrophilic compounds for the synthesis of various heterocyclic compounds [62,63,64].
In 2019, Tang et al. [65] developed a recyclable nickel-catalyzed C–H/O–H dual functionalization of mandelic acids 173 with free phenols 174. Various of 3-aryl benzofuran-2((3H)-ones 175 were obtained with 31–97% yields under solvent-free conditions. The authors proposed a possible pathway for dual activation of phenols (Scheme 30). Intermediate 176 was generated from mandelic acid in the presence of Ni(OTf)2. Then, an inter-molecular Friedel-Crafts alkylation of 176 with phenol took place to give intermediate 177, which underwent intramolecular esterification to give the corresponding product 175. This method can be enlarged and applied to synthesize antioxidant Irganox HP-136 in an excellent yield.
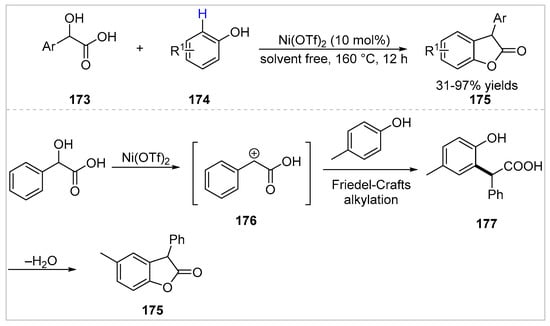
Scheme 30.
Nickel-catalyzed C–H/O–H dual functionalization of phenols with mandelic acids.
In 2020, Li et al. [66] developed a base promoted diastereoselective [3 + 3] cycloaddition reaction of 2-arylideneindan-1,3-diones 178 with free β-naphthols 179. Various functionalized pentacyclic indeno[1,2-b]chromen-(4bH)-ones 180 were synthesized with 47–99% yields under mild conditions (Scheme 31). Splenocyte cells were treated with the synthesized compounds, and cell viability was determined by CCK-8 assay. These tested compounds showed selective inhibitive activity against ConA-induced T-cell proliferation.
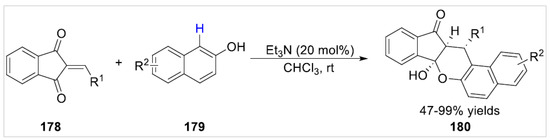
Scheme 31.
Base promoted diastereoselective [3 + 3] cycloaddition reaction of 2-arylideneindan-1,3-diones with 2-naphthols.
Subsequently, the Ren group [67] reported a silver(I)-mediated cascade reaction of 2-(1-alkynyl)-2-alken-1-ones 181 with free 2-naphthols 182. A series of ortho-functionalization-cyclization 1,2-dihydronaphtho[2,1-b]furans 183 were obtained with 53–84% yields (Scheme 32). The controlled experiments indicated that the silver trifluoroacetate first coordinated with the triple bond of 2-(phenylethynyl)cyclohex-2-en-1-one to give the incomplex 184, which then formed furan intermediate 185. The naphthalen-2-ol attacked the intermediate 185 to afford furan compound 186 and regenerated silver trifluoroacetate. In the presence of AgTFA and oxygen, the furan ring in compound 186 was further oxidized to 2-buten-1,4-dione intermediate 187. Finally, the 1,2-dihydronaphtho[2,1-b]furan 183 was obtained through a highly regio- and diastereoselective oxa-Michael addition.
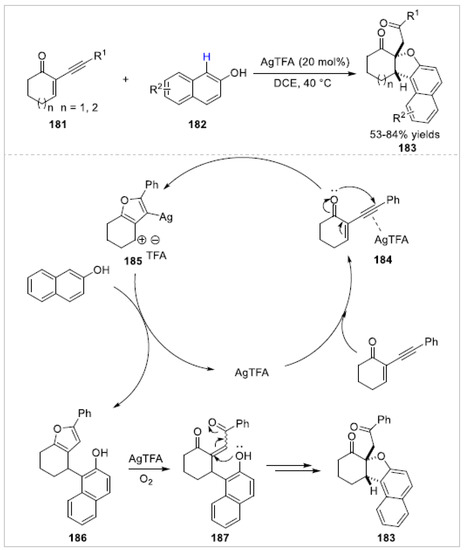
Scheme 32.
Silver(I)-mediated cascade reaction of 2-(1-alkynyl)-2-alken-1-ones with 2-naphthols.
In addition, there were some reactions in which C–H functionalization outside the original phenol ring. For example, in 2019, Yao group [68] reported a palladium-catalyzed remote 1,n-arylamination of unactivated terminal alkenes with aryl iodides and arylamines. In 2020, Wu’s group [69] developed a quadruple C–H activation coupled to hydro functionalization and C–H silylation/borylation based on weakly coordinated palladium catalyst. The mechanisms of both reactions involved the coordination of phenol with palladium and activation of C–H bond outside the original phenol ring.
6. Conclusions
This review has described recent progress in regioselective C–H bond functionalization of free phenols, which are readily available chemical feedstocks in organic synthesis. We have identified a number of methodologies which have been made to overcome longstanding challenges of selectivity in the field of the C–H activation of free phenols without directing groups. These include the ortho-, meta-, and para- selective C–H functionalization of free phenols, which have been successfully achieved by transition-metal catalysis such as Cu, Co, Bi, Pd, Ag, Au and In, electrocatalysis, photocatalysis, and so on, leading to the productions of many useful functionalized phenols.
Although significant progress has been made, there are still some challenges to the regioselective C–H bond functionalization of free phenols, including as below: (1) there are few reaction types, especially the meta- selective C–H functionalization of free phenols, which remains an important area for future development; (2) how to make proper use of electrocatalysis or photocatalysis to avoid the use of a lot of oxidants and chemical reagents, which will be more efficient and environmentally friendly in organic synthesis; (3) the asymmetric C–H bond functionalization of free phenols is rarely developed, which is a very interesting area of research. With regard to these challenges, we hope that the explanation of the regioselective C–H bond functionalization of free phenols in this review will provide useful guidance for further development methods for C–H activation of free phenols to proceed in efficient, simple, mild and environmentally friendly conditions.
Author Contributions
Y.L. wrote the Abstract, Introduction, C2-Functionalization, and C3-Functionalization. J.S. wrote C4-Functionalization, Ortho-Functionalization-Cyclization Process, and the Conclusion. Y.H. and Z.L. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge financial support from the Scientific Research Foundation for High-level Talents of Anhui University of Science and Technology (Grant No. 13220021).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chem. Int. Ed. 2011, 50, 586–621. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Ackermann, L. Cobalt-Catalyzed Direct Arylation and Benzylation by C–H/C–O Cleavage with Sulfamates, Carbamates, and Phosphates. Angew. Chem. Int. Ed. 2012, 51, 8251–8254. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.; Rio, D.D.; Clifford, M.N. Bioavailability of dietary flavonoids and phenolic compounds. Mol. Asp. Med. 2010, 31, 446–467. [Google Scholar] [CrossRef] [PubMed]
- Tyman, J.H.P. Synthetic and Natural Phenols; Studies in Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 2004; Volume 52. [Google Scholar]
- Demchenko, A.P.; Tang, K.-C.; Chou, P.-T. Excited-state proton coupled charge transfer modulated by molecular structure and media polarization. Chem. Soc. Rev. 2013, 42, 1379–1408. [Google Scholar] [CrossRef]
- Fan, H.; Peng, J.; Hamann, M.T.; Hu, J.-F. Lamellarins and Related Pyrrole-Derived Alkaloids from Marine Organisms. Chem. Rev. 2008, 108, 264–287. [Google Scholar] [CrossRef]
- Lee, D.-H.; Kwon, K.-H.; Yi, C.S. Dehydrative C–H Alkylation and Alkenylation of Phenols with Alcohols: Expedient Synthesis for Substituted Phenols and Benzofurans. J. Am. Chem. Soc. 2012, 134, 7325–7328. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-J.; Li, C.-J.; Zeng, H. -Y. Dearomatization-Rearomatization Strategy for ortho-Selective Alkylation of Phenols with Primary Alcohols. Angew. Chem. Int. Ed. 2021, 60, 4043–4048. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Naveen, T.; Maji, A.; Manna, S.; Maiti, D. Palladium-Catalyzed Synthesis of Benzofurans and Coumarins from Phenols and Olefins. Angew. Chem. Int. Ed. 2013, 52, 12669–12673. [Google Scholar] [CrossRef]
- Gaster, E.; Vainer, Y.; Regev, A.; Narute, S.; Sudheendran, K.; Werbeloff, A.; Shalit, H.; Pappo, D. Significant Enhancement in the Efficiency and Selectivity of Iron-Catalyzed Oxidative Cross-Coupling of Phenols by Fluoroalcohols. Angew. Chem. Int. Ed. 2015, 54, 4198–4202. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Lumb, J.-P. Phenol-Directed C-H Functionalization. ACS Catal. 2019, 9, 521–555. [Google Scholar] [CrossRef]
- Xu, X.; Luo, J.F. Transition Metal-Catalyzed Directing-Group-Assisted C–H Activation of Phenols. Chem. Sus. Chem. 2019, 12, 4601–4616. [Google Scholar] [CrossRef]
- Mamari, H.H.; Štefane, B.; Žugelj, H.B. Metal-Catalyzed C–H Bond Functionalization of Phenol Derivatives. Tetrahedron 2020, 76, 130925. [Google Scholar] [CrossRef]
- Youn, S.W.; Cho, C.-G. Transition-metal-catalyzed ortho-selective C–H functionalization reactions of free phenols. Org. Biomol. Chem. 2021, 19, 5028–5047. [Google Scholar] [CrossRef] [PubMed]
- Bashir, M.A.; Wei, J.; Wang, H.F.; Zhong, F.R.; Zhai, H.B. Recent advances in catalytic oxidative reactions of phenols and naphthalenols. Org. Chem. Front. 2022, 9, 5395–5413. [Google Scholar] [CrossRef]
- Truong, T.; Daugulis, O. Divergent reaction pathways for phenol arylation by arynes: Synthesis of helicenes and 2-arylphenols. Chem. Sci. 2013, 4, 531–535. [Google Scholar] [CrossRef]
- Yang, J.-F.; Wang, R.-H.; Wang, Y.-X.; Yao, W.-W.; Liu, Q.-S.; Ye, M. LigandAccelerated Direct C−H Arylation of BINOL: A Rapid One-Step Synthesis of Racemic 3,3′ -Diaryl BINOLs. Angew. Chem. Int. Ed. 2016, 55, 14116–14120. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.-L.; Shao, N.-Q.; Zhang, J.; Jia, R.-P.; Wang, D.-H. Cu(II)-Catalyzed ortho-Selective Aminomethylation of Phenols. J. Am. Chem. Soc. 2017, 139, 12390–12393. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Chandna, N.; Dubey, P.; Singh, A.K.; Jain, N. GO–Cu7S4 catalyzed ortho-aminomethylation of phenol derivatives with N,N-dimethylbenzylamines: Site-selective oxidative CDC. Chem. Commun. 2018, 54, 7511–7514. [Google Scholar] [CrossRef]
- Yu, C.J.; Patureau, F.W. Cu-catalyzed cross-dehydrogenative ortho-aminomethylation of Phenols. Angew. Chem. Int. Ed. 2018, 57, 11807–11811. [Google Scholar] [CrossRef]
- Xie, J.; Chen, M.; Peng, L.-L.; Wu, J.Q.; Zhou, Q.; Zhou, C.S.; Xiong, B.Q.; Liu, Y. Facile preparation of Cu(II)-modified nitrogen-rich covalent organic polymer for cross-dehydrogenative ortho-aminomethylation of phenols. Catal. Commun. 2021, 159, 106348. [Google Scholar] [CrossRef]
- Ma, B.; Tang, Z.Q.; Zhang, J.L.; Liu, L. Copper-catalysed ortho-selective C–H bond functionalization of phenols and naphthols with a-aryl-a-diazoesters. Chem. Commun. 2020, 56, 9485–9488. [Google Scholar] [CrossRef]
- Wang, H.; Xie, Y.; Li, Y.L.; Yang, Y.; Long, J.L.; Zhang, H. Copper-Catalyzed Tandem Cross-Coupling/Annulation of Phenols with Ketoximes through Dual C H Functionalization: Synthesis of Substituted 2-Aryl-1H-indoles. Asian J. Org. Chem. 2021, 10, 1382–1385. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, F.; Wang, L.-L.; Guo, J.; Xu, Y.-Q.; Chen, Z.-S.; Ji, K.G. Cu(II)-Catalyzed C2-site functionalization of p-aminophenols: An approach for selective cross-dehydrogenative aminations. Org. Chem. Front. 2022, 9, 1010–1015. [Google Scholar] [CrossRef]
- Rostami, A.; Khakyzadeh, V.; Zolfigol, M.A.; Rostami, A. Co(II)-catalyzed regioselective clean and smooth synthesis of 2-(aryl/alkylthio)phenols via sp2 C-H bond activation. Mol. Catal. 2018, 452, 260–263. [Google Scholar] [CrossRef]
- Khaef, S.; Rostami, A.; Khakyzadeh, V.; Zolfigol, M.A.; Taherpour, A.A.; Yarie, M. Regioselective Ortho-C-H sulfenylation of free phenols catalyzed by Co(II)-immobilized on silica-coated magnetic nanoparticles. Mol. Catal. 2020, 484, 110772. [Google Scholar] [CrossRef]
- Reiss, H.; Shalit, H.; Vershinin, V.; More, N.Y.; Forckosh, H.; Pappo, D. Cobalt(II)[salen]-Catalyzed Selective Aerobic Oxidative CrossCoupling between Electron-Rich Phenols and 2-Naphthols. J. Org. Chem. 2019, 84, 7950–7960. [Google Scholar] [CrossRef]
- Gao, H.H.; Zha, Z.G.; Zhang, Z.L.; Ma, H.Y.; Wang, Z.Y. A simple and efficient approach to realize difunctionalization of arylketones with malonate esters via an electrochemical oxidation. Chem. Commun. 2014, 50, 5034–5036. [Google Scholar] [CrossRef]
- Li, Y.N.; Gao, H.H.; Zhang, Z.L.; Qian, P.; Bi, M.X.; Zha, Z.G.; Wang, Z.Y. Electrochemical synthesis of α-enaminones from aryl ketones. Chem. Commun. 2016, 52, 8600–8603. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-N.; Wang, B.; Huang, Y.-K.; Hu, J.-S.; Sun, J.-N. Recent advances in metal catalyst- and oxidant-free electrochemical C-H bond functionalization of nitrogen-containing heterocycles. Front. Chem. 2022, 10, 967501. [Google Scholar] [CrossRef]
- Lian, F.; Xu, K.; Zeng, C.-C. The Synergism of Sequential Paired Electrosynthesis with Halogen Bonding Activation for the Cyclization of Organochlorides with Olefins. Sci. China Chem. 2023, 66, 540–547. [Google Scholar] [CrossRef]
- Wang, H.M.; Gao, X.L.; Lv, Z.C.; Abdelilah, T.; Lei, A.W. Recent Advances in Oxidative R1-H/R2-H Cross-Coupling with Hydrogen Evolution via Photo-/Electrochemistry. Chem. Rev. 2019, 119, 6769–6787. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.A.; Niu, L.B.; Wang, S.C.; Liu, J.M.; Lei, A.W. Visible Light-Induced C(sp3)-H Oxidative Arylation with Heteroarenes. Org. Lett. 2019, 21, 2441–2444. [Google Scholar] [CrossRef]
- Lips, S.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Regioselective Metal- and Reagent-Free Arylation of Benzothiophenes by Dehydrogenative Electrosynthesis. Angew. Chem. Int. Ed. 2018, 130, 13509–13513. [Google Scholar] [CrossRef]
- Yue, Y.Y.; Chao, J.L.; Wang, Z.Y.; Yang, Y.; Ye, Y.Q.; Sun, C.Y.; Guo, X.H.; Liu, J.M. Electrooxidative double C–H/C–H coupling of phenols with 3-phenylbenzothiophenes: Facile access to benzothiophene derivatives. Org. Biomol. Chem. 2021, 19, 7156–7160. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, S.Y.; Liu, Y.C.; Cong, H.J.; Lei, A.W. Electrochemical Oxidative C-H Amination of Phenols: Access to Triarylamine Derivatives. Angew. Chem. Int. Ed. 2018, 57, 4737–4741. [Google Scholar] [CrossRef]
- Feng, P.J.; Ma, G.J.; Chen, X.G.; Wu, X.; Lin, L.; Liu, P.; Chen, T.F. Electro-oxidative and Regioselective C-H Azolation of Phenol and Aniline Drivatives. Angew. Chem. Int. Ed. 2019, 58, 8400–8404. [Google Scholar] [CrossRef]
- Wang, Q.-Q.; Jiang, Y.-Y.; Zeng, C.C.; Sun, B.G. Electrocatalytic Synthesis of Non-Symmetric Biphenols Mediated by Tri(p-bromophenyl)amine: Selective Oxidative Cross-Coupling of Different Phenols and Naphthols. Chin. J. Chem. 2019, 37, 352–358. [Google Scholar] [CrossRef]
- Röckl, J.L.; Schollmeyer, D.; Franke, R.; Waldvogel, S.R. Dehydrogenative Anodic C-C Coupling of Phenols Bearing ElectronWithdrawing Groups. Angew. Chem. Int. Ed. 2020, 59, 315–319. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, F.J.; Yan, K.L.; Feng, W.F.; Sun, X.J.; Yang, J.J.; Wen, J.W. Electrochemical-In-Situ-Oxidative Sulfonylation of Phenols with Sulfinic Acids as an Access to Sulfonylated Hydroquinones. Adv. Synth. Catal. 2021, 363, 3485–3490. [Google Scholar] [CrossRef]
- Zhu, D.-L.; Jiang, S.; Young, D.J.; Wu, Q.; Li, H.-Y.; Li, H.-X. Visible-light-driven C(sp2)–H arylation of phenols with arylbromides enabled by electron donor–acceptor excitation. Chem. Commun. 2022, 58, 3637–3640. [Google Scholar] [CrossRef]
- Wu, J.Z.; Kozlowski, M.C. Visible-Light-Induced Oxidative Coupling of Phenols and Alkenylphenols with a Recyclable, Solid Photocatalyst. Org. Lett. 2023, 25, 907–911. [Google Scholar] [CrossRef]
- Wang, G.Q.; Gao, L.Z.; Chen, H.; Liu, X.T.; Cao, J.; Chen, S.D.; Cheng, X.; Li, S.H. Chemoselective Borane-Catalyzed Hydroarylation of 1,3-Dienes with Phenols. Angew. Chem. Int. Ed. 2019, 58, 1694–1699. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.M.; Huang, J.; Lu, C.H.; Jiang, H.F.; Huang, L.B. B(C6F5)3-Catalyzed Hydroarylation of Terminal Alkynes with Phenols. Adv. Synth. Catal. 2021, 363, 3962–3967. [Google Scholar] [CrossRef]
- Wang, X.Y.; Wang, S.C.; Gao, Y.M.; Sun, H.; Liang, X.; Bu, F.X.; Abdelilah, T.; Lei, A.W. Oxidant-Induced Azolation of Electron-Rich Phenol Derivatives. Org. Lett. 2020, 22, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.Q.; Li, D.D.; Yue, Y.D.; Peng, D.; Liu, L. Brønsted acid catalysed chemo- and ortho-selective aminomethylation of phenol. Org. Biomol. Chem. 2021, 19, 5777–5781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-H.; Wang, B.; Ding, Y.; Loh, T.-K.; Tian, J.-S. Aqueous C–H aminomethylation of phenols by iodine catalysis. Chem. Commun. 2023, 59, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Bedford, R.B.; Limmert, M.E. Catalytic Intermolecular Ortho-Arylation of Phenols. J. Org. Chem. 2003, 68, 8669–8682. [Google Scholar] [CrossRef]
- Huang, C.H.; Chattopadhyay, B.; Gevorgyan, V. Silanol: A Traceless Directing Group for Pd-Catalyzed o-Alkenylation of Phenols. J. Am. Chem. Soc. 2011, 133, 12406–12409. [Google Scholar] [CrossRef]
- Cazorla, C.; De Vries, T.S.; Vedejs, E. P-Directed Borylation of Phenols. Org. Lett. 2013, 15, 984–987. [Google Scholar] [CrossRef]
- Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A.F.; Yu, J.-Q. Pd(II)-Catalyzed ortho- or meta-C–H Olefination of Phenol Derivatives. J. Am. Chem. Soc. 2013, 135, 7567–7571. [Google Scholar] [CrossRef]
- Hua, Y.D.; Asgari, P.; Avullala, T.; Jeon, J. Catalytic Reductive ortho-C–H Silylation of Phenols with Traceless, Versatile Acetal Directing Groups and Synthetic Applications of Dioxasilines. J. Am. Chem. Soc. 2016, 138, 7982–7991. [Google Scholar] [CrossRef]
- Senior, A.; Ruffell, K.; Ball, L.T. meta-Selective C–H arylation of phenols via regiodiversion of electrophilic aromatic substitution. Nat. Chem. 2023, 15, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, V.V.; Bhanage, B.M. Palladium-Catalyzed Aerobic Oxidative Carbonylation of C-H Bond of Phenol for the Synthesis p-hydroxybenzoate. Eur. J. Org. Chem. 2018, 22, 2877–2881. [Google Scholar] [CrossRef]
- Adak, T.; Schulmeister, J.; Dietl, M.C.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Gold-Catalyzed Highly Chemo- and Regioselective C-H Bond Functionalization of Phenols with Haloalkynes. Eur. J. Org. Chem. 2019, 24, 3867–3876. [Google Scholar] [CrossRef]
- Zhao, D.; Luo, J.Y.; Liu, L.; Liu, Y.Y. Regiospecific and site-selective C–H allylation of phenols with vinyldiazo compounds catalyzed by In(III). Org. Chem. Front. 2021, 8, 6252–6258. [Google Scholar] [CrossRef]
- Dahms, B.; Kohlpaintner, P.J.; Wiebe, A.; Breinbauer, R.; Schollmeyer, D.; Waldvogel, S.R. Selective Formation of 4,4’-Biphenols by Anodic Dehydrogenative Cross- and Homo-Coupling Reaction. Chem. Eur. J. 2019, 25, 2713–2716. [Google Scholar] [CrossRef]
- Taily, I.M.; Saha, D.; Banerjee, P. Direct Synthesis of Paracetamol via Site-Selective Electrochemical Ritter-type C−H Amination of Phenol. Org. Lett. 2022, 24, 2310–2314. [Google Scholar] [CrossRef]
- Kumar, R.; Taily, I.M.; Banerjee, P. Electrochemical sulfinylation of phenols with sulfides: A metal- and oxidant-free cross-coupling for the synthesis of aromatic sulfoxides. Chem. Commun 2023, 59, 310–313. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Biremond, T.; Perry, G.J.P.; Procter, D.J. Para-coupling of phenols with C2/C3-substituted benzothiophene S-oxides. Tetrahedron 2020, 76, 131315. [Google Scholar] [CrossRef]
- Liu, Z.L.; Li, G.H.; Yao, T.F.; Zhang, J.L.; Liu, L. Triflic Acid-Catalyzed Chemo- and Site-Selective C H Bond Functionalization of Phenols with 1,3-Dienes. Adv. Synth. Catal. 2021, 363, 2740–2745. [Google Scholar] [CrossRef]
- Li, G.T.; Li, Z.K.; Gu, Q.; You, S.L. Asymmetric Synthesis of 4-Aryl-3,4-dihydrocoumarins by N-Heterocyclic Carbene Catalyzed Annulation of Phenols with Enals. Org. Lett. 2017, 19, 1318–1321. [Google Scholar] [CrossRef]
- Narute, S.; Pappo, D. Iron Phosphate Catalyzed Asymmetric Cross-Dehydrogenative Coupling of 2-Naphthols with β-Ketoesters. Org. Lett. 2017, 19, 2917–2920. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.; Srivastava, A.; Yadav, D.; Singh, M.S. Copper-Catalyzed One-Pot Cross-Dehydrogenative Thienannulation: Chemoselective Access to Naphtho[2,1-b]thiophene-4,5-diones and Subsequent Transformation to Benzo[a]thieno[3,2-c]phenazines. J. Org. Chem. 2018, 83, 2173–2181. [Google Scholar] [CrossRef]
- Tang, Z.; Tong, Z.; Xu, Z.H.; Au, C.T.; Qiu, R.H.; Yin, S.F. Recyclable nickel-catalyzed C–H/O–H dual functionalization of phenols with mandelic acids for the synthesis of 3-aryl benzofuran-2(3H)-ones under solvent-free conditions. Green Chem. 2019, 21, 2015–2022. [Google Scholar] [CrossRef]
- Li, N.; Tu, L.; Cheng, G.G.; Sa, H.L.; Li, Z.H.; Feng, T.; Zheng, Y.S.; Liu, J.K. Diastereoselective [3 + 3] cycloaddition reaction of 2-arylideneindan-1,3-diones with β-naphthols: Efficient assemble of immunosuppressive pentacyclic chromanes. Tetrahedron Lett. 2020, 61, 151579–151582. [Google Scholar] [CrossRef]
- Li, Z.H.; Peng, J.Y.; He, C.L.; Xu, J.F.; Ren, H.J. Silver(I)-mediated cascade reaction of 2-(1-alkynyl)-2-alken-1-ones with 2-naphthols. Org. Lett. 2020, 22, 5768–5772. [Google Scholar] [CrossRef]
- Han, C.H.; Fu, Z.Y.; Guo, S.J.; Fang, X.X.; Lin, A.J.; Yao, H.Q. Palladium-catalyzed remote 1,n-arylamination of unactivated terminal alkenes. ACS Catal. 2019, 9, 4196–4202. [Google Scholar] [CrossRef]
- Tang, B.C.; Lin, W.X.; Chen, X.L.; He, C.; Ma, J.T.; Wu, Y.D.; Lan, Y.; Wu, A.X. Quadruple C-H activation coupled to hydrofunctionalization and C-H silylation/borylation enabled by weakly coordinated palladium catalyst. Nat. Commun. 2020, 11, 5662–5673. [Google Scholar] [CrossRef]

































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).